
Abstract
The mucosal immune system and the microbiota in the intestinal tract have recently been shown to play a key role in the pathogenesis of inflammatory bowel disease (IBD). Both of these can be influenced by food. Thus, we propose dietary intervention as a therapeutic option for IBD. In this review, we discuss the interaction of the intestinal mucosal immune system and the intestinal microbiota in the context of IBD. In addition, we discuss the impact of food components on immune responses in IBD. Finally, we address the current evidence of how this interaction (i.e., immune system–microbiota) can be modulated by food components, pre/probiotics, and fecal microbiota transplantation (FMT) and how these approaches can support intestinal homeostasis. By gathering the vast amount of literature available on the impact of food on IBD, we aim to distinguish between scientifically sound data and theories, which have not been included in this review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The role of the immune system and the intestinal microbiota in IBD
IBD is a group of chronic disorders characterized by relapsing inflammation of the gastrointestinal (GI) tract. The most common entities are Crohn’s Disease (CD) and Ulcerative Colitis (UC). While CD is characterized by a segmental transmural inflammation and granulomatous lesions of the whole GI tract, inflammation in UC is mostly restricted to the colon and the rectum. Both incidence and prevalence of IBD are currently increasing worldwide [1].
Although the etiology of IBD has not yet been fully elucidated, more than a hundred genetic risk loci have been identified so far. These genetic studies point toward an important role of the immune system and its interaction with the microbiota in the development of IBD. The majority of these loci are indeed linked to loss of barrier function (e.g., GNA12), to host interaction with bacteria (e.g., NOD2), or to immune pathways (e.g., IL-23) implicated in the control of cytokines, such as interleukin (IL)-17 and IL-22 [2]. Notably, these cytokines are profoundly involved in the regulation of the intestinal mucosal barrier homeostasis and are triggered by signals from the intestinal microbiota [3,4,5]. This reinforces the idea that a deregulated immune response to intestinal microbiota is involved in IBD pathogenesis [6].
In this section, we will refresh the knowledge on the intestinal microbiota, how it interacts with the immune system, and furthermore the role of the key cytokines (IL-23, IL-17, IL-22, IL-10), which result from this interaction in the context of IBD.
Intestinal microbiota
The human body coexists with around 1013 bacterial organisms, which colonize different sides of the body, such as the intestine and the skin [7]. In the intestine, we refer to these bacteria as the intestinal microbiota. The dominant phyla are the Firmicutes (64%) that mostly comprise bacteria of the Clostridia class, followed by Bacteroidetes, which account for approximately 23% of the intestinal flora, Proteobacteria, and Actinobacteria [8, 9]. The physiological metabolic activity of the microbiota is beneficial for the host. The intestinal microbiota promotes, for example, the fermentation of indigestible carbohydrates to, e.g., short-chain fatty acids (SCFA) and absorption of complex lipids or vitamin synthesis [10,11,12].
An altered microbiota has been associated with IBD, and a variety of extra-intestinal diseases, such as obesity, type 1 and type 2 diabetes, asthma, and liver disease highlighting the systemic impact of the intestinal flora [13, 14]. Sequencing analysis of intestinal biopsies or fecal samples of IBD patients showed a different composition of mucosa-associated and fecal microbiota compared to healthy controls with a reduced microbial biodiversity as a hallmark of this disease [15]. More detailed analysis revealed that certain bacteria species protect from IBD while others appear to be pathogenic.
Various studies demonstrated a marked reduction of Firmicutes within the microbiota of patients with IBD [16,17,18,19]. Within the Firmicutes-phylum, in particular, the species Faecalibacterium prausnitzii was shown to be protective in chemically induced colitis in mice. This observation has been further sustained by human twin studies [20, 21].
Bacterial species from the Proteobacteria phylum have mainly been reported as holding pathogenic potential in the development of IBD. For example, Escherichia coli (E. coli) was shown to be more abundant in patients with active IBD [22]. In particular, adhesive-invasive E. coli (AIEC) is enriched in the inflamed ileal mucosa of IBD patients and might trigger local inflammation [23, 24].
Moreover, sulfate-reducing bacteria (SRB), most of which belong to the Deltaproteobacteria class (Proteobacteria phylum), are increased in IBD and have been shown to promote a chemically induced colitis in mice underlining a possible role in IBD pathogenesis [25, 26]. However, also species from other bacterial phyla, such as Bacteroidetes, have shown pathogenic properties. For example, in a mouse model that is susceptible for colitis, the fecal microbiota was characterized by expansion of the Prevotella genus and the TM7 phylum [27].
Interaction between the immune system and the microbiota
The immune system has several sensors, which detect the microbiota and its changes and consequently can promote intestinal inflammation or maintain homeostasis. For the maintenance of a physiological interaction, a highly regulated network of microbial sensor systems has evolved.
The mucosal immune system recognizes the conserved microbe-associated molecular patterns (MAMP) via pattern recognition receptors (PRR), such as Toll-like receptors (TLR) and nucleotide oligomerization domain (NOD)-like receptors (NLR). These specialized receptors respond to a variety of triggers, including unmethylated double-stranded DNA, single-stranded RNA, and bacterial products [28]. Ligand-receptor interaction induces the recruitment of signaling adaptors, such as myeloid differentiation primary response gene 88 (MyD88) and TIR domain-containing adapter-inducing interferon-β (TRIF). Toll-like receptors are expressed by various cells of the immune system including dendritic cells (DCs), macrophages, T cells and B cells, and also by epithelial cells [29].
A variety of bacterial products have been reported as TLR ligands, such as lipopolysaccharide (LPS) from gram-negative bacteria, peptidoglycans from gram-positive bacteria, or flagellin derived from flagellated bacterial species [30]. Sensing of these bacterial components by individual TLRs triggers pro-inflammatory cytokines, such as TNF-α and can induce also type I interferons [31]. The NLR receptor family features a wide range of functions. Besides bacterial products, NLRs also sense cell stress signals, for example, via reactive oxygen species (ROS) or via oxidized mitochondrial DNA [32, 33]. Downstream signals of NLR activation have also been shown to induce production of anti-microbial peptides as well as goblet cell mucus production thereby directly promoting mucosal barrier defense [34, 35].
Bacterial products are also sensed by inflammasomes, which are multiprotein complexes assembling with the adapter protein apoptosis-associated speck-like protein (ASC) and procaspase-1. In response to PAMPs, activated caspase-1 cleaves the pro-forms of the pro-inflammatory cytokines IL-1β and IL-18 [28]. Impaired microbial sensing has been shown to impact the microbial composition and to promote inflammation: global deletion of inflammasome components, such as ASC or NLRP6, a member of the NLR family, associates with dysbiosis with microbial shifts, and a subsequent colitis [27].
Resulting cytokine: IL-23 mediates the interaction between the microbiota and the immune system
Intestinal immune responses and intestinal tissue homeostasis are regulated by cytokines, which are produced when the abovementioned sensory systems are engaged. Within these cytokines IL-23 has a prominent role in the intestine, specifically in IBD as highlighted by genome-wide association studies (GWAS) [2, 36]. Interleukin -23 is an important downstream signal of TLR-activation, and IL-23 signaling has been shown in mouse and human studies to promote IBD [37,38,39]. Important triggers of IL-23 production are bacterial products, such as flagellin. Flagellin is produced by bacteria belonging to the clostridia class of the Firmicutes phylum, but also by members of the Proteobacteria phylum, such as Escherichia coli. Interestingly, patients with CD show flagellin-specific circulating antibodies in contrast to healthy controls underlining the immunogenicity of flagellin in the context of intestinal inflammation [40]. One of the cellular sources of IL-23 are dendritic cells located in the lamina propria of the epithelial layer, which produce IL-23 upon sensing bacterial-derived flagellin via TLR-5 [41]. Recently, also CXCR1+ CXCR2+ neutrophils located in the inflamed colonic mucosa of patients with IBD have been identified as a cellular source of IL-23 [42]. IL-23 promotes an immune response characterized by the induction of other two key cytokines IL-17 and IL-22. These cytokines are secreted by both innate and adaptive immune cells, and they will be further discussed in the following paragraphs.
Resulting cytokine: IL-17 and its protective and pathogenic properties
In particular, the role of IL-17 in IBD became the subject of controversial debate. Among the IL-17 family members, the biological role of IL-17A and IL-17F are best characterized. On the one hand, in line with the GWAS, IL-23-dependent secretion of IL-17 was shown to promote T cell-mediated colitis [43, 44]. Recently, also IL-17A derived from group 3 innate lymphoid cells (ILC) in response to IL-23 has been shown to mediate intestinal inflammation in an innate colitis model [45]. Likewise, IL-17F has been reported to play a pathogenic role in colitis development with either redundant or different functions compared to IL-17A [43, 46]. On the other hand, there are studies reporting a protective effect of IL-17A in various colitis models, such as dextran sulfate sodium (DSS)-induced or T cell-mediated colitis models [47, 48]. Insights in mechanisms underlying the protective capacity of IL-17A have recently been delineated: during DSS-colitis IL-17A-dependent regulation of tight junctions can limit excessive intestinal permeability and therefore promote barrier integrity. Of note, in this model, IL-17A was produced by γδ T cells in an IL-23-independent manner [49]. Beside ILC, also TH17 cells can produce IL-17. These cells are a subtype of effector T helper cells, which express high levels of IL-17A and IL-17F and which are highly enriched in the inflamed mucosal tissue of IBD patients [50, 51]. Interestingly, some commensal bacteria, such as segmented filamentous bacteria (SFB), can induce TH17 cells [52].
In conclusion, these findings suggest that IL-17A can have dual functions, which might depend on the cellular source, on the environment, such as the microbiota, and on the type of intestinal pathology. Of note, IL-17A blockade using an antibody was not effective in IBD in humans [53]. However, by understanding the context-dependent effect of IL-17A, one may be able to identify a subset of IBD patients, which might benefit from this therapy.
Resulting cytokine: IL-22 and its dual role at mucosal surfaces
IL-22 acts also downstream of IL-23 and plays a central role in promoting barrier homeostasis by regulating genes encoding molecules associated with tissue repair or antimicrobial properties [54]. Both adaptive immune cells and cells of the innate immune system can produce IL-22, and early studies demonstrated that adaptive and innate IL-22 protects from IBD [55]. In particular, IL-22 produced by innate lymphoid cells is essential for intestinal barrier control in a murine model for enteropathogenic E. coli infection [56]. Innate -derived IL-22 is also crucial in controlling SFB [57].
However, as shown for the IL-17 family, IL-22 can also mediate pathogenic activity in intestinal inflammation. For example, T cell-derived IL-22 promotes colitis upon transfer of memory cells into Rag1-deficient recipients [58]. Furthermore, IL-22-mediated antimicrobial response suppresses the commensal flora and indirectly favors the colonization of enteropathogenic bacteria, such as Salmonella enterica [59].
In conclusion, IL-22 has pathogenic and protective properties. Therefore, a tight control is essential. This control is exerted on at least two levels. First, as mentioned above, the production of IL-22 is regulated by IL-23. Second, there is an endogenous inhibitor of IL-22, namely IL-22 binding protein (IL-22BP). IL-22BP binds and blocks IL-22 activity. IL-22BP can be produced by DCs, granulocytes, and T cells [60,61,62]. Interestingly, T cell-derived IL-22BP was shown to be upregulated in IBD and to promote IBD via blockade of the beneficial effects of IL-22 [62].
Resulting cytokines: IL-10 and immune tolerance
Intestinal homeostasis is the result of a fine-tuned balance of factors promoting immune responses and signals mediating immune tolerance. A key player in limiting immune responses is the anti-inflammatory cytokine IL-10 [63]. Polymorphisms leading to a loss of function of the IL-10 receptor or IL-10 itself are associated with early-onset IBD reinforcing the theory of a pathogenic loss of immune tolerance to intestinal microbes [64, 65]. Microbial products can drive IL-10 expression by macrophages and DCs. By various mechanisms, such as limiting antigen presentation and the expression of costimulatory molecules or by down regulation of pro-inflammatory cytokines, IL-10 mediates immune tolerance [66, 67]. For example, germ-free mice colonized with the human commensal species Bacteroides fragilis show induction of IL-10-producing regulatory T (Treg) cells via lipopolysaccharide A (LPA) [68]. In line with these results, administration of LPA itself has been shown to be protective in different experimental models of colitis [68, 69]. Various clostridia species have also been reported to induce IL-10-producing Treg cells, and oral inoculation of these bacterial strains mediates amelioration of chemically induced colitis underlining the protective role of IL-10 in these models [70, 71]. Also, signaling of the NLR family member NOD2 induces IL-10 expression, and NOD2 mutations are associated with Crohn’s disease [72, 73].
Overall, IL-10 is a crucial player in controlling inflammatory responses in the intestine. In particular, understanding the microbiota-derived signals promoting Treg cell responses will help to optimize flora-based therapeutic approaches for IBD.
Impact of food components on the intestinal microbiota and on the local immune response in IBD
Epidemiological studies support the role of environmental factors in pathogenesis of IBD. In particular, the association between dietary intake and incidence of IBD points to food components as a potential pathogenic agent. Indeed, certain food components, such as polyunsaturated fatty acids (PUFA), omega-6 fatty acids, and meat seem to predispose for IBD, whereas fibers and fruits or vegetables were associated with a decreased risk of CD or CU, respectively [74].
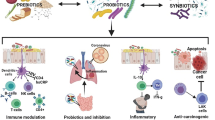
While there is little evidence of food components having a direct impact on IBD, they do impact in an indirect fashion (Fig. 1). Food components essentially influence the intestinal homeostasis by modulation of the microbiota and subsequent activation of the immune system. Indeed, a controlled-feeding study of ten donors showed that the microbiota responds by compositional change within 24 h of initiating a low fat/high fiber or high fat/low fiber diet [75]. Wu et al. showed that different diets are associated with specific bacterial clusters of the microbiota. Diets based on protein and animal fat compared to carbohydrates were associated with two enterotypes according to the levels of Bacteroides and Prevotella, both of which are genera, which are involved in colitis development [75]. These observations are also supported by animal studies showing that a high fat diet was associated with an increase in Firmicutes and a reduction of species from the Bacteroides phylum [76]. Of note, high-fat diets were also shown to promote chemically induced colitis in mice and were associated with expression of inflammatory cytokines and a decreased frequency of regulatory T cells [77]. On the other hand, high-fat diet was also reported to decrease intestinal TH17 cells [78]. Collectively, these data indicate that basic dietary components, such as fat and carbohydrates, can specifically modulate the microbial composition of the intestine.
Further, important modulators of the microbiota are dietary fibers. A diet low in fibers (considered the Western diet), results in a loss of the microbial diversity as observed in IBD patients [79]. Fermentable fiber is metabolized by the intestinal microbiota, mainly by clostridia species, to SCFA, such as acetate and butyrate. SCFA are strong modulators of the intestinal immune system, and their effects are coordinated by specific metabolic programs of immune cells [80].

Schematic overview of interactions between diet, microbiota and immune system in the intestine
The intestinal microbiota is also involved in the synthesis and the metabolism of various vitamins, such as B-group vitamins and vitamin K. On the other hand, vitamins have been shown to modulate the microbiota, and to act directly on the immune system; vitamin A and its down-stream metabolite retinoic acid (RA) are involved in multiple immune processes, such as Treg homeostasis and IgA antibody production [81, 82].
Deciphering the role of food components in modulating the intestinal immune system might help to use this potential to expand treatment options for IBD. In this section, we summarize current evidence of how certain food components (carbohydrates, dietary fiber, fat, and vitamins) modulate the intestinal immune system.
Carbohydrates, dietary fiber, and fat
Several studies showed a positive, but still not consistent, association between high carbohydrate intake and an increased risk of IBD [74]. Based on sequencing analysis, carbohydrate consumption significantly correlates with abundance of the Firmicutes phylum and is associated with the Prevotella-enterotype [75]. Currently, there is a lack of studies investigating the specific effect of carbohydrates or withdrawal of them from diet in IBD. Thus, the exact role of carbohydrates in IBD is currently not clear.
However, there are several studies pointing toward a role of fat intake in IBD, although this role is still controversial. A positive association has been reported between high intake of saturated fats, mono- and polyunsaturated fatty acids (PUFA), and an increased risk of CD, whereas high total fats correlated with an increased CU risk [74]. Likewise, high-fat diets promote chemically induced colitis in mice and are associated with a decrease in the frequency of regulatory T cells [77]. Furthermore, milk-derived saturated fat can increase the availability of organic sulfur by pathological conjugation of hepatic bile acids, thereby promoting sulfate-reducing bacteria and an associated colitis in a mouse model [83].
In contrast to the pathogenic properties, beneficial effects of fat have also been reported. In particular, omega-3 (3-n) PUFA might play a protective role in IBD [84]. Indeed, during chronic colitis, transgenic mice synthesizing (n-3) PUFA de novo show reduced polarization of TH17 cells. A possible mechanism, by which PUFA control TH17 cell pathogenicity is the modulation of cholesterol-derived ligands for RORγt, the major transcription factor of TH17 cells and group 3 ILC [85].
Finally, dietary fiber has been linked to IBD. In a meta-analysis, high intake of dietary fiber was associated with a decreased risk of IBD [74]. As mentioned above, the metabolism of fiber requires the intestinal microbiota. Fermentable fiber is metabolized mainly by clostridia species, to SCFA, such as acetate and butyrate, which seem to exert the protective properties in IBD. Interestingly, fecal SCFA levels were shown to be decreased in IBD patients [86]. Furthermore, a protective role of SCFA is supported by animal studies. Indeed, orally administered SCFA protected mice from T cell-mediated colitis or intestinal lesions induced by cytostatic agents [87, 88]. Several potential mechanisms explain these protective properties. First, SCFA show direct anti-inflammatory activity. SCFA can, for example, suppress NF-κB and trigger expression of the Vitamin D receptor (VDR, see below) [89, 90]. Furthermore, SCFA were shown to modulate oxidative stress and to promote barrier function by enhancing tight junction assembly [91]. Of note, the beneficial effects of fiber on intestinal inflammation also involve activation of the NLRP3 inflammasome by hyperpolarization of the epithelial membrane [92].
Second, SCFA have indirect protective properties, which are mediated via regulatory CD4+ T cells. Indeed, butyrate and propionate were shown to promote extra-thymic Treg generation [93]. Furthermore, interaction of SCFA and G-protein-coupled receptors (GPRs), such as GPR43, has been shown to mediate enhancement of frequency and function of Tregs [88]. Besides this interaction with GPR, also the distinct metabolic programs of immune cells explain the effect of SCFA on regulatory CD4+ T cell generation. Compared to effector T cells, such as TH17 cells, which are reliant on glucose metabolism, the generation of regulatory CD4+ T cells has been shown to be selectively dependent on lipid oxygenation [80]. The increased expression of genes regulating fatty acid oxidation enables Tregs to effectively use SCFA as an energy source [94]. Of note, the protective effect of colonization by Clostridia from clusters IV and XIVa with induction of regulatory T cells might also be mediated by SCFA but underlying mechanisms still remain to be revealed [95].
In conclusion, several studies underline the central role of metabolites in controlling the balance of inflammatory and regulatory immune responses based on immune cell-specific metabolic programs.
Vitamins D and A
Vitamin deficiency is a common feature of IBD. Malnutrition due to decreased food intake, increased requirements, malabsorption, and maldigestion as possible causes are being discussed. A recent retrospective study in IBD patients showed high frequencies of Vitamins D and A deficiency [96]. There is now growing evidence for immunological action of these vitamins, for both adaptive and innate immunity.
Indeed, several studies demonstrated that vitamin D enhances the development of TH2 cells, a T helper cell subset characterized by the production of the anti-inflammatory cytokines IL-4, IL-5, and IL-13. Furthermore, the suppression of TH17 cells and the induction of IL-10-producing Treg cells in response to vitamin D have been reported [97, 98]. Interestingly, TLR triggering has been shown to promote Vitamin D receptor expression and downstream signals including the production of anti-microbial peptides [99].
Furthermore, Vitamin D might influence IBD via interactions with the microbiota. Indeed, absence of the Vitamin D receptor can directly impact the intestinal microbiota with an increase in Bacteroides, which was associated with an increased susceptibility toward DSS-induced colitis [90].
In line with the mouse studies, there are also human data supporting the role of Vitamin D in IBD. First, VDR gene polymorphisms are linked to increased IBD susceptibility [90]. Second, in a prospective study, low Vitamin D levels were associated with increased risk of surgery and hospitalization of IBD patients, and oral Vitamin D supplementation was associated with a reduced risk of relapse in Chrohn’s disease (CD) in a randomized double-blind placebo-controlled study [100, 101]. In line with these results, a recent cross-sectional study also showed a positive correlation of Vitamin D deficiency and IBD disease activity [102].
Vitamin A is a group of nutritional organic compounds. The exact role of Vitamin A in IBD is currently unknown. However, there are some data linking vitamin A-derived metabolites to the immune response in the intestine. One example is retinol, which is one principal compound of the vitamin A group that is metabolized to retinoic acid (RA) by the sequential action of retinol dehydrogenase (RDH) and retinaldehyde dehydrogenase (RALDH) enzymes [103]. Dendritic cells are the major source of RA in the intestine that is produced in response to inflammatory signals. Local RA induces the conversion of naïve CD4+ T cells into Treg cells and inhibits the differentiation of TH17 cells thereby promoting immune tolerance [82, 104]. Furthermore, in a recent study, RA has been shown to induce IL-22 secretion by γδ T cells and innate lymphoid cells with protective effects in murine colitis models. By directly targeting the IL-22 promotor, RA enhanced production of antimicrobial peptides [105]. Thus, RA might impact IBD via IL-17 and IL-22-mediated effects.
Therapeutic opportunities: pro/prebiotics, diet intervention, and fecal microbiota therapy
To maintain homeostasis, a fine-tuned balance between immune tolerance and immune activation toward the intestinal microbiota is critical. Regarding IBD patients, observations have shown that this critical balance is deregulated favoring chronic immune activation. There are several possible ways to reset this balance with the aim of treating IBD patients. The first is to target the microbiota directly by microbiota transplantation; the second is to target the microbiota indirectly by diet (Fig. 1). In addition, there are also several therapies, which aim to modify the microbiota composition, such as the use of probiotic bacteria, and different diets have already been tested in IBD.
This section will provide a summary of the most recent evidence relating to the role of probiotics, prebiotics, diets, and fecal microbiota transplantation in the treatment of IBD.
Probiotics
An option to influence the composition of intestinal microbiota is the use of probiotics. Specific microorganisms are deemed probiotic based on their scientifically proven effect on the promotion of health or prevention and treatment of a specific disease. Probiotics, such as Escherichia coli Nissle 1917 and VSL#3 seem to be efficient in UC as outlined below. However, there is not sufficient evidence to support the use of probiotics in daily clinical practice in Crohn’s disease [106].
Patients with CU in remission treated with Escherichia coli Nissle 1917 compared to immunosuppressive therapy with 5-aminosalicylic acid showed a comparable clinical relapse rate [107]. Likewise, this probiotic shows the same efficacy in maintaining remission compared to 5-aminosalicylic acid [108]. Furthermore, VSL#3, which contains four strains of Lactobacillus was shown to induce a significantly higher rate of remission compared to placebo in mild-to-moderately active UC [109].
Probiotics show signs of exerting their effects via at least two mechanisms. First, probiotics are able to enhance mucosal barrier function. They were shown to promote secretion of IgA and mucins [110] and tight junction function [111, 112]. Second, probiotics interact with the local immune system. An example is that some probiotic strains can enhance regulatory T cell responses, downregulate the expression of pro-inflammatory cytokines, such as TNF-α and IFN-γ [113, 114], and induce the production of a variety of anti-inflammatory cytokines, e.g., IL-10 and TGF-β [115].
Finally, probiotics impact the IL-22-mediated antimicrobial response. It has been shown that tryptophan is metabolized by a subset of commensal Lactobacilli, a bacterial genus contained in many probiotic products, to indole-3-aldehyde (IAld). IAld induced AhR-dependent IL-22 production by ILC and mediated both antifungal activity and amelioration of chemically induced colitis [116].
Prebiotics
Prebiotics are indigestible substances, mostly carbohydrates that can be metabolized by beneficial bacterial strains hence enhancing their activity. Prebiotics in clinical use include fructo-oligosaccharides (FOSs), inulin, galacto-oligosaccharides (GOSs), and soybean oligosaccharides that stimulate mainly bifidobacteria species [117]. There is not much clinical evidence of therapeutic efficacy of prebiotics in IBD.
Patients with active CD were randomized and treated with fructo-oligosaccharides (FOSs) versus placebo with no significant difference in clinical response [118]. In a more recent study, De Preter et al. observed a significantly reduced clinical disease activity after 4 weeks of treatment with a prebiotic combination of oligofructose and inulin [119]. In this trial, an increase of fecal butyrate was shown after prebiotic treatment. This SCFA has been shown to induce T cell apoptosis and suppression of IFN-γ-mediated inflammation in colonic epithelial cells [120]. Therefore, the observed impact of prebiotics is probably linked to promotion of probiotic effects including bifidobacteria-mediated increase in IL-10 expression [121].
Diets: elemental and polymeric
Exclusive enteral nutrition (EEN) is a dietary therapy comprised of a period of 6–8 weeks of exclusive liquid feeding with either elemental or polymeric formulae. Elemental diets contain all essential nutrients and protein in the form of free amino acids whereas polymeric formulae comprise non-hydrolyzed proteins and long-chain triglycerides. These formulae were developed to improve nutrient absorption and to limit mucosal antigen exposure.
Interestingly, several studies demonstrated effectiveness of EEN in the treatment of CD equivalent to the use of corticosteroids mainly in pediatric patients [122, 123]. In contrast, results of trial testing the efficacy in treatment of adults, were apparently controversial [124, 125].
One possible reason for the discrepancy between the efficacy of ENT in pediatric and adult patients might be the higher chance of non-compliance by adult patients, which in turn might be related to an increased intolerance of the formula observed in adult patients.
The exact mechanism of EEN in IBD is unknown, but several possibilities are being explored. First, the effects might be linked to the intestinal microbiota. During the dietary treatment, profound modification of all predominant bacterial groups were observed with a decrease in microbiota diversity [126]. Interestingly, a reduction of the protective commensal Faecalibacterium prausnitzii was reported, which might help to explain a therapy failure in some cases [127]. Second, results from in vitro experiments showed that the components of EEN reduced inflammatory responses, which is supported by a human study showing reduced mucosal expression of IL-1β and IFN-γ after EEN treatment [128,129,130]. Finally, also promotion of barrier function has been reported: EEN based on a polymeric formula prevented TNF-α-mediated tight junction dysfunction in vitro [131].
Overall, conflicting results regarding the clinical effectiveness and the loss of protective microbiota species point out that a more defined formula or more stringent diet is needed to effectively use this therapy for adult IBD patients. For pediatric IBD patients, EEN is an established therapy [132]. Even though it has proven effective, understanding the molecular and cellular mechanisms by which EEN promotes its beneficial effects will help to improve the outcome of pediatric IBD by creating optimized formulae.
Fecal microbiota therapy
The idea of modulating dysbiosis to treat IBD gave rise to the proposal of direct microbiota transplantation with a donor microbiota as a potential therapeutic option (Fig. 1). Fecal microbiota therapy has been successfully used in treating refractory Clostridium difficile infection, a colitis triggered by antibiotic-induced dysbiosis [133]. Low frequencies of adverse events have been reported [134]. Fecal microbiota transfer has also shown therapeutic efficacy in inflammatory diseases that are associated with an altered microbiota, such as diabetes and obesity [135].
Data evaluating FMT in IBD still present some limitations. Initial evidence of the role of microbiota transplant in IBD was shown in case reports as having lead to a relief of symptoms in disease refractory to sulfasalazine [136, 137]. In a following retrospective case series, FMT has been shown to induce remission after 4 months of treatment [138]. Further cases have been reported. The proven efficacy of FMT in adults with IBD and a recent phase 1 trial showed a clinical response rate of 67% and clinical remission of 33% of children and young adults after FMT, respectively [139,140,141]. A more recent study reported the efficacy in CU patients based on intensive dosing of FMT derived from multiple donors [142]. The intense application of 40 FMT during 8 weeks and pooling stool of up to seven donors was the factor which distinguished this trial from recent studies that failed to induce remission, although fecal microbiota changes could be observed [143, 144].
The regimen used by Paramsothy et al. [142] showed a higher microbiota diversity after the fecal transplant, which was also observed 8 weeks after the treatment. However, the invasive and intense form of FMT application is challenging for the patient, and it is still not clear how long patients need to be treated in order to maintain remission.
The role of FMT in the treatment of IBD remains to be fully defined, and more controlled clinical trials are necessary to critically assess its therapeutic effect. It is currently not clear whether antibiotic pretreatment is necessary and which method of administration is the most effective. In the abovementioned study of Pramsothy et al., [142] specific taxa were associated with a positive outcome and others with a negative outcome. These results might be helpful to improve the design of the FMT. Standardized microbiota samples that can be ingested in the form of a pill might display a future option.
Future perspectives
Taken together, the abovementioned studies point to the fact that the local immune response to intestinal microbiota could be one key mechanism involved in IBD pathogenesis. Numerous risk loci and independent observations described above point to a model where deregulation of the interaction between the immune system and the microbiota favors chronic intestinal inflammation. However, further insights are needed to better decipher the molecular and cellular mechanisms and to develop future therapeutic approaches without the current limitations, such as lack of long-term remission. We hope to have highlighted the need for a unified, scientifically sound basis for future development in this growing field. The identification of food components with an anti-inflammatory function can help to further control IBD and serve as a proof of concept for an approach to induce and maintain remission that could potentially be extended to other immune-mediated diseases. Keeping in mind that each patient’s immune system and microbiota may respond to food components differently, individual dietary approaches will be essential.
References
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142:46–54.e42 quiz e30
Khor B, Gardet A, Xavier RJ (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474:307–317
Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR (2008) Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349
Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, Flavell RA (2013) IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol 190:5306–5312
Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Horst RT, Jansen T, Jacobs L, Bonder MJ, Kurilshikov A, Fu J, Joosten LA, Zhernakova A, Huttenhower C, Wijmenga C, Netea MG, Xavier RJ (2016) Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 167:1897
Major G, Spiller R (2014) Irritable bowel syndrome, inflammatory bowel disease and the microbiome. Curr Opin Endocrinol Diabetes Obes 21:15–21
Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308:1635–1638
Sartor RB (2008) Microbial influences in inflammatory bowel diseases. Gastroenterology 134:577–594
Rios-Covian D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilan CG, Salazar N (2016) Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol 7:185
Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Boren J, Oresic M, Backhed F (2010) The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res 51:1101–1112
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI (2011) Human nutrition, the gut microbiome and the immune system. Nature 474:327–336
Kelsen JR, Wu GD (2012) The gut microbiota, environment and diseases of modern society. Gut Microbes 3:374–382
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, Zhou J, Ni S, Liu L, Pons N, Batto JM, Kennedy SP, Leonard P, Yuan C, Ding W, Chen Y, Hu X, Zheng B, Qian G, Xu W, Ehrlich SD, Zheng S, Li L (2014) Alterations of the human gut microbiome in liver cirrhosis. Nature 513:59–64
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J (2006) Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55:205–211
Sha S, Xu B, Wang X, Zhang Y, Wang H, Kong X, Zhu H, Wu K (2013) The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn Microbiol Infect Dis 75:245–251
Andoh A, Kuzuoka H, Tsujikawa T, Nakamura S, Hirai F, Suzuki Y, Matsui T, Fujiyama Y, Matsumoto T (2012) Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J Gastroenterol 47:1298–1307
Aomatsu T, Imaeda H, Fujimoto T, Takahashi K, Yoden A, Tamai H, Fujiyama Y, Andoh A (2012) Terminal restriction fragment length polymorphism analysis of the gut microbiota profiles of pediatric patients with inflammatory bowel disease. Digestion 86:129–135
Strober W (2013) The impact of the gut microbiome on mucosal inflammation. Trends Immunol 34:423–430
Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L, Tysk C, Jansson JK (2009) Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis 15:653–660
Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P, Langella P (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105:16731–16736
Lopez-Siles M, Martinez-Medina M, Busquets D, Sabat-Mir M, Duncan SH, Flint HJ, Aldeguer X, Garcia-Gil LJ (2014) Mucosa-associated Faecalibacterium prausnitzii and Escherichia coli co-abundance can distinguish irritable bowel syndrome and inflammatory bowel disease phenotypes. Int J Med Microbiol 304:464–475
Barnich N, Darfeuille-Michaud A (2007) Adherent-invasive Escherichia coli and Crohn’s disease. Curr Opin Gastroenterol 23:16–20
Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, Berg D, Schukken Y, Scherl E, Simpson KW (2007) Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J 1:403–418
Rowan F, Docherty NG, Murphy M, Murphy B, Calvin Coffey J, O’Connell PR (2010) Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum 53:1530–1536
Fava F, Danese S (2011) Intestinal microbiota in inflammatory bowel disease: friend of foe? World J Gastroenterol 17:557–566
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757
Ignacio A, Morales CI, Camara NO, Almeida RR (2016) Innate sensing of the gut microbiota: modulation of inflammatory and autoimmune diseases. Front Immunol 7:54
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Uematsu S, Akira S (2008) Toll-like receptors (TLRs) and their ligands. Handb Exp Pharmacol. https://doi.org/10.1007/978-3-540-72167-3_1:1-20
Kumar H, Kawai T, Akira S (2009) Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388:621–625
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414
Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, Elinav E, Finlay BB, Flavell RA (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156:1045–1059
Uehara A, Fujimoto Y, Fukase K, Takada H (2007) Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol 44:3100–3111
Kim SW, Kim ES, Moon CM, Park JJ, Kim TI, Kim WH, Cheon JH (2011) Genetic polymorphisms of IL-23R and IL-17A and novel insights into their associations with inflammatory bowel disease. Gut 60:1527–1536
Pidasheva S, Trifari S, Phillips A, Hackney JA, Ma Y, Smith A, Sohn SJ, Spits H, Little RD, Behrens TW, Honigberg L, Ghilardi N, Clark HF (2011) Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One 6:e25038
Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A (2006) IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med 203:2485–2494
Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ (2006) Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med 203:2473–2483
Duck LW, Walter MR, Novak J, Kelly D, Tomasi M, Cong Y, Elson CO (2007) Isolation of flagellated bacteria implicated in Crohn’s disease. Inflamm Bowel Dis 13:1191–1201
Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, Littman DR, Pamer EG (2012) Interleukin 23 production by intestinal CD103(+) CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36:276–287
Kvedaraite E, Lourda M, Idestrom M, Chen P, Olsson-Akefeldt S, Forkel M, Gavhed D, Lindforss U, Mjosberg J, Henter JI, Svensson M (2015) Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut. https://doi.org/10.1136/gutjnl-2014-309014
Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF (2009) RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136:257–267
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D (2006) IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 116:1310–1316
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464:1371–1375
Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C (2008) Regulation of inflammatory responses by IL-17F. J Exp Med 205:1063–1075
Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y (2004) Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol 110:55–62
O'Connor W Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA (2009) A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol 10:603–609
Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, Li X, Cua DJ (2015) Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43:727–738
Monk JM, Jia Q, Callaway E, Weeks B, Alaniz RC, McMurray DN, Chapkin RS (2012) Th17 cell accumulation is decreased during chronic experimental colitis by (n-3) PUFA in Fat-1 mice. J Nutr 142:117–124
Galvez J (2014) Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm 2014:928461
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498
Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP (2012) Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61:1693–1700
Sonnenberg GF, Fouser LA, Artis D (2011) Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12:383–390
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA (2008) Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29:947–957
Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D (2011) CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34:122–134
Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, Fu YX, Zhou L (2013) Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39:386–399
Kamanaka M, Huber S, Zenewicz LA, Gagliani N, Rathinam C, O'Connor W Jr, Wan YY, Nakae S, Iwakura Y, Hao L, Flavell RA (2011) Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med 208:1027–1040
Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, Raffatellu M (2014) The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 40:262–273
Martin JC, Beriou G, Heslan M, Chauvin C, Utriainen L, Aumeunier A, Scott CL, Mowat A, Cerovic V, Houston SA, Leboeuf M, Hubert FX, Hemont C, Merad M, Milling S, Josien R (2014) Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol 7:101–113
Martin JC, Beriou G, Heslan M, Bossard C, Jarry A, Abidi A, Hulin P, Menoret S, Thinard R, Anegon I, Jacqueline C, Lardeux B, Halary F, Renauld JC, Bourreille A, Josien R (2016) IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunol 9:539–549
Pelczar P, Witkowski M, Perez LG, Kempski J, Hammel AG, Brockmann L, Kleinschmidt D, Wende S, Haueis C, Bedke T, Witkowski M, Krasemann S, Steurer S, Booth CJ, Busch P, Konig A, Rauch U, Benten D, Izbicki JR, Rosch T, Lohse AW, Strowig T, Gagliani N, Flavell RA, Huber S (2016) A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 354:358–362
Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK (2010) The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol 11:854–861
Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hatscher N, Pfeifer D, Sykora KW, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C (2009) Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361:2033–2045
Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, Pfeifer D, Kreipe H, Pfister ED, Baumann U, Puchalka J, Bohne J, Egritas O, Dalgic B, Kolho KL, Sauerbrey A, Buderus S, Gungor T, Enninger A, Koda YK, Guariso G, Weiss B, Corbacioglu S, Socha P, Uslu N, Metin A, Wahbeh GT, Husain K, Ramadan D, Al-Herz W, Grimbacher B, Sauer M, Sykora KW, Koletzko S, Klein C (2012) Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143:347–355
Paul G, Khare V, Gasche C (2012) Inflamed gut mucosa: downstream of interleukin-10. Eur J Clin Investig 42:95–109
Saraiva M, O'Garra A (2010) The regulation of IL-10 production by immune cells. Nat Rev Immunol 10:170–181
Round JL, Mazmanian SK (2010) Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107:12204–12209
Mazmanian SK, Round JL, Kasper DL (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331:337–341
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500:232–236
Hedl M, Abraham C (2011) Secretory mediators regulate Nod2-induced tolerance in human macrophages. Gastroenterology 140:231–241
Philpott DJ, Girardin SE (2009) Crohn’s disease-associated Nod2 mutants reduce IL10 transcription. Nat Immunol 10:455–457
Hou JK, Abraham B, El-Serag H (2011) Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol 106:563–573
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science 334:105–108
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14
Ma X, Torbenson M, Hamad AR, Soloski MJ, Li Z (2008) High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin Exp Immunol 151:130–138
Garidou L, Pomie C, Klopp P, Waget A, Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S, Dray C, Iacovoni JS, Courtney M, Collet X, Amar J, Servant F, Lelouvier B, Valet P, Eberl G, Fazilleau N, Douin-Echinard V, Heymes C, Burcelin R (2015) The gut microbiota regulates intestinal CD4 T cells expressing RORgammat and controls metabolic disease. Cell Metab 22:100–112
Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL (2016) Diet-induced extinctions in the gut microbiota compound over generations. Nature 529:212–215
Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186:3299–3303
Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, von Andrian UH (2006) Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314:1157–1160
Nolting J, Daniel C, Reuter S, Stuelten C, Li P, Sucov H, Kim BG, Letterio JJ, Kretschmer K, Kim HJ, von Boehmer H (2009) Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. J Exp Med 206:2131–2139
Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487:104–108
Turner D, Shah PS, Steinhart AH, Zlotkin S, Griffiths AM (2011) Maintenance of remission in inflammatory bowel disease using omega-3 fatty acids (fish oil): a systematic review and meta-analyses. Inflamm Bowel Dis 17:336–345
Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK (2015) CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163:1413–1427
Huda-Faujan N, Abdulamir AS, Fatimah AB, Anas OM, Shuhaimi M, Yazid AM, Loong YY (2010) The impact of the level of the intestinal short chain fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J 4:53–58
Ramos MG, Bambirra EA, Nicoli JR, Cara DC, Vieira EC, Alvarez-Leite J (1999) Protection by short-chain fatty acids against 1-beta-D-arabinofuranosylcytosine-induced intestinal lesions in germfree mice. Antimicrob Agents Chemother 43:950–953
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573
Tedelind S, Westberg F, Kjerrulf M, Vidal A (2007) Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J Gastroenterol 13:2826–2832
Wu S, Zhang YG, Lu R, Xia Y, Zhou D, Petrof EO, Claud EC, Chen D, Chang EB, Carmeliet G, Sun J (2015) Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 64:1082–1094
Elamin EE, Masclee AA, Dekker J, Pieters HJ, Jonkers DM (2013) Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J Nutr 143:1872–1881
Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, Ian McKenzie C, Hijikata A, Wong C, Binge L, Thorburn AN, Chevalier N, Ang C, Marino E, Robert R, Offermanns S, Teixeira MM, Moore RJ, Flavell RA, Fagarasan S, Mackay CR (2015) Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun 6:6734
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455
Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC (2015) Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125:194–207
McDermott AJ, Huffnagle GB (2014) The microbiome and regulation of mucosal immunity. Immunology 142:24–31
Alkhouri RH, Hashmi H, Baker RD, Gelfond D, Baker SS (2013) Vitamin and mineral status in patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 56:89–92
Reich KM, Fedorak RN, Madsen K, Kroeker KI (2014) Vitamin D improves inflammatory bowel disease outcomes: basic science and clinical review. World J Gastroenterol 20:4934–4947
Zhang H, Wu H, Liu L, Li H, Shih DQ, Zhang X (2015) 1,25-dihydroxyvitamin D3 regulates the development of chronic colitis by modulating both T helper (Th)1 and Th17 activation. APMIS 123:490–501
Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL (2006) Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773
Jorgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, Bartels LE, Kelsen J, Christensen LA, Dahlerup JF (2010) Clinical trial: vitamin D3 treatment in Crohn’s disease—a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther 32:377–383
Ananthakrishnan AN, Cagan A, Gainer VS, Cai T, Cheng SC, Savova G, Chen P, Szolovits P, Xia Z, De Jager PL, Shaw SY, Churchill S, Karlson EW, Kohane I, Plenge RM, Murphy SN, Liao KP (2013) Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease. Inflamm Bowel Dis 19:1921–1927
Torki M, Gholamrezaei A, Mirbagher L, Danesh M, Kheiri S, Emami MH (2015) Vitamin D deficiency associated with disease activity in patients with inflammatory bowel diseases. Dig Dis Sci 60:3085–3091
Napoli JL (2012) Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta 1821:152–167
Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK (2008) Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 181:2277–2284
Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, Bracken AP, Mills KH (2013) Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210:1117–1124
Butterworth AD, Thomas AG, Akobeng AK (2008) Probiotics for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD006634.pub2:Cd006634
Kruis W, Fric P, Pokrotnieks J, Lukas M, Fixa B, Kascak M, Kamm MA, Weismueller J, Beglinger C, Stolte M, Wolff C, Schulze J (2004) Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 53:1617–1623
Rembacken BJ, Snelling AM, Hawkey PM, Chalmers DM, Axon AT (1999) Non-pathogenic Escherichia coli versus mesalazine for the treatment of ulcerative colitis: a randomised trial. Lancet 354:635–639
Sood A, Midha V, Makharia GK, Ahuja V, Singal D, Goswami P, Tandon RK (2009) The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol 7:1202–1209 1209.e1201
Kabeerdoss J, Devi RS, Mary RR, Prabhavathi D, Vidya R, Mechenro J, Mahendri NV, Pugazhendhi S, Ramakrishna BS (2011) Effect of yoghurt containing Bifidobacterium lactis Bb12(R) on faecal excretion of secretory immunoglobulin A and human beta-defensin 2 in healthy adult volunteers. Nutr J 10:138
Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, Wells JM (2010) Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. Am J Physiol Gastrointest Liver Physiol 298:G851–G859
Kotzampassi K, Giamarellos-Bourboulis EJ (2012) Probiotics for infectious diseases: more drugs, less dietary supplementation. Int J Antimicrob Agents 40:288–296
Zhao HM, Huang XY, Zuo ZQ, Pan QH, Ao MY, Zhou F, Liu HN, Liu ZY, Liu DY (2013) Probiotics increase T regulatory cells and reduce severity of experimental colitis in mice. World J Gastroenterol 19:742–749
Resta-Lenert S, Barrett KE (2006) Probiotics and commensals reverse TNF-alpha- and IFN-gamma-induced dysfunction in human intestinal epithelial cells. Gastroenterology 130:731–746
de Moreno de Leblanc A, Del Carmen S, Zurita-Turk M, Santos Rocha C, van de Guchte M, Azevedo V, Miyoshi A, Leblanc JG (2011) Importance of IL-10 modulation by probiotic microorganisms in gastrointestinal inflammatory diseases. ISRN Gastroenterol 2011:892971
Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39:372–385
Thomas DW, Greer FR (2010) Probiotics and prebiotics in pediatrics. Pediatrics 126:1217–1231
Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL, Kamm MA, Sanderson JD, Knight SC, Forbes A, Stagg AJ, Whelan K, Lindsay JO (2011) Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn’s disease. Gut 60:923–929
De Preter V, Joossens M, Ballet V, Shkedy Z, Rutgeerts P, Vermeire S, Verbeke PK (2013) Metabolic profiling of the impact of oligofructose-enriched inulin in Crohn’s disease patients: a double-blinded randomized controlled trial. Clin Transl Gastroenterol 4:e30
Zimmerman MA, Singh N, Martin PM, Thangaraju M, Ganapathy V, Waller JL, Shi H, Robertson KD, Munn DH, Liu K (2012) Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am J Physiol Gastrointest Liver Physiol 302:G1405–G1415
Lindsay JO, Whelan K, Stagg AJ, Gobin P, Al-Hassi HO, Rayment N, Kamm MA, Knight SC, Forbes A (2006) Clinical, microbiological, and immunological effects of fructo-oligosaccharide in patients with Crohn’s disease. Gut 55:348–355
Gorard DA, Hunt JB, Payne-James JJ, Palmer KR, Rees RG, Clark ML, Farthing MJ, Misiewicz JJ, Silk DB (1993) Initial response and subsequent course of Crohn’s disease treated with elemental diet or prednisolone. Gut 34:1198–1202
Borrelli O, Cordischi L, Cirulli M, Paganelli M, Labalestra V, Uccini S, Russo PM, Cucchiara S (2006) Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: a randomized controlled open-label trial. Clin Gastroenterol Hepatol 4:744–753
Zachos M, Tondeur M, Griffiths AM (2007) Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD000542.pub2:Cd000542
Triantafillidis JK, Vagianos C, Papalois AE (2015) The role of enteral nutrition in patients with inflammatory bowel disease: current aspects. Biomed Res Int 2015:197167
Leach ST, Mitchell HM, Eng WR, Zhang L, Day AS (2008) Sustained modulation of intestinal bacteria by exclusive enteral nutrition used to treat children with Crohn’s disease. Aliment Pharmacol Ther 28:724–733
Gerasimidis K, Bertz M, Hanske L, Junick J, Biskou O, Aguilera M, Garrick V, Russell RK, Blaut M, McGrogan P, Edwards CA (2014) Decline in presumptively protective gut bacterial species and metabolites are paradoxically associated with disease improvement in pediatric Crohn’s disease during enteral nutrition. Inflamm Bowel Dis 20:861–871
Meister D, Bode J, Shand A, Ghosh S (2002) Anti-inflammatory effects of enteral diet components on Crohn’s disease-affected tissues in vitro. Dig Liver Dis 34:430–438
de Jong NS, Leach ST, Day AS (2007) Polymeric formula has direct anti-inflammatory effects on enterocytes in an in vitro model of intestinal inflammation. Dig Dis Sci 52:2029–2036
Fell JM, Paintin M, Arnaud-Battandier F, Beattie RM, Hollis A, Kitching P, Donnet-Hughes A, MacDonald TT, Walker-Smith JA (2000) Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn’s disease. Aliment Pharmacol Ther 14:281–289
Nahidi L, Day AS, Lemberg DA, Leach ST (2012) Differential effects of nutritional and non-nutritional therapies on intestinal barrier function in an in vitro model. J Gastroenterol 47:107–117
Day AS, Lopez RN (2015) Exclusive enteral nutrition in children with Crohn’s disease. World J Gastroenterol 21:6809–6816
van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ (2013) Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368:407–415
Gough E, Shaikh H, Manges AR (2011) Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 53:994–1002
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143:913–916.e917
Bennet JD, Brinkman M (1989) Treatment of ulcerative colitis by implantation of normal colonic flora. Lancet 1:164
Borody TJ, George L, Andrews P, Brandl S, Noonan S, Cole P, Hyland L, Morgan A, Maysey J, Moore-Jones D (1989) Bowel-flora alteration: a potential cure for inflammatory bowel disease and irritable bowel syndrome? Med J Aust 150:604
Borody TJ, Warren EF, Leis S, Surace R, Ashman O (2003) Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol 37:42–47
Anderson JL, Edney RJ, Whelan K (2012) Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther 36:503–516
Kunde S, Pham A, Bonczyk S, Crumb T, Duba M, Conrad H Jr, Cloney D, Kugathasan S (2013) Safety, tolerability, and clinical response after fecal transplantation in children and young adults with ulcerative colitis. J Pediatr Gastroenterol Nutr 56:597–601
Cui B, Li P, Xu L, Zhao Y, Wang H, Peng Z, Xu H, Xiang J, He Z, Zhang T, Nie Y, Wu K, Fan D, Ji G, Zhang F (2015) Step-up fecal microbiota transplantation strategy: a pilot study for steroid-dependent ulcerative colitis. J Transl Med 13:298
Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D, Leong RWL, Connor S, Ng W, Paramsothy R, Xuan W, Lin E, Mitchell HM, Borody TJ (2017) Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet 389:1218–1228
Kump PK, Grochenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM, Deutschmann A, Wenzl HH, Petritsch W, Krejs GJ, Gorkiewicz G, Hogenauer C (2013) Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis 19:2155–2165
Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, Novacek G, Trauner M, Loy A, Berry D (2013) Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol 108:1620–1630
Acknowledgements
This work was supported by the ERC (StG 715271 to N.G. and StG 337215 to S.H.)
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Dietary Control of Immunometabolism - Guest Editors: Joerg Heeren and Ludger Scheja
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Witkowski, M., Witkowski, M., Gagliani, N. et al. Recipe for IBD: can we use food to control inflammatory bowel disease?. Semin Immunopathol 40, 145–156 (2018). https://doi.org/10.1007/s00281-017-0658-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-017-0658-5