
Abstract
Immune-mediated inflammation in the retina is regulated by a combination of anatomical, physiological and immuno-regulatory mechanisms, referred to as the blood–retina barrier (BRB). The BRB is thought to be part of the specialised ocular microenvironment that confers protection or “immune privilege” by deviating or suppressing destructive inflammation. The barrier between the blood circulation and the retina is maintained at two separate anatomical sites. These are the endothelial cells of the inner retinal vasculature and the retinal pigment epithelial cells on Bruch’s membrane between the fenestrated choroidal vessels and the outer retina. The structure and regulation of the tight junctions forming the physical barrier are described. For leukocyte migration across the BRB to occur, changes are needed in both the leukocytes themselves and the cells forming the barrier. We review how the blood–retina barrier is compromised in various inflammatory diseases and discuss the mechanisms controlling leukocyte subset migration into the retina in uveoretinitis in more detail. In particular, we examine the relative roles of selectins and integrins in leukocyte interactions with the vascular endothelium and the pivotal role of chemokines in selective recruitment of leukocyte subsets, triggering adhesion, diapedesis and migration of inflammatory cells into the retinal tissue.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune-mediated inflammation in the retina is regulated by a combination of anatomical, physiological and immuno-regulatory mechanisms, referred to as the blood–retina barrier (BRB). The BRB is thought to be part of the specialised ocular microenvironment that confers protection or “immune privilege” by deviating or suppressing destructive inflammation [120, 121]. These mechanisms are designed to prevent normal immune surveillance and delete or inactivate cells migrating across the BRB to mitigate the effects of deleterious immune responses [15, 99]. Nevertheless, retinal inflammation does occur, and in addition to well-defined inflammatory diseases such as uveoretinitis, immune mechanisms affecting the integrity of the BRB and leukocyte infiltration of the retina are implicated in other ocular diseases such as diabetic retinopathy [76] and age-related macular degeneration (ARMD) [72].
The barrier between the blood circulation and the retina is maintained at two separate anatomical sites. These are the endothelial cells of the inner retinal vasculature and the retinal pigment epithelial cells (RPE) on Bruch’s membrane between the fenestrated choroidal vessels and the outer retina. For leukocyte migration across the BRB to occur, changes are needed in both the leukocytes themselves and the cells forming the barrier [9, 137]. The retina is an extension of the central nervous system (CNS), and as such, many of the molecular structures making up the inner BRB are similar to those found in the vascular blood–brain barrier (BBB) [1]. However, there are also important differences between the tissues, including vascular heterogeneity in the brain and numbers of microglia in the retina.
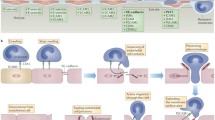
Much research has been carried out into mechanisms controlling inflammatory cell adhesion and extravasation of leukocytes from the circulation into neural tissues, and a paradigm invoking sequential, separate events involving interacting pairs of selectins and their ligands, chemokines and cell adhesion molecules (CAM) has evolved to be one of the central tenets in immunology (Fig. 1) [12]. Less is understood about the process of extravasation, and there is evidence for leukocyte extravasation in the CNS both through intercellular endothelial tight junctions (paracellular route) and for transcellular migration through the endothelial cell itself [45].

Mechanisms involved in leukocyte trafficking across the blood-retina barrier at the endothelium. Initial interactions between leukocyte and endothelium are usually mediated by selectins that induce tethering and rolling (1). If G-protein-coupled receptors on the leukocyte engage appropriate chemokines on surface of endothelium, then the leukocyte may become activated (2), leading to conformational changes in integrin molecules allowing firm adhesion to the endothelium and leukocyte spreading (3). Diapedesis across the endothelium and into the retina can then take place (4). This is triggered by additional chemokine and cytokine signals and gradients and mediated by matrix metalloproteinases secreted by the leukocyte and alterations in the signalling and regulatory molecules of the TJ that control the interaction between the membranous component and the cytoskeleton of the endothelial cell, leading to breakdown of the BRB (5). How inflammatory responses in the retina are initiated or resolved is still controversial. There is evidence that resident and/or infiltrating cells may influence the progression (+Ve signal?) or resolution (−Ve signal?) of the inflammatory response
To date, many studies have been based on neutrophil migration, or undefined mixed leukocyte populations, but recent research, including our own, has revealed that different leukocyte subsets have distinct requirements for endothelial cell interactions and subsequent migration. These requirements may also vary depending on the type of tissue, whether normal surveillance or inflammatory trafficking is involved and if inflammatory on the type of stimulus. Mononuclear cells including monocytes and T lymphocytes are primarily involved in the pathologies observed in various retinal inflammations and will be the focus for this review. Understanding these processes and whether mechanisms really differ between peripheral tissues and ocular or other CNS sites such as the brain is important as every stage of the process, from initial engagement to migration into the tissue, is a potential target for immunotherapy to control retinal damage [53].
Leukocyte adhesion cascade
Adhesion molecules in the leukocyte adhesion cascade
The process of the inflammatory response and how blood leukocytes are recruited from the blood to the tissues has been described as the leukocyte adhesion cascade. Rolling of leukocytes on the luminal endothelial cell wall is usually the first step in the adhesion cascade that culminates in leukocyte extravasation, and the mechanisms underlying the various stages involved has been recently reviewed [82]. Selectins, such as L selectin, expressed on microvilli of most leukocytes and P and E selectin, which may be expressed by activated or inflamed endothelium, mediate both tethering and rolling [119]. These molecules interact with ligands such as P-selectin glycoprotein ligand-1 (PSGL-1), expressed on other leukocytes or endothelium only when correctly glycosylated [89]. This initial tethering and fast rolling allows adherence under flow, and shear stress forces support the adhesive and signalling interactions taking place that then mediate slow rolling. Integrins are also involved in rolling, and α4β1-integrin (VLA4) dependent rolling and LFA-1/ICAM-1 interactions have been shown to be important for firm adhesion and lymphocyte trafficking in the CNS [44, 59]. Leukocyte rolling via low-affinity interactions can then be converted to firm adhesion as chemokine-receptor G-protein dependent activation takes place, resulting in conformational changes in the binding domains of LFA-1 to high-affinity state and in membrane clustering of the integrins giving increased avidity [6, 78]. This leads to firm adhesion and is the prelude to spreading, crawling and migration through the endothelium, either by the paracellular route involving release of endothelial adherens junctional proteins [87, 113, 126] or the transcellular route via small continuous membrane associated structures or vesiculo-vacuolar organelles.
Chemokines in the leukocyte adhesion cascade
Leukocyte migration is controlled to a large extent by members of a family of chemoattractants, the chemokines [109]. The panel of chemokine receptors that a leukocyte carries enables it to respond to chemokine signals either from normal tissue thus controlling immune surveillance or from an inflammatory site. Forty-three human chemokines have now been officially named, and these do not include isoforms, polymorphisms, splice variants and those chemokines encoded by viruses. There are currently 18 chemokine-specific G-protein-coupled receptors [92]. As these chemokines can be allocated to four groups based on the position of 2 N-terminal conserved cysteines, this has been used as the basis for their systematic nomenclature [7, 143]. The two main groups of chemokines are now designated either CCL, where there is no amino acid separating the cysteines, or CXCL, where there is one amino acid between the cysteine molecules.
Chemokines can bind to more than one receptor, and receptors can bind to more than one chemokine. Receptor affinity varies between the ligands but does not necessarily determine the functional potency of the chemokine [32, 130]. In response to ligand binding, the receptors trigger numerous secondary mediators, which initiate functional responses such as cell migration. The resulting functions may differ depending on the cell type and the prevailing microenvironment [77, 111]. The multiple different combinations of chemokines and receptors, expressed either simultaneously or sequentially [13], plus the cell type and the local microenvironment ensure that the chemokine communication system can deliver a precise and accurate communication message to the required cell based on local circumstances, which may be rapidly changing.
Chemokines add additional specificity to the adhesive interactions initiated by the adhesion molecules during the recruitment of leukocytes from the circulation. Chemokines produced within the tissue are transported by transcytosis to the luminal surface of the endothelium. The transport across the endothelium is active and may involve the chemokine binding protein DARC [110]. Once at the luminal surface, the chemokines are displayed by glycosaminoglycans (GAGs) [124] at the tips of endothelial processes [95, 96]. This prevents the desensitisation of leukocyte chemokine receptors by chemokines in the blood stream and focusses the action of the chemokines on leukocyte adhesion and emigration. Chemokines have a GAG-binding site as well as specific receptor binding site [5], and GAG binding has been shown to be important in vivo [104].
Chemokines presented on the endothelium stimulate firm adhesion by increasing integrin affinity and avidity [6, 20, 26, 74] and then the spreading of cells, their migration to the endothelial cell junction [129] and the initiation of leukocyte transmigration [23, 24]. More than one chemokine is likely to be needed for the complete process of leukocyte extravasation with specific chemokines at distinct steps [142]. It is thought that chemokines do not need to form a gradient across the endothelium to enable adherent cells to move from the luminal surface into the tissue as surface immobilised chemokines and a shear force may be sufficient for transendothelial migration [24].
Normal retina and immune surveillance
In normal physiology, one function of the immune system is to recognise and destroy cells that are abnormal (e.g. transformed tumour cells) or that display foreign or non-self antigens (e.g. virus-infected cells). This entails recirculation of relatively long-lived lymphocytes through the blood stream and lymphatics, moving from one lymph node to another and to peripheral inflammatory sites. In contrast, myeloid monocytes and dendritic cells, generated in the bone marrow, recirculate in the blood for only a few days before migrating into the tissues, forming extensive networks of phagocytic antigen presenting cells (APC). Some of these cells differentiate into long lived, highly specialised cells such as the osteoclasts of bone and microglia of the nervous system. In the tissues, APC may be stimulated by ingesting apoptotic tissue cells or by contact with microbes. They then migrate via the lymphatics to local draining lymph nodes where interaction with T and B lymphocytes can occur, and either tolerance to self-antigens or immunity to altered-self or foreign antigens is generated. Immune surveillance must be regulated in vulnerable tissues such as the brain and retina where tissue repair cannot take place, so structures that anatomically separate the central nervous system have evolved. Thus, the BRB may protect the delicate neural tissue from local, damaging inflammation but at the cost of defective peripheral tolerance to retinal antigens. Consequently, the retina remains vulnerable to autoimmune inflammation, increasing the importance of the BRB in excluding, eliminating or suppressing infiltrating inflammatory cells [15, 99].
Structure and function of the blood–retina barrier
The inner BRB formed at the endothelium is supported by perivascular cells such as smooth muscle cells, pericytes or retinal macrophages [53]. Astrocyte foot process contact is known to be essential for formation of intercellular tight junctions (TJ) and maintenance of structural integrity [1]. Other complex interactions between other constituents of the microenvironment surrounding the endothelial cells, such as the basement membrane, nerve endings, microglial cells and the extracellular fluid, are all required for the proper functioning of the barrier. The outer BRB formed at the RPE cell layer is simpler, the barrier being maintained by tight junctions between polarised epithelial cells on Bruch’s membrane. Barrier function develops during embryogenesis, and once formed, is highly selective excluding molecules above 300 kDa. It is also effective in excluding most migrating cells, repopulation of the retina with bone-marrow-derived cells such as microglia taking from 6 to 12 months [4, 132], the rate of haematopoetic-derived cell turnover probably being dependent upon integrity of the RPE cell layer [134].
In normal retina, two distinct molecular structures form the physical BRB; these are the TJ and the adherens junctions (Fig. 1). These closely resemble TJ found in neural tissue in the brain [46, 102]. The TJ are highly organised pericellular structures that appear as multilammellar, usually continuous strands containing several integral membrane proteins that seal adjacent endothelial or epithelial cells, creating distinct membrane domains that restrict soluble and ion transport to apical and basal surfaces and block paracellular passage of macromolecular fluids and cells. The two major components are the claudins and the occludins, which are linked via zonula occludin (ZO) proteins to signalling proteins and the actin cytoskeleton. The junctional adhesion molecules (JAMs) are also located in the TJ, JAM A and C being particularly highly expressed in the RPE consistent with a role in establishing and maintaining cell polarity [33, 90]. Adherens junctions are formed mainly by cadherins (mainly VE cadherin), which interact with catenins to bind the cytoskeleton. PECAM/CD31 expressed by endothelial cells in the junction also binds to the cytoskeleton via catenins and has been implicated in the migration of monocytic myeloid cells [98]. The general structure and regulation of neural tight junctions in retina and brain has been recently reviewed [46, 63].
Leukocyte infiltration of normal retina
Despite an apparently intact BRB, lymphocytes have been shown to infiltrate the normal retina. If both the leukocytes and the endothelium are normal, leukocytes do not cross the blood–retina barrier [135, 137], but if lymphocytes are activated, they are able to initiate a transient breakdown in the BRB, enabling sampling of the retinal environment and possibly further recruitment of inflammatory cells [103, 139]. The mechanisms controlling this sampling or immune surveillance of the retina in the absence of retinal inflammation are not clear, and similar observations have been made in the CNS [65, 66]. Using scanning laser ophthalmoscopy, we have shown that circulating, activated T cells induce changes in the retinal vasculature that allow T cell firm adhesion to venule endothelium without rolling (secondary tethering), inferring a role for chemokine or integrin signalling in T cell capture and subsequent diapedesis in the absence of selectin-mediated rolling. Furthermore, recirculation of as few as 1 × 105 activated cells for 8–16 h is sufficient to allow a permissive endothelium to develop, allowing T cell adhesion and diapedesis. This compares with a minimum of 1 h for diapedesis when both leukocytes and endothelium are activated [138]. Interestingly, retinal antigens have been identified as having chemotactic properties for T cells via chemokine receptors, providing a mechanism for recruitment of lymphocytes to the retina in the absence of inflammation [68, 101]. Whether this occurs in vivo and contributes to induction of autoimmune inflammation remains to be proven but is consistent with the observation that migration of activated retinal antigen-specific T cells into the retina is not enhanced compared with non-specifically activated T cells [103].
Leukocyte infiltration of the retina during inflammation
Various inflammatory conditions in the eye involve dysfunction of the BRB, and in addition to overt inflammation of the retina in diseases such as uveoretinitis, immune dysfunction leading to vascular permeability and angiogenic changes is linked to pathologies as diverse as retinopathy of prematurity, ARMD and diabetic retinopathy. In premature infants, where the retinal vasculature is underdeveloped, changes in oxygen level in the retina can signal release of vascular endothelial growth factor (VEGF) and development of abnormal vessels [21], and anti-VEGF therapies for ARMD, although possibly harmful to normal circulation, are widely used [88]. VEGF mediates increased vaso-permeability through p38 MAP kinase and ERK1/ERK2-dependent mechanisms that alter occludin expression. VEGF activity, in turn, is regulated by eNOS-dependent dephosphorylation of the VEGF receptor-2, indicating close regulation of these pathways in normal retina [11, 14]. Decreased localisation of occludin at both endothelial and RPE cell borders has been shown to be linked to increased serine and threonine phosphorylation in occludin, which in turn reduces binding to ZO-1 and connection with the cytoskeleton [46].
Angiogenesis and inflammation
VEGF can be secreted by many retinal cell types under stress, including pericytes, astrocytes and RPE, and although not normally considered an inflammatory mediator, VEGF is chemotactic for monocytes, adhesion and migration through endothelium being mediated by increased integrin expression at sites of angiogenesis [64]. VEGF164, in particular, has been shown to induce inflammation and cellular immunity during pathological but not physiological ischaemia-induced sites of retinal angiogenesis [70]. Thus, pathologies involving VEGF are increasingly considered to have an inflammatory component. However, in the absence of adaptive immune responses to antigen in these non-infectious conditions, the inflammatory response is limited to the immediate vicinity by the naturally immunosuppressive ocular microenvironment, avoiding the mononuclear cell influx that characterises infectious or autoimmune inflammation [15, 120].
Diabetic retinopathy caused by systemic changes in metabolic and cardiovascular control is a well-described complication of diabetes and also linked to ischaemic release of VEGF, leukostasis and BRB breakdown [69, 105]. Hyperglycemia and advanced glycation end products also promote pathology in a number of cell types including endothelium, neurons and glia. Thus, retinopathy may be considered a chronic inflammation involving all major cell types within the retina [52]. What first triggers retinal microvascular changes and breakdown of the BRB in diabetic retinopathy is unclear. In a rat model of diabetes, retinal vascular endothelial cells and RPE showed elevated levels of the matrix metalloproteinases (MMP) MMP-2, MMP-9 and MMP-14 with specific degradation of occludin [56]. Advanced glycation end products also increased leukostasis through NFκB upregulation of integrins as early events [97].
There is also a direct relationship between the VEGF and prostaglandin-cyclo-oxygenase (COX-2) pathways. These pathways have vasoactive effects and interact with the nitric oxide synthase pathway (NOS), which in turn also increases vasodilation [57, 128]. Endothelial NOS (eNOS) is constitutively expressed by endothelium and is important for maintaining normal vascular physiology, in part by maintaining smooth muscle tone, but the inducible isoform (iNOS) has also been shown to have a predominant role in leukostasis and BRB breakdown in diabetic iNOS knock-out mice [80]. Expression of ICAM-1 and protein levels of all isoforms of NOS were increased in diabetic mouse retinas whilst occludin and ZO-1 protein decreased. These effects were prevented by the NOS inhibitor N(G)-nitro-l-arginine methyl ester (l-NAME) and absent in the iNOS knock-out diabetic mouse.
Leukocyte infiltration of the retina in uveoretinitis
Endogenous posterior uveoretinitis (EPU) or more correctly, posterior intraocular inflammation, can take many clinical forms such as multifocal choroiditis, sympathetic ophthalmia and pars planitis. These clinical forms have features in common, including retinal vasculitis, macular edema, focal chorioretinal infiltrates and inflammatory cells and exudates in the vitreous, which develop as a result of BRB breakdown. EPU appears to be autoimmune- or immune-mediated rather than infectious in nature. However, it is becoming clear that infectious agents may be linked to autoimmunity through molecular mimicry, polyclonal activation and failure of bystander suppression [49].
Experimental autoimmune uveoretinitis (EAU) is a Th1-type organ-specific autoimmune disease induced by immunisation with retinal antigens such as S antigen and interphotorecepter retinoid-binding protein (IRBP), at distant sites, in susceptible strains of rats or mice. It serves as an animal model of posterior intraocular inflammation, closely mimicking effects seen in the human situation [17, 50]. The BRB is breached in EAU, allowing leukocytes, both CD4+ T lymphocytes and macrophages to move into the retina, resulting in extensive tissue damage [38, 83]. Macrophages are thought to be the main effector cell responsible for tissue damage. EAU can also be induced by adoptive transfer of 5 × 105 retinal antigen-specific T cells [16, 17, 50, 103, 114, 122], and in particular, Th1 cells [18].
Mechanisms of leukocyte migration across the blood–retina barrier in uveoretinitis
Adhesion molecules involved in leukocyte–endothelial cell interactions
Appropriate adhesion molecule expression on both leukocytes and cells of the BRB is clearly important in enabling the migration of leukocytes into the inflamed retina.
During the initial stages of the inflammatory reaction, the endothelium is activated by locally produced cytokines, TNFα and IL-1β in particular. The cytokines up-regulate adhesion molecules, particularly the selectins [108] and chemokines on the luminal surface of the endothelium. Leukocytes in the circulation then respond to the activated endothelium. In vitro studies have shown clear differences in adhesion molecule expression between human RPE and retinal endothelial cells [85]. ICAM is constitutively expressed at high levels on RPE cells, whereas endothelial cells expressed ICAM-1 only after induction with IFNγ. Binding of CD4+ lymphocytes to RPE was dependent on ICAM-1, although after maximal stimulation, there was also an ICAM-1-independent component. Migration of T lymphocytes across retinal endothelial cells in the rat used both LFA-1-dependent and LFA-1-independent mechanisms [58, 61, 94]. Migration of T lymphocytes across unstimulated RPE was dependent on ICAM-1 and LFA-1, whereas after activation of RPE with IFNγ, migration was dependent on both ICAM-1 and VCAM-1 [37].
Receptors for various integrins and adhesion molecules are up-regulated on human RPE and vascular endothelial cells in vivo during ocular inflammation such as sympathetic ophthalmia. These include enhanced or de novo expression of ICAM-1, VCAM-1, CD62E and CD44, which in vitro, may be induced by the cytokines IL-1 and TNF-α. Kuppner et al. [79] compared adhesion molecule expression in acute and fibrotic sympathetic ophthalmia with normal human eyes. Expression of several integrins of the VLA family, which form receptors for extracellular matrix proteins, were found to be expressed by the RPE. However, VLA-4, the ligand for VCAM-1 and fibronectin, VLA-5 (fibronectin) and VLA-6 (laminin), which were increased on endothelium in the inflamed eyes, were not up-regulated on RPE cells. In contrast, ICAM-1 and CD44 expressions were greatly enhanced. Electron microscopy studies have also shown differences in the manner in which leukocytes cross the retinal endothelium and the RPE [40]. Inflammatory cell diapedesis through Bruch’s membrane involved separation of its constitutive layers, migration through the pores in the membrane and between the RPE cells without apparent significant physical disruption of the RPE cell layer. Retinal vascular diapedesis, however, involved changes in post-capillary venules to high endothelial cell morphology, indicating endothelial cell activation [93], with migration appearing to be via the paracellular route [60]. As RPE cells fail to express iNOS during inflammation whilst endothelium does, [19, 67] it has been proposed that the RPE maintains a predominantly immunosuppressive role during inflammation [84].
Mechanisms of leukocyte subset migration into the retina
To study the role of adhesion molecules and leukocyte induced breakdown of the BRB, we have developed scanning laser ophthalmoscopy (SLO) methods for use in rodent EAU models. In this technique, syngeneic fluorescently labelled (eGFP CFSE or calcein AM) splenocytes, T helper cell subsets or monocytes are injected into the tail vein of normal or transgenic mice at various disease stages. These transferred cells can then be tracked in vivo in real-time and their interaction with the retinal vascular endothelium quantified in terms of rolling efficiency, rolling velocity and adhesion. These images can then be analysed to investigate the role of various molecules involved in leukocyte recruitment at the BRB [131]. This can be followed by confocal microscopy of retinal wholemounts, which allows both the quantification of cells entering the retina and further immunostaining to reveal changes in phenotype of endothelial cells and trafficking leukocytes.
The earliest changes observed are 24 h prior to detectable leukocyte infiltration. Vascular changes occur, which reduce shear stress in retinal veins and venules from approximately 30 to 20 dyn/cm2. Although still considerably higher than in normal fenestrated endothelium, as shear stress falls, rolling and sticking efficiencies of leukocytes increase in the post-capillary venules, correlating with selective upregulation of P and E selectin and ICAM-1 on these vessels, particularly at sites of extravasation [137]. This effect is not observed in arteries or arterioles, although ICAM, VCAM, PECAM, E selectin, P selectin and CD44 are also up-regulated on other retinal endothelium as EAU progresses [135, 140]. As CD44 is widely expressed on most leukocyte subsets, it is required for leukocyte rolling under flow and is involved in early stages of extravasation in non-neuronal tissues [34, 35]. We investigated the role of CD44 in retinal inflammation in our model. Treatment of a mixed leukocyte population with neutralizing antibody against CD44 significantly suppressed the rolling of these cells on inflamed retinal venules and reduced infiltration into the retina. The importance of CD44 in leukocyte recruitment in EAU was also supported by the fact that EAU severity was diminished by administration of the anti-CD44 antibody at the stage of disease in which leukocytes are first seen to infiltrate the retina [140].
Both Th1 cells and Th2 subsets of T lymphocyte are implicated in the pathogenesis of uveitis [10, 27]. So to investigate the role of adhesion molecules in the trafficking of different T cell subsets across the BRB in Th1-mediated EAU, naive CD4+ T lymphocytes were isolated from lymph node cells and polarised in vitro into Th1- and Th2-like cells. Surface PSGL-1 and LFA-1 were up-regulated on both populations but expressed at higher levels on Th1-like cells, whereas CD44 expression was up-regulated to a greater extent on Th2 cells. Pretreatment of the polarised T lymphocyte populations with anti-PSGL-1 inhibited rolling and infiltration of Th1-like cells but not Th2, providing direct in vivo evidence for the inability of Th2 to respond to P/E selectin despite increased expression of PSGL-1. Anti-LFA-1 pretreatment inhibited infiltration of both Th1- and Th2-like cells, but this inhibition was more substantial for the Th-1 cells. Thus, preferential recruitment of Th1 cells in EAU is mediated by PSGL-1:P/E selectin, whereas non-selective trafficking of activated T cells (both Th1 and Th2) across the BRB is mediated by CD44:CD44r and LFA-1:ICAM-1 [137].
Monocytes are also necessary for full expression of disease in EAU. We have established a model for studying bone-marrow-derived monocyte trafficking in vivo, and found that, whereas T cells, whether antigen specific or not, roll on inflamed endothelium and rapidly migrate into the retina, monocytes can only traffic into the inflammatory site once they have acquired a specific phenotype [136]. This phenotype was consistent with the CCR2+ phenotype described by others but was also found to be a constitutive time-limited property of the transferred monocytes and independent of an existing inflammation. Monocytes purified from bone marrow failed to roll on inflamed endothelium until they had undergone in vivo recirculation for at least 24 h, with maximum retinal infiltration not occurring until 48-h post-transfer. Of note was the ability of transferred monocytes to undergo further differentiation during trafficking into the retina. Transferred cells matured into classical tissue macrophages or CD11c+ dendritic cells that persisted within the retina for several days. Several striking contrasts between inflammatory monocyte and Th1 cell trafficking were observed. Monocyte rolling in inflamed venules was faster and CD62-L dependent rather than LFA-1 dependent. While PSGL-1 was found to play a role in regulating diapedesis to the inflammatory site, CD62-L was shown to have a key role in regulating recruitment of monocytes to the lymphoid tissue from the circulation during inflammation (manuscript submitted for publication). These observations reveal that different molecular mechanisms are involved in leukocyte subset adhesion to endothelium and migration into the tissues.
Mechanisms of blood–retina barrier breakdown in EAU
Whether BRB breakdown is necessary before lymphocytes and monocytes can infiltrate or whether infiltration results in BRB breakdown and free movement of leukocytes between the circulation and the retina is controversial [60, 86]. Blood flow within the CNS and, by extension the retina, is regulated by interactions between neurons, glia and the microvasculature, contact between the microvessel wall and perivascular endfeet of astrocytes, mediated by agrin, being important for maintaining barrier function in the brain [1]. Other cellular components include pericytes and perivascular macrophages, contributing to what has been termed the “neurovascular unit”. Astrocytes can secrete a range of cytokines and chemical mediators that up-regulate TJ and polarised expression of transporters and other enzyme systems that control transendothelial transport of molecules and ions to maintain a metabolic barrier [62]. In BBB disruption, agrin is lost from the abluminal surface of the endothelial cells adjacent to the astrocytic endfeet and contact lost. In the brain, bradykinin activation of astrocytes has been implicated in BBB breakdown in stroke or trauma, but the molecular signals responsible for BRB breakdown in autoimmune inflammation are still not understood [2].
Using our in vivo cell trafficking model, we have studied the relationship between changes in vascular permeability, adhesion molecule expression and leukocyte migration into the retina. As described above, circulation of activated lymphocytes induces changes in the endothelium of the retinal venules, allowing transient breakdown of the BRB. Where T cells are non-antigen specific, this occurs in the absence of cell rolling, without any reduction in shear stress and without generalised inflammation of the retina occurring. In contrast, in EAU where retinal autoreactive T cells are present, cell rolling occurs, and extravasation of T cells in the venules (but not arterioles) occurs together with BRB breakdown in these venules as evidenced by dye leakage and an influx of inflammatory cells [135].
BRB breakdown involves changes in levels of expression and localisation of TJ proteins occludin-1, claudins and ZO-1 [116]. To elucidate mechanisms of BRB and its regulation by inflammatory cells and mediators in vivo, we examined wholemount retinas and choroids from normal and EAU mice. Using confocal microscopy, we showed that in normal retina, TJ proteins were evenly distributed along endothelial cell margins with increased expression in tri-cellular corners, hypothesised to be a “weak spot” in the TJ network. In EAU, disruption of TJ proteins only occurred in the venules where leukocyte infiltration was occurring. Western blot analysis confirmed that claudin 1/3 and occludin proteins were reduced. ZO-1 protein expression was not reduced but redistributed from the cell membrane and the TJs to the cytoplasm [133]. Examination of venules in retinas in early EAU (with few infiltrating cells) showed that loss of occludin-1 from venule TJ occurred at the point of contact with adherent leukocytes undergoing transendothelial migration. In the absence of other migrating inflammatory cells, TJ appeared to reform. In contrast, in areas of the retina where there was significant inflammatory cell infiltration, confocal microscopy revealed that occludin-1 was lost from venules but not capillaries and arterioles. This correlated with loss of astrocyte foot process contact with the endothelium of the venule [133]. These studies are consistent with a major role for venule endothelium in leukocyte recruitment to the retina and imply that breakdown of the BRB in uveitis is an active event triggered by adherent leukocytes rather than a passive event driven solely by local cytokine microenvironment.
The role of chemokines in leukocyte migration across the BRB in EAU
Chemokines are vital for the development of a focussed immune response, and certain chemokines are associated with inflammatory responses and the attraction of inflammatory cells to the site of infection or injury [91, 111]. Chemokines can be produced by both resident cells at the inflammatory site, such as epithelial cells, endothelial cells, fibroblasts and resident macrophages and dendritic cells as well as early infiltrating macrophages and neutrophils and then at later stages, infiltrating T cells. Chemokine production may be via toll-like receptors [36, 71, 115] or via inflammatory cytokines such as IL-1, TNFα and IFNγ, in the case of a Th1-type response or IL-4, IL-5 and IL-13 in the case of a Th2-type response [8, 107, 109]. Thus, the inflammatory stimulus will generate a chemokine “fingerprint”, which will be the basis for determining the tailored leukocyte response [22, 111].
Production of chemokines is up-regulated in the murine eye in EAU and generally shows, as expected, a Th1-like pattern of expression; for example, CCL2, CCL3 and CCL5 [28]. CCL3 and CCL2 were associated with retinal and choroidal vessels as well as with infiltrating cells, whereas CCL5 was associated predominantly with infiltrating cells [28]. RT-PCR analysis found expression of mRNA transcripts for these and several other chemokines including CXCL9, CXCL10 CCL6, CCL9, CCL19 and CCL22 is also up-regulated, although some of these studies were carried out with whole eyes [31, 51]. Recipients receiving Th1 cells had a similar pattern of expression [51]. Laser capture microdissection showed that both RPE and infiltrating leukocytes expressed chemokine transcripts in distinct but overlapping patterns [51]. In vitro, human cells of the BRB, both RPE and retinal microvascular endothelial cells respond to pro-inflammatory cytokines IL-1β, TNFα and IFNγ by producing substantial levels of CCL2, CCL5, CXCL8, CXCL10 and CXCL1 [29, 41, 42]. Receptors for these chemokines are also up-regulated in EAU. Expression of mRNA transcripts for CCR1, CCR5, CXCR3, CXCR2, CCR6, CCR8 and CCR2 was detected in whole eyes [51], and cell surface expression of CCR5, CXCR3 and CCR2 on infiltrating cells was detected by immunohistochemistry (pers. comm. IJC).
We have investigated the importance of CCL3 in recruitment of leukocytes in vivo at the inflamed BRB using SLO. We showed that CCL3, a ligand for CCR5, was involved in leukocyte recruitment at the BRB and was linked to the inflammatory process and disease pathogenesis in EAU [30]. The effect of short-term anti-CCL3 treatment was examined by injecting anti-CCL3 antibody into mice with EAU 1 h prior to tracking a labelled population of activated leukocytes (splenocytes from mice with EAU) in real-time using SLO. This treatment inhibited leukocyte slowing and accumulation and subsequent extravasation of leukocytes at the blood–retina barrier. This was effective predominantly in the post-capillary venules, which have been shown to be the main site of passage of leukocytes across the BRB. Long-term anti-CCL3 treatment also prevented decreased leukocyte velocity and reduced disease severity as measured clinically, histologically and in terms of BRB breakdown [30].
To define this further, we examined the trafficking of Th1-like cells, polarised in vitro. These cells, which expressed high levels of CCR5, were labelled and adoptively transferred and their trafficking monitored in vivo at an early disease stage in EAU using SLO. Treatment of the cells with antibody against CCR5 prior to transfer resulted in a reduction in their infiltration into the retina. However, rolling velocity, rolling efficiency and adherence of the cells to retinal endothelium was not reduced. CCR5 is clearly important for Th1 cell recruitment at the BRB and acts at the level of transendothelial migration rather than at the earlier stage of rolling on the endothelium [31]. This is consistent with other work on the arrest of monocytes or Th-1-like T cells by CCL5 (RANTES) immobilised to activated endothelium under flow conditions. Arrest was mediated predominantly by CCR1, whereas CCR5 was responsible for spreading. This was irrespective of the degree of expression of the receptors on the different cell types [127].
Interestingly, studies in which CCR1/CCR5 was blocked in EAU have emphasised the fact that the action of a chemokine and its receptor in cell recruitment at the BRB may differ dramatically depending on the antigen used to induce uveitis and the stage of the disease and the resulting microenvironment. In Lewis rats in which EAU was induced with S-antigen peptide, treatment with the CCR1/CCR5 receptor antagonist Met-RANTES was effective in reducing uveitis in the efferent phase, possibly inhibiting migration of activated T cells or monocytes into the retina [39]. When EAU was induced with IRBP peptide, however, Met-RANTES appeared to affect activation of T cells, reducing intraocular inflammation if administered early in the initiation phase of the response but moderately enhancing uveitis if given during the efferent phase [39]. Antibody against RANTES given during the efferent phase in C57BL/6 mice immunised with IRBP peptide also led to exacerbation of EAU, and this was attributed to a change in the ratio of T cell subsets recruited favouring CD4 over CD8 T cells [117].
Monocyte trafficking across the BRB has also been examined, and in particular, the role of two chemokine receptors which have been shown to be involved in monocyte recruitment, CCR2 and CX3CR1. This type of experiment is constrained by the number of monocytes, which can be isolated from the blood. However, when in vitro-cultured bone marrow monocytes, non-activated peritoneal monocytes and freshly isolated bone marrow monocyte precursors were compared, only the latter precursors continued to circulate and trafficked efficiently to the inflamed retina in EAU [136]. These were, therefore, labelled and used to investigate trafficking in EAU, and interestingly, it was found that these cells could not initially migrate across the BRB and required 24–48 h in vivo before they started to infiltrate the inflamed retina as described above. This coincided with an increase in expression of CCR2 on these cells. This was only a transient increase, and CCR2 expression and the migratory phenotype were lost 72 h after adoptive transfer [136]. However, evidence from further studies indicates that although CCR2 may improve the efficiency of monocyte transendothelial migration at the inflamed BRB, it is not essential for monocyte emigration at this site (manuscript in preparation).
This would confirm findings from other inflammatory sites in which it has been shown that although CCR2-positive monocytes may be recruited preferentially to an inflammatory site, CCR2-negative monocytes are also able to traffic to these sites [106, 112, 123]. CCR2 also has a role in facilitating the egress of monocytes from the bone marrow, with more monocytes present in the bone marrow in CCR2−/− mice and fewer circulating even before infection [112], but its role in monocyte recruitment to an inflammatory site is, in addition to this, and may be mediated by CCL7 (MCP-3) and CCL2 (MCP-1) [125]. In some inflammatory situations, CCR2 is clearly more critical for monocyte recruitment than we show at the BRB; for example, for recruitment into the peritoneum in response to thioglycolate [125]. Thus, reliance on CCR2 for recruitment of monocytes may vary depending on the particular inflammatory stimulus and the microenvironment that is generated.
CX3CR1 has also been implicated in the trafficking of monocytes, and although it has been proposed to be important for immune surveillance [55], there are also reports that it has a role in recruitment in an inflammatory situation; for example, in atherosclerosis [25, 81], crescentic glomerulonephritis [47] and in cerebral ischaemia [118]. It is possible that this receptor is important for the recruitment of a subpopulation of inflammatory CCR2-negative monocytes. However, recently, it has been shown in an atherosclerosis model that although CX3CR1-positive monocytes can enter atherosclerotic plaques, CX3CR1 was not necessary for this entry. In contrast, CCR2-positive monocytes, which also express substantial levels of CX3CR1 [55], required CX3CR1 in addition to CCR2 for the accumulation of monocytes in these lesions [123]. In EAU, our studies to date indicate that CX3CR1 is not essential for the trafficking of monocytes into the inflamed retina (manuscript in preparation).
Conclusions and implications for design of novel therapies
Current treatment for uveitis is still dependent in the most part on systemic, non-specific immunosuppression. Additional therapies such as cyclosporin A, tacrolimus and rapamycin may act by interfering with the production and action of cytokines, particularly interleukin-2, targeting T cell function. Mycophenolate mofetil, an antimetabolite affecting purine synthesis, is also valuable. Other immunosuppressants such as azathioprine, cyclophosphamide and chlorambucil have severe side effects restricting their use. Blockade of TNF-α is also effective at least in the short term and now used clinically in different forms. Despite the usefulness of these therapies, they are all relatively non-specific, and in recent years, the quest for specific, tailored therapies for uveitis has been paramount.
The ability of leukocytes to migrate across the BRB depends on many different factors, the importance of which will vary depending on the leukocyte subset, the specific site and the microenvironment created as a result of the inflammatory stimulus and genetic background. Therapies designed to block the passage of cells into the inflamed retina will, therefore, need to be tailored to the situation to increase their potential for success. Understanding in depth the stages in the process of leukocyte migration across the BRB will be fundamental for this. It is clear that there is a degree of redundancy in adhesion molecule use as well as in cytokine, chemokine and integrin signalling, and different inflammatory cell subsets will require different strategies to block their recruitment and effector functions. However, some therapies targeting key intracellular signalling pathways may be beneficial. For instance, lymphocyte trafficking is dependent upon endothelial cell G-protein signalling, and recently, lovastatin has been shown to reduce retinal disease in the mouse model of EAU. Lovastatin inhibited the synthesis of precursors required for prenylation and post-translational activation of endothelial Rho GTPase, an essential step in ICAM-1-mediated leukocyte migration [3, 54]. Although different statins may have different effects in different diseases, they are proven safe drugs and provide attractive therapeutic options.
Some therapies that target specific aspects of leukocyte migration are already in use and in clinical trials in some inflammatory diseases. Although targeting some adhesion molecules has not always proved successful [100, 141], there are some promising results. These include natalizumab, directed against VLA-4, which is in use for MS and Crohn’s despite a low risk of progressive multifocal leukoencephalopathy, a viral infection of the CNS [73]; and efalizumab, a recombinant humanised antibody, which binds to LFA-1 preventing binding to ICAM-1, is in use for the treatment of psoriasis [75]. The possibility of targetting chemokines and their receptors to prevent recruitment of inflammatory cells has also received much attention from pharmaceutical companies. For example, a number of compounds with anti-CCR2 activity have been developed and patented with use planned in multiple sclerosis and atherosclerosis [48]. An antagonist of CCR5 (maraviroc) is also available, which has been designed for use in HIV [43]. These drugs may have potential use in inflammatory conditions involving the BRB.
References
Abbott NJ (2002) Astrocyte–endothelial interactions and blood–brain barrier permeability. J Anat 200:629–638
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53
Adamson P, Wilbourn B, Etienne-Manneville S, Calder V, Beraud E, Milligan G, Couraud PO, Greenwood J (2002) Lymphocyte trafficking through the blood–brain barrier is dependent on endothelial cell heterotrimeric G-protein signaling. FASEB J 16:1185–1194
Albini TA, Wang RC, Reiser B, Zamir E, Wu GS, Rao NA (2005) Microglial stability and repopulation in the retina. Br J Ophthalmol 89:901–903
Ali S, Palmer ACV, Banerjee B, Fritchley SJ, Kirby JA (2000) Examination of the function of RANTES, MIP-1 alpha, and MIP-1 beta following interaction with heparin-like glycosaminoglycans. J Biol Chem 275:11721–11727
Alon R, Feigelson S (2002) From rolling to arrest on blood vessels: leukocyte tap dancing on endothelial integrin ligands and chemokines at sub-second contacts. Semin Immunol 14:93–104
Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, Matsushima K, Murphy P, Nomiyama H, Oppenheim J, Rot A, Schall T, Tsang M, Thorpe R, Van Damme J, Wadhwa M, Yoshie O, Zlotnik A, Zoon K (2002) Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res 22:1067–1068
Baggiolini M, Dewald B, Moser B (1997) Human chemokines: an update. Annu Rev Immunol 15:675–705
Bakker AB, Hoek RM, Cerwenka A, Blom B, Lucian L, McNeil T, Murray R, Phillips LH, Sedgwick JD, Lanier LL (2000) DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity 13:345–353
Banerjee S, Savant V, Scott RA, Curnow SJ, Wallace GR, Murray PI (2007) Multiplex bead analysis of vitreous humor of patients with vitreoretinal disorders. Invest Ophthalmol Vis Sci 48:2203–2207
Cai J, Jiang WG, Ahmed A, Boulton M (2006) Vascular endothelial growth factor-induced endothelial cell proliferation is regulated by interaction between VEGFR-2, SH-PTP1 and eNOS. Microvasc Res 71:20–31
Callahan MK, Ransohoff RM (2004) Analysis of leukocyte extravasation across the blood–brain barrier: conceptual and technical aspects. Curr Allergy Asthma Rep 4:65–73
Campbell JJ, Foxman EF, Butcher EC (1997) Chemoattractant receptor cross talk as a regulatory mechanism in leukocyte adhesion and migration. Eur J Immunol 27:2571–2578
Campbell M, Collery R, McEvoy A, Gardiner TA, Stitt AW, Brankin B (2006) Involvement of MAPKs in endostatin-mediated regulation of blood–retinal barrier function. Curr Eye Res 31:1033–1045
Caspi RR (2006) Ocular autoimmunity: the price of privilege? Immunol Rev 213:23–35
Caspi RR, Chan C-C, Wiggert B, Chader GJ (1990) The mouse as a model of experimental autoimmune uveoretinitis (EAU). Curr Eye Res 9:169–174
Caspi RR, Roberge FG, Chan C, Wiggert B, Chader GJ, Rozenszajn LA, Lando Z, Nussenblatt RB (1988) A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol 140:1490–1495
Caspi RR, Silver PB, Chan C-C, Sun B, Agarwal RK, Wells J, Oddo S, Fujino Y, Najafian F, Wilder RL (1996) Genetic susceptibility to experimental autoimmune uveoretinitis in the rat is associated with an elevated Th1 response. J Immunol 157:2668–2675
Chakravarthy U, Stitt AW, McNally J, Bailie JR, Hoey EM, Duprex P (1995) Nitric oxide synthase activity and expression in retinal capillary endothelial cells and pericytes. Curr Eye Res 14:285–294
Chan JR, Hyduk SJ, Cybulsky MI (2001) Chemoattractants induce a rapid and transient upregulation of monocyte alpha 4 integrin affinity for vascular cell adhesion molecule 1 which mediates arrest: an early step in the process of emigration. J Exp Med 193:1149–1158
Chen J, Smith LE (2007) Retinopathy of prematurity. Angiogenesis 10:133–140
Chensue SW (2001) Molecular machinations: chemokine signals in host–pathogen interactions. Clin Microbiol Rev 14:821–835
Cinamon G, Grabovsky V, Winter E, Franitza S, Feigelson S, Shamri R, Dwir O, Alon R (2001) Novel chemokine functions in lymphocyte migration through vascular endothelium under shear flow. J Leukoc Biol 69:860–866
Cinamon G, Shinder V, Alon R (2001) Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol 2:515–522
Combadiere C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debre P, Tedgui A, Murphy PM, Mallat Z (2003) Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 107:1009–1016
Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C (2000) Chemokines trigger immediate beta 2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity 13:759–769
Crane IJ, Forrester JV (2005) Th1 and Th2 lymphocytes in autoimmune disease. Crit Rev Immunol 25:75–102
Crane IJ, McKillop-Smith S, Wallace CA, Lamont GR, Forrester JV (2001) Expression of the chemokines MIP-1a, MCP-1 and RANTES in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 42:1547–1552
Crane IJ, Wallace CA, McKillop-Smith S, Forrester JV (2000) Control of chemokine production at the blood–retina barrier. Immunology 101:426–433
Crane IJ, Xu H, Manivannan A, McKillop-Smith S, Lamont G, Wallace C, Sharp PF, Forrester JV (2003) Effect of anti-macrophage inflammatory protein-1a on leukocyte trafficking and disease progression in experimental autoimmune uveoretinitis. Eur J Immunol 33:402–410
Crane IJ, Xu H, Wallace C, Manivannan A, Mack M, Liversidge J, Marquez G, Sharp P, Forrester JV (2006) Involvement of CCR5 in the passage of Th1-type cells across the blood–retina barrier in experimental autoimmune uveitis. J Leukoc Biol 79:435–443
D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, Sozzani S, Allavena P, Mantovani A (2000) Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nat Immunol 1:387–391
Daniele LL, Adams RH, Durante DE, Pugh EN Jr, Philp NJ (2007) Novel distribution of junctional adhesion molecule-C in the neural retina and retinal pigment epithelium. J Comp Neurol 505:166–176
DeGrendele HC, Estess P, Picker LJ, Siegelman MH (1996) CD44 and its ligand hyaluronate mediate rolling under physiologic flow: a novel lymphocyte-endothelial cell primary adhesion pathway. J Exp Med 183:1119–1130
DeGrendele HC, Estess P, Siegelman MH (1997) Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science 278:672–675
Devaney JM, Greene CM, Taggart CC, Carroll TP, O’Neill SJ, McElvaney NG (2003) Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett 544:129–132
Devine L, Lightman SL, Greenwood J (1996) Role of LFA-1, ICAM-1, VLA-4 and VCAM-1 in lymphocyte migration across retinal pigment epithelial monolayers in vitro. Immunology 88:456–462
Dick AD, McMenamin PG, Korner H, Scallon BJ, Ghrayeb J, Forrester JV, Sedgwick JD (1996) Inhibition of tumour necrosis factor activity minimizes target organ damage in experimental autoimmune uveoretinitis despite quantitatively normal activated T cell traffic to the retina. Eur J Immunol 26:1018–1025
Diedrichs-Mohring M, Nelson PJ, Proudfoot AEI, Thurau SR, Wildner G (2005) The effect of the CC chemokine receptor antagonist Met-RANTES on experimental autoimmune uveitis and oral tolerance. J Neuroimmunol 164:22–30
Dua HS, McKinnon A, McMenamin PG, Forrester JV (1991) Ultrastructural pathology of the ‘barrier sites’ in experimental autoimmune uveitis and experimental autoimmune pinealitis. Br J Ophthalmol 75:391–397
Elner VM, Burnstine MA, Strieter RM, Kunkel SL, Elner SG (1997) Cell-associated human retinal pigment epithelium interleukin-8 and monocyte chemotactic protein-1: immunochemical and in-situ hybridization analyses. Exp Eye Res 65:781–789
Elner SG, DelMonte D, Bian ZM, Lukacs NW, Elner VM (2006) Differential expression of retinal pigment epithelium (RPE) IP-10 and interleukin-8. Exp Eye Res 83:374–379
Emmelkamp JM, Rockstroh JK (2007) CCR5 antagonists: comparison of efficacy, side effects, pharmacokinetics and interactions—review of the literature. Eur J Med Res 12:409–417
Engelhardt B, Ransohoff RM (2005) The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol 26:485–495
Engelhardt B, Wolburg H (2004) Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol 34:2955–2963
Erickson KK, Sundstrom JM, Antonetti DA (2007) Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis 10:103–117
Feng LL, Chen SZ, Garcia GE, Xia YY, Siani MA, Botti P, Wilson CB, Harrison JK, Bacon KB (1999) Prevention of crescentic glomerulonephritis by immunoneutralization of the fractalkine receptor CX3CR1. Kidney Int 56:612–620
Feria M, Diaz-Gonzalez F (2006) The CCR2 receptor as a therapeutic target. Expert Opin Ther Pat 16:49–57
Forrester JV (2000) Autoimmunity and autoimmune diseases of the eye. In: Pleyer U, Zierhut M, Behrens-Baumann W (eds) Immuno-ophthalmology, Dev Ophthalmol. Karger, Basel, pp 167–185
Forrester JV, Liversidge W, Dua HS, Dick A, Harper F, McMenamin PG (1992) Experimental autoimmune uveoretinitis: a model system for immunointervention: a review. Curr Eye Res 11:33–40
Foxman EF, Zhang MF, Hurst SD, Muchamuel T, Shen DF, Wawrousek EF, Chan CC, Gery I (2002) Inflammatory mediators in uveitis: differential induction of cytokines and chemokines in Th1-versus Th2-mediated ocular inflammation. J Immunol 168:2483–2492
Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW (2002) Diabetic retinopathy: more than meets the eye. Surv Ophthalmol 47(Suppl 2):S253–S262
Ge S, Song L, Pachter JS (2005) Where is the blood–brain barrier… really? J Neurosci Res 79:421–427
Gegg ME, Harry R, Hankey D, Zambarakji H, Pryce G, Baker D, Adamson P, Calder V, Greenwood J (2005) Suppression of autoimmune retinal disease by lovastatin does not require Th2 cytokine induction. J Immunol 174:2327–2335
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82
Giebel SJ, Menicucci G, McGuire PG, Das A (2005) Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood–retinal barrier. Lab Invest 85:597–607
Greenwood J (1991) Mechanisms of blood–brain barrier breakdown. Neuroradiology 33:95–100
Greenwood J, Calder VL (1993) Lymphocyte migration through cultured endothelial cell monolayers derived from the blood–retinal barrier. Immunology 80:401–406
Greenwood J, Amos CL, Walters CE, Couraud PO, Lyck R, Engelhardt B, Adamson P (2003) Intracellular domain of brain endothelial intercellular adhesion molecule-1 is essential for T lymphocyte-mediated signaling and migration. J Immunol 171:2099–2108
Greenwood J, Howes R, Lightman S (1994) The blood–retinal barrier in experimental autoimmune uveoretinitis. Leukocyte interactions and functional damage. Lab Invest 70:39–52
Greenwood J, Wang Y, Calder VL (1995) Lymphocyte adhesion and transendothelial migration in the central nervous system: the role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology 86:408–415
Haseloff RF, Blasig IE, Bauer HC, Bauer H (2005) In search of the astrocytic factor(s) modulating blood–brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol 25:25–39
Hawkins BT, Davis TP (2005) The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185
Heil M, Clauss M, Suzuki K, Buschmann IR, Willuweit A, Fischer S, Schaper W (2000) Vascular endothelial growth factor (VEGF) stimulates monocyte migration through endothelial monolayers via increased integrin expression. Eur J Cell Biol 79:850–857
Hickey WF (2001) Basic principles of immunological surveillance of the normal central nervous system. Glia 36:118–124
Hickey WF, Hsu BL, Kimura H (1991) T-lymphocyte entry into the central nervous system. J Neurosci Res 28:254–260
Hoey S, Grabowski PS, Ralston SH, Forrester JV, Liversidge J (1997) Nitric oxide accelerates the onset and increases the severity of experimental autoimmune uveoretinitis through an IFN-gamma-dependent mechanism. J Immunol 159:5132–5142
Howard OMZ, Dong HF, Su SB, Caspi RR, Chen X, Plotz P, Oppenheim JJ (2005) Autoantigens signal through chemokine receptors: uveitis antigens induce CXCR3- and CXCR5-expressing lymphocytes and immature dendritic cells to migrate. Blood 105:4207–4214
Ishida S, Usui T, Yamashiro K, Kaji Y, Ahmed E, Carrasquillo KG, Amano S, Hida T, Oguchi Y, Adamis AP (2003) VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci 44:2155–2162
Ishida S, Usui T, Yamashiro K, Kaji Y, Amano S, Ogura Y, Hida T, Oguchi Y, Ambati J, Miller JW, Gragoudas ES, Ng YS, D’Amore PA, Shima DT, Adamis AP (2003) VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med 198:483–489
Janeway CA, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Jha P, Bora PS, Bora NS (2007) The role of complement system in ocular diseases including uveitis and macular degeneration. Mol Immunol 44:3901–3908
Johnson KP (2007) Natalizumab (Tysabri) treatment for relapsing multiple sclerosis. Neurologist 13:182–187
Johnston B, Butcher EC (2002) Chemokines in rapid leukocyte adhesion triggering and migration. Semin Immunol 14:83–92
Joshi A, Bauer R, Kuebler P, White M, Leddy C, Compton P, Garovoy M, Kwon P, Walicke P, Dedrick R (2006) An overview of the pharmacokinetics and pharmacodynamics of efalizumab: a monoclonal antibody approved for use in psoriasis. J Clin Pharmacol 46:10–20
Kastelan S, Zjacic-Rotkvic V, Kastelan Z (2007) Could diabetic retinopathy be an autoimmune disease? Med Hypotheses 68:1016–1018
Kenakin T, Onaran O (2002) The ligand paradox between affinity and efficacy: can you be there and not make a difference? Trends Pharmacol Sci 23:275–280
Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301:1720–1725
Kuppner MC, Liversidge J, McKillop-Smith S, Lumsden L, Forrester JV (1993) Adhesion molecule expression in acute and fibrotic sympathetic ophthalmia. Curr Eye Res 12:923–934
Leal EC, Manivannan A, Hosoya K, Terasaki T, Cunha-Vaz J, Ambrosio AF, Forrester JV (2007) Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood–retinal barrier breakdown in diabetic retinopathy. Invest Ophthalmol Vis Sci 48:5257–5265
Lesnik P, Haskell CA, Charo IF (2003) Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J Clin Invest 111:333–340
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689
Liversidge J, Forrester JV (1988) Experimental autoimmune uveitis (EAU): immunophenotypic analysis of inflammatory cells in chorio retinal lesions. Curr Eye Res 7:1231–1241
Liversidge J, Forrester JV (1999) Regulation of immune responses by the retinal pigment epithelium. In: Marmor MF, Wolfensberger TJ (eds) Retinal pigment epithelium, current aspects of function and disease. Oxford University Press, Oxford, pp 511–527
Liversidge J, Sewell HF, Forrester JV (1990) Interactions between lymphocytes and cells of the blood–retina barrier: mechanisms of T lymphocyte adhesion to human retinal capillary endothelial cells and retinal pigment epithelial cells in vitro. Immunology 71:390–396
Luna JD, Chan CC, Derevjanik NL, Mahlow J, Chiu C, Peng B, Tobe T, Campochiaro PA, Vinores SA (1997) Blood–retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res 49:268–280
Luscinskas FW, Ma S, Nusrat A, Parkos CA, Shaw SK (2002) The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol Rev 186:57–67
Maharaj AS, D’Amore PA (2007) Roles for VEGF in the adult. Microvasc Res 74:100–113
Maly P, Thall A, Petryniak B, Rogers CE, Smith PL, Marks RM, Kelly RJ, Gersten KM, Cheng G, Saunders TL, Camper SA, Camphausen RT, Sullivan FX, Isogai Y, Hindsgaul O, von Andrian UH, Lowe JB (1996) The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell 86:643–653
Mandell KJ, Berglin L, Severson EA, Edelhauser HF, Parkos CA (2007) Expression of JAM-A in the human corneal endothelium and retinal pigment epithelium: localization and evidence for role in barrier function. Invest Ophthalmol Vis Sci 48:3928–3936
Mantovani A (1999) The chemokine system: redundancy for robust outputs. Immunol Today 20:254–257
Mantovani A, Bonecchi R, Locati M (2006) Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol 6:907–918
McMenamin PG, Forrester JV, Steptoe RJ, Dua HS (1992) Ultrastructural pathology of experimental autoimmune uveitis. Quantitative evidence of activation and possible high endothelial venule-like changes in retinal vascular endothelium. Lab Invest 67:42–55
Mesri M, Liversidge J, Forrester JV (1996) Prostaglandin E2 and monoclonal antibody to lymphocyte function-associated antigen-1 differentially inhibit migration of T lymphocytes across microvascular retinal endothelial cells in rat. Immunology 88:471–477
Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A (1997) Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 91:385–395
Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA (2002) Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood 100:3853–3860
Moore TC, Moore JE, Kaji Y, Frizzell N, Usui T, Poulaki V, Campbell IL, Stitt AW, Gardiner TA, Archer DB, Adamis AP (2003) The role of advanced glycation end products in retinal microvascular leukostasis. Invest Ophthalmol Vis Sci 44:4457–4464
Muller WA, Randolph GJ (1999) Migration of leukocytes across endothelium and beyond: molecules involved in the transmigration and fate of monocytes. J Leukoc Biol 66:698–704
Niederkorn JY (2006) See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol 7:354–359
Norman MU, Kubes P (2005) Therapeutic intervention in inflammatory diseases: a time and place for anti-adhesion therapy. Microcirculation 12:91–98
Oppenheim JJ, Dong HF, Plotz P, Caspi RR, Dykstra M, Pierce S, Martin R, Carlos C, Finn O, Koul O, Howard OM (2005) Autoantigens act as tissue-specific chemoattractants. J Leukoc Biol 77:854–861
Pachter JS, De Vries HE, Fabry Z (2003) The blood–brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol 62:593–604
Prendergast RA, Iliff CE, Coskuncan NM, Caspi RR, Sartani G, Tarrant TK, Lutty GA, McLeod DS (1998) T cell traffic and the inflammatory response in experimental autoimmune uveoretinitis. Invest Ophthalmol Vis Sci 39:754–762
Proudfoot AEI, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TNC, Kosco-Vilbois MH (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA 100:1885–1890
Qaum T, Xu Q, Joussen AM, Clemens MW, Qin W, Miyamoto K, Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP (2001) VEGF-initiated blood–retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci 42:2408–2413
Quinones MP, Ahuja SK, Jimenez F, Schaefer J, Garavito E, Rao A, Chenaux G, Reddick RL, Kuziel WA, Ahuja SS (2004) Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J Clin Invest 113:856–866
Rollins BJ (1997) Chemokines. Blood 90:909–928
Rosen SD (2004) Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol 22:129–156
Rossi D, Zlotnik A (2000) The biology of chemokines and their receptors. Annu Rev Immunol 18:217–243
Rot A (2005) Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev 16:687–694
Rot A, von Andrian UH (2004) Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 22:891–928
Serbina NV, Pamer EG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7:311–317
Shaw SK, Bamba PS, Perkins BN, Luscinskas FW (2001) Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol 167:2323–2330
Silver PB, Chan C-C, Wiggert B, Caspi RR (1999) The requirement for pertussis to induce EAU is strain-dependent: B10.RIII, but not B10. A mice develop EAU and Th1 responses to IRBP without pertussis treatment. Invest Ophthalmol Vis Sci 40:2898–2905
Smiley ST, King JA, Hancock WW (2001) Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 167:2887–2894
Song L, Pachter JS (2004) Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res 67:78–89
Sonoda KH, Sasa Y, Qiao H, Tsutsumi C, Hisatomi T, Komiyama S, Kubota T, Sakamoto T, Kawano YI, Ishibashi T (2003) Immunoregulatory role of ocular macrophages. The macrophages produce RANTES to suppress experimental autoimmune uveitis. J Immunol 171:2652–2659
Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y (2002) Mice deficient in fractalkine are less susceptible to cerebral ischemia–reperfusion injury. J Neuroimmunol 125:59–65
Stein JV, Cheng G, Stockton BM, Fors BP, Butcher EC, von Andrian UH (1999) L-selectin-mediated leukocyte adhesion in vivo: microvillous distribution determines tethering efficiency, but not rolling velocity. J Exp Med 189:37–50
Streilein JW (2003) Ocular immune privilege: the eye takes a dim but practical view of immunity and inflammation. J Leukoc Biol 74:179–185
Streilein JW, Ksander BR, Taylor AW (1997) Immune deviation in relation to ocular immune privilege. J Immunol 158:3557–3560
Sun B, Rizzo LV, Sun S-H, Chan C-C, Wiggert B, Wilder RL, Caspi RR (1997) Genetic susceptibility to experimental autoimmune uveitis involves more than a predisposition to generate a T Helper-1-like or a T Helper-2-like response. J Immunol 159:1004–1011
Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu JH, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194
Tanaka Y, Adams DH, Shaw S (1993) Proteoglycans on endothelial cells present adhesion-inducing cytokines to leukocytes. Immunol Today 14:111–115
Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF (2007) Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117:902–909
Vestweber D (2000) Molecular mechanisms that control endothelial cell contacts. J Pathol 190:281–291
Weber C, Weber KSC, Klier C, Gu S, Wank R, Horuk R, Nelson PJ (2001) Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and Th1-like/CD45RO+ T cells. Blood 97:1144–1146
Wilkinson-Berka JL (2004) Vasoactive factors and diabetic retinopathy: vascular endothelial growth factor, cycoloxygenase-2 and nitric oxide. Curr Pharm Des 10:3331–3348
Worthylake RA, Burridge K (2001) Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr Opin Cell Biol 13:569–577
Xanthou G, Duchesnes CE, Williams TJ, Pease JE (2003) CCR3 functional responses are regulated by both CXCR3 and its ligands CXCL9, CXCL10 and CXCL11. Eur J Immunol 33:2241–2250
Xu H, Manivannan A, Goatman K, Liversidge J, Sharp PF, Forrester JV, Crane I (2002a) Improved leukocyte tracking in mouse retinal and choroidal circulation. Exp Eye Res 74:403–410
Xu H, Chen M, Mayer EJ, Forrester JV, Dick AD (2007a) Turnover of resident retinal microglia in the normal adult mouse. Glia 55:1189–1198
Xu H, Dawson R, Crane IJ, Liversidge J (2005a) Leukocyte diapedesis in vivo induces transient loss of tight junction protein at the blood–retina barrier. Invest Ophthalmol Vis Sci 46:2487–2494
Xu H, Dawson R, Forrester JV, Liversidge J (2007b) Identification of novel dendritic cell populations in normal mouse retina. Invest Ophthalmol Vis Sci 48:1701–1710
Xu H, Forrester JV, Liversidge J, Crane IJ (2003a) Leukocyte trafficking in experimental autoimmune uveitis: breakdown of blood–retinal barrier and upregulation of cellular adhesion molecules. Invest Ophthalmol Vis Sci 44:226–234
Xu H, Manivannan A, Dawson R, Crane IJ, Mack M, Sharp P, Liversidge J (2005b) Differentiation to the CCR2+ inflammatory phenotype in vivo is a constitutive, time-limited property of blood monocytes and is independent of local inflammatory mediators. J Immunol 175:6915–6923
Xu H, Manivannan A, Goatman KA, Jiang HR, Liversidge J, Sharp PF, Forrester JV, Crane IJ (2004a) Reduction in shear stress, activation of the endothelium, and leukocyte priming are all required for leukocyte passage across the blood–retina barrier. J Leukoc Biol 75:224–232
Xu H, Manivannan A, Jiang HR, Liversidge J, Sharp PF, Forrester JV, Crane IJ (2004b) Recruitment of IFN-gamma-producing (Th1-like) cells into the inflamed retina in vivo is preferentially regulated by P-selectin glycoprotein ligand 1:P/E-selectin interactions. J Immunol 172:3215–3224
Xu H, Manivannan A, Liversidge J, Sharp PF, Forrester JV, Crane IJ (2003b) Requirements for passage of T lymphocytes across non-inflamed retinal microvessels. J Neuroimmunol 142:47–57
Xu H, Manivannan A, Liversidge J, Sharp PF, Forrester JV, Crane IJ (2002b) Involvement of CD44 in leukocyte trafficking at the blood–retinal barrier. J Leukoc Biol 72:1133–1141
Yonekawa K, Harlan IJ (2005) Targeting leukocyte integrins in human diseases. J Leukoc Biol 77:129–140
Zernecke A, Weber KSC, Erwig LP, Kluth DC, Schroppel B, Rees AJ, Weber C (2001) Combinatorial model of chemokine involvement in glomerular monocyte recruitment: role of CXC chemokine receptor 2 in infiltration during nephrotoxic nephritis. J Immunol 166:5755–5762
Zlotnik A, Yoshie O (2000) Chemokines: a new classification system and their role in immunity. Immunity 12:121–127
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Crane, I.J., Liversidge, J. Mechanisms of leukocyte migration across the blood–retina barrier. Semin Immunopathol 30, 165–177 (2008). https://doi.org/10.1007/s00281-008-0106-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-008-0106-7