
Abstract
Purpose
Oral temozolomide is approved in many countries for malignant glioma and for melanoma in some countries outside the USA. This study evaluated the exposure equivalence and safety of temozolomide by intravenous infusion and oral administration.
Methods
Subjects with primary central nervous system malignancies (excluding central nervous system lymphoma) received 200 mg/m2 of oral temozolomide on days 1, 2 and 5. On days 3 and 4, subjects received 150 mg/m2 temozolomide either as a 90-min intravenous infusion on one day or by oral administration on an alternate day.
Results
Ratio of log-transformed means (intravenous:oral) of area under the concentration–time curve and maximum concentration of drug after dosing for temozolomide and 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide (MTIC) met exposure equivalence criteria (90% confidence interval = 0.8–1.25). Treatment-emergent adverse events were consistent with those reported previously in subjects with recurrent glioma treated with oral temozolomide, except for mostly mild and transient injection site reactions with intravenous administration.
Conclusions
This study demonstrated an exposure equivalence of a 90-min intravenous infusion of temozolomide and an equivalent oral dose.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DNA alkylating agents have historically played an important role in systemic chemotherapy for cancer, including brain tumors. The first-generation alkylating agent dacarbazine (DTIC) requires enzymatic conversion to the active cytotoxic metabolite 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide (MTIC) in the liver [1]. There are concerns that the therapeutic potential of DTIC in central nervous system (CNS) malignancies is limited because MTIC may not efficiently penetrate the blood–brain barrier. Temozolomide, an oral alkylating agent, overcomes these limitations. Temozolomide is rapidly absorbed following oral administration, with T max values of approximately 1 h, and undergoes spontaneous pH-dependent hydrolysis to MTIC at physiologic pH with a half-life (t ½) of approximately 1.8 h. MTIC is characterized by the formation of rate-limited pharmacokinetics (PK), with an observed in vivo half-life similar to that of temozolomide. MTIC subsequently degrades by pH-dependent hydrolysis to a reactive methyl-diazonium cation and AIC (5-aminoimidazole-4-carboxamide). Based on data from a limited number of subjects, the systemic bioavailability of oral temozolomide appears to be nearly 100% [2, 3]. Additionally, in a 14C-AME study, the mean recovery of radioactivity in feces was approximately 1% of the administered dose, suggesting that temozolomide is completely absorbed following oral administration. [4] Because temozolomide is lipophilic, it efficiently crosses the blood–brain barrier and is bioavailable to the CNS (20–30% of plasma exposure) [2, 5].
Oral temozolomide is approved in many countries for the treatment of malignant glioma, and is approved for the treatment of melanoma in some regions. Oral temozolomide cannot be administered to all patients, specifically patients with difficulty swallowing capsules. This includes: patients with oropharyngeal dysfunction resulting from increased intracranial pressure/brain stem involvement; patients unable to take oral medications because of gastrointestinal obstruction, intractable nausea and vomiting, or other comorbidities affecting systemic absorption of the drug; and pediatric patients. An intravenous formulation provides a reasonable alternative for these patients.
An exposure equivalence approach was used to develop an intravenous formulation for temozolomide. Previous studies in nonhuman primates [6], dogs (unpublished data) and rats [7] demonstrated similar plasma PK profiles of temozolomide following oral or intravenous administration. Based on these data, a pilot clinical study was conducted to compare the PK profiles of equivalent doses of temozolomide, administered orally or by a 60-min intravenous infusion. The study demonstrated that the area under the curve (AUC) of oral and intravenous temozolomide were similar. However, the ratio (intravenous:oral) of maximum concentration of drug after dosing (C max) did not meet the criteria for exposure equivalence. The key parameter that required optimization was the rate of intravenous infusion so as to better match gastrointestinal absorption kinetics and achieve similar C max values to that achieved via oral administration. Subsequently, Monte Carlo simulations to evaluate virtual crossover exposure equivalence trials using a population PK model derived from a previous population study of oral temozolomide [8] were conducted and suggested that a 90-min intravenous infusion could achieve exposure equivalence with respect to C max (unpublished data). The goal of this pivotal, randomized crossover study was to examine the exposure equivalence and safety profile of a 90-min intravenous infusion of temozolomide compared with an equivalent oral dose.
Methods
Eligibility
Eligible subjects had a diagnosis of a primary CNS tumor (excluding CNS lymphoma), were at least 18 years of age and had a Karnofsky performance score of 70 of higher. All subjects were required to have adequate hematologic, hepatic and renal function. Subjects were excluded if they had impaired gastrointestinal absorption, vomiting or any other medical condition that would compromise the intake of oral medication. Subjects were excluded if they had received chemotherapy or biologic anticancer therapy within 4 weeks before study entry, or mitomycin C or nitrosourea therapy within 6 weeks before study entry. This study was conducted in accordance with good clinical practice (GCP) and in compliance with the World Medical Association Declaration of Helsinki with respect to written informed consent and the protection of rights of human subjects.
Study design
This multicenter, open-label, randomized, crossover study compared the PK of intravenous and oral temozolomide. As this study was conducted in the context of treating subjects with primary CNS malignancies, oral temozolomide was administered at the highest approved dose (200 mg/m2) on days 1, 2 and 5. On days 3 and 4, temozolomide was administered orally on one day and by 90-min intravenous infusion on an alternate day at a dose of 150 mg/m2 (the approved dose of temozolomide for the first cycle of treatment). Subjects were assigned, according to a computer-generated random code, to receive intravenous temozolomide either on day 3 or day 4 with oral temozolomide on an alternate day. All daily oral doses were rounded down to the nearest 5 mg. The doses to be administered on days 3 and 4, intravenous versus oral, were identical. If vomiting occurred during oral dosing, the subject was not redosed. For PK sampling on days 3 and 4, subjects were to fast for a minimum of 8 h before each dose of temozolomide and to continue fasting for 4 h afterward.
The primary objective was to evaluate exposure equivalence of a 90-min intravenous infusion to an equivalent oral dose of temozolomide based on the ratio of the log-transformed means for AUC and C max for both temozolomide and MTIC. Based on regulatory guidelines, exposure equivalence was defined as a 90% confidence interval (CI) for the ratio of the means based on log-transformed data within the range of 80–125% [9, 10]. Secondary end points included local tolerability and safety. Adverse events were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0 over a 28-day period beginning from administration of the first dose of temozolomide.
The PK of temozolomide and MTIC following intravenous and oral administration was determined from serial blood samples taken on days 3 and 4, just before dosing (0 h), and at 0.25, 0.50, 1.00, 1.25, 1.5 (for intravenous dose, within 5 min after the end of infusion), 1.75, 2.00, 2.50, 3.00, 4.00, 6.00 and 8.00 h after initiation of infusion or administration of the oral dose. Plasma temozolomide and MTIC samples were collected and procured as previously described [4, 11, 12]. Briefly, blood samples for MTIC were collected in prechilled heparinized tubes and immediately centrifuged for 10 min at 3,000 rpm at 4°C. The resulting plasma was immediately frozen in a dry ice methanol bath and then stored at -70°C until assayed. Blood samples for temozolomide were collected in prechilled heparinized tubes and then centrifuged for 10 min at 3,000 rpm at 4°C. Immediately following centrifugation, 50 μL of 8.5% phosphoric acid was added to each mL of plasma. Samples were then vortexed and stored at −20°C until assayed. Plasma concentrations of temozolomide and MTIC were determined by liquid chromatography, followed by tandem mass spectroscopy (LC-MS/MS). The lower limit of quantitation (LLOQ) for temozolomide and MTIC were 20 and 5 ng/mL, respectively. These methods were validated for selectivity, sensitivity, precision and accuracy. The stability of temozolomide in plasma after the addition of phosphoric acid and the stability of MTIC in plasma without phosphoric acid under various conditions were established. The temozolomide plasma assay was linear over the range of 20–30,000 ng/mL. The accuracy ranged from −6.8 to −2.1%, and the precision was 9.1–10%. The internal standard was ethazolastone. The MTIC plasma assay was linear over the range of 5–4,000 ng/mL. The accuracy ranged from –3.3 to 0.8% and the precision was 3.1–9.4%. The internal standard was dacarbazine.
The study protocol was written such that data from subjects/samples could be prospectively excluded from the primary analysis in case of protocol violations, unsuccessful dosing or possible sample procurement errors. For instance, subjects who vomited within 4 h of oral dosing on pharmacokinetic days, whose dose on days 3 and 4 were not within 10% of the recommended dose, or subjects whose intravenous infusion duration was not within 10% of 90 min were excluded. Additionally, if anomalous temozolomide or MTIC concentrations were observed (i.e., concentrations below LLOQ, a zero concentration between two non-LLOQ concentrations, or an LLOQ between two non-zero concentrations), the pH of the respective PK sample was checked to ensure that the sample was properly procured. If the sample was not at the recommended pH necessary to stabilize the analyte, the sample was excluded from analyses.
Noncompartmental analyses were conducted on individual concentration–time data. Log-transformed PK parameters (AUC and C max) for temozolomide and MTIC were subjected to a crossover analysis of variance (ANOVA) model, extracting the effects due to treatment, sequence, subject within sequence and period. Assuming an intrasubject variability (coefficient of variation) of 20% and modeling and simulation results, a target enrollment of 20 subjects was selected to provide a minimum of 90% power for the 90% CI of the ratio of the treatment means for derived AUC and C max for temozolomide and MTIC to fall within the 80–125% confidence range.
Results
Subject disposition
A total of 22 subjects were enrolled at three centers. Subject demographics are presented in Table 1. All 22 subjects were randomized and received 5 days of treatment with temozolomide (once-daily oral dosing of temozolomide for 4 days and a single intravenous dose of temozolomide for 1 day, either on day 3 or day 4). Eleven subjects received intravenous treatment on day 3, and 11 subjects received intravenous treatment on day 4. Pharmacokinetic data from three subjects were excluded from the primary analysis as per the prospectively defined exclusion criteria. Two subjects had anomalous temozolomide/MTIC levels [one subject had predose temozolomide and MTIC concentrations that were 45 and 15%, respectively, of the corresponding C max values; the other subject had MTIC concentrations on day 4 that were at or below assay LLOQ for all samples (in case of the latter subject, it was determined that the samples were not procured at the appropriate pH)] and one subject had an interrupted infusion schedule. Thus, 19 subjects were included in the pharmacokinetic analyses.
Pharmacokinetic assessments
The C max, AUC and t ½ for intravenous and orally administered temozolomide were similar and showed low intersubject variability (coefficient of variation [CV] = 12–21%; Table 2). In contrast, the intersubject variability for MTIC C max, AUC(I) and AUC(t f) ranged from 53 to 62%. Individual subject data for the C max of temozolomide are shown in Fig. 1a, and the mean values following intravenous and oral administration were 7.4 and 7.7 μg/mL, respectively. Individual subject data for the C max of MTIC are shown in Fig. 1b. The individual AUC(I) values of temozolomide and MTIC following intravenous and oral administration are shown in Fig. 1c, d.

Paired individual C max for temozolomide (a) and MTIC (b) following intravenous (i.v.) and oral (p.o.) administration. Paired individual AUC(I) for temozolomide (c) and MTIC (d) following intravenous and oral administration. C max maximum concentration of drug after dosing; MTIC 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide, AUC(I) area under the concentration–time curve from 0 h to infinity
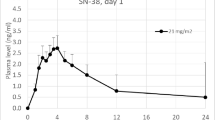
The ratios of the model-based (least-squares) geometric means (intravenous:oral) for the parameters of C max, AUC(I) and AUC(t f) for both temozolomide and MTIC were within ±6% of unity (Table 3). The mean plasma concentration–time profiles for temozolomide and MTIC following temozolomide administration orally or a 90-min intravenous infusion were identical (Fig. 2a, b). The 90% CIs for the ratio of the log-transformed means for C max and AUC (for both temozolomide and MTIC) were within the range for exposure equivalence (80–125%). ANOVA of log-transformed PK parameters (C max and AUC) did not demonstrate any effects due to sequence of administration or period.

Mean plasma concentration–time profiles for temozolomide (a) and MTIC (b) following intravenous (i.v.) and oral (p.o.) administration. MTIC 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide
Safety evaluation (n = 22)
The type and frequency of acute systemic toxicities experienced on days 3 and 4 were similar for intravenous and oral administration (Table 4). All of these adverse events were CTC grade 1 or 2. The most commonly reported adverse events on days 3 and 4 were headache, dizziness, nausea and vomiting. The most commonly reported adverse events occurring throughout the entire 28-day study period are also shown in Table 4. Four subjects experienced 12 severe or life-threatening treatment-emergent adverse events including hematologic toxicities (which is the known dose-limiting toxicity for temozolomide), hydrocephalus and appendicitis (both occurring in the same subject and deemed unrelated to temozolomide), and headache and convulsions (which were consistent with the subject’s underlying disease and deemed unrelated to temozolomide).
Local toxicities were evaluated by subject-reported adverse events and a local tolerability score. Ten subjects reported 11 injection site reactions following intravenous administration. Nearly all events were mild and transient (one subject reported moderate infusion site pain). Ten events reported in nine subjects resolved on the day of the infusion (five events with a duration of 1–6 min, four events with a duration of 15–62 min, and one event with a duration of 131 min), and one event resolved the following day. Injection site reactions included infusion/injection site pain (n = 4), infusion site swelling (n = 1), injection site warmth (n = 2), injection site irritation (n = 2), injection site erythema (n = 1) and pruritus (n = 1). No thrombophlebitis was reported. All subjects with a local tolerability score greater than 0 also had a treatment-emergent injection site adverse event reported. Three subjects required local treatment for these events, consisting of application of ice to the injection site, the use of a pressure bandage, and/or change of injection site location. No subject was unable to complete the infusion or study treatment because of an adverse event at the injection site, although one subject had the injection site changed twice.
Discussion
This pivotal exposure equivalence study was designed to evaluate the bioavailability and safety of a 90-min intravenous infusion of temozolomide compared with an equivalent oral dose. The goal was to establish comparable systemic exposure (C max and AUC) to both temozolomide and its active degradation product MTIC, following intravenous and oral administration. A crossover design was used to decrease variability and the number of subjects required to be enrolled. The crossover design used in this study is analogous to the approach used in other studies that have examined the exposure equivalence of intravenous versus oral administration of chemotherapy agents [13, 14]. In this study, 150 mg/m2 temozolomide, the approved dose of temozolomide for the first cycle of treatment in patients with recurrent glioma, including refractory anaplastic astrocytoma (AA), was the dose chosen for intravenous administration.
The intravenous dose was administered by a 90-min infusion either on day 3 or day 4 in the middle of a 5-day treatment course. Given that temozolomide and MTIC have similar half-lives of approximately 1.8 h [4, 12, 15–17], once-daily dosing on days 3 and 4 with a 24-h washout period (corresponding to approximately 13 half-lives between each dose) was sufficient. In fact, predose (0 h) concentrations for all PK-evaluable subjects in this study were below the LLOQ for both temozolomide and MTIC.
The results of this study showed that a 90-min intravenous infusion and an equivalent oral dose of temozolomide met the exposure equivalence criteria based on the ratio of mean AUC and C max for both temozolomide and MTIC. Treatment-emergent adverse events were consistent with those reported previously in patients with recurrent glioma treated with oral temozolomide, except for local reactions because of intravenous administration. Injection site reactions were mostly mild and transient. No new safety concerns emerged.
The intrasubject variability in AUC and C max for both temozolomide and MTIC was low (CV ≤ 13%; Table 3). The intersubject variability in PK parameters for temozolomide was also low following both intravenous and oral administration. This was not unexpected, as gastrointestinal absorption of temozolomide is rapid and bioavailability is high. The intersubject variability in C max and AUC for MTIC was higher than for temozolomide. Nonetheless, systemic exposure to MTIC was similar within individual subjects receiving intravenous versus oral administration, and the intrasubject variability of MTIC was low. The reason for the higher intersubject variability in MTIC is unclear. However, the low intrasubject variability (8–13%) suggests that random issues with sample handling were not a cause of the higher intersubject variability. In addition, the data were consistent across clinical sites.
In the present study, the PK profile of temozolomide was independent of the route of administration. This is consistent with previous studies evaluating the PK profile of oral temozolomide, which demonstrated that temozolomide exhibits linear pharmacokinetics over the therapeutic dose range [2, 4, 16–19] and that the PK profile of temozolomide is independent of the route of administration (i.e., intravenous, oral or hepatic intra-arterial infusion) [2]. Other studies have demonstrated that total body clearance of temozolomide is linear [8, 16] and independent of dose [16]. Moreover, the PK characteristics of temozolomide have been shown to be independent of dosing schedules [17]. The results of the present study are also consistent with earlier studies that compared the PK of temozolomide administered by intravenous, oral or intra-hepatic routes in a few patients using different formulations [2]. Therefore, the exposure equivalence data obtained from this study allow for direct extrapolation across the range of therapeutically meaningful doses and administration schedules.
In conclusion, this study demonstrated the exposure equivalence of a 90-min intravenous infusion with oral administration of temozolomide. Intravenous administration of temozolomide was generally well tolerated. In clinical practice, oral temozolomide is administered using a variety of doses and schedules. Based on the data from this study and the known PK characteristics of temozolomide, intravenous administration would result in an equivalent exposure compared with oral administration at any given dose and schedule. Potential applications include use in patients in whom oral administration is not feasible because of the inability to swallow, nausea, vomiting or impaired gastrointestinal absorption and in pediatric patients.
References
Tsang LL, Quarterman CP, Gescher A et al (1991) Comparison of the cytotoxicity in vitro of temozolomide and dacarbazine, prodrugs of 3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother Pharmacol 27:342–346
Marzolini C, Decosterd LA, Shen F et al (1998) Pharmacokinetics of temozolomide in association with fotemustine in malignant melanoma and malignant glioma patients: comparison of oral, intravenous, and hepatic intra-arterial administration. Cancer Chemother Pharmacol 42:433–440
Newlands ES, Blackledge GR, Slack JA et al (1992) Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br J Cancer 65:287–291
Baker SD, Wirth M, Statkevich P et al (1999) Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin Cancer Res 5:309–317
Ostermann S, Csajka C, Buclin T et al (2004) Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res 10:3728–3736
Patel M, McCully C, Godwin K et al (2003) Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J Neurooncol 61:203–207
Reyderman L, Statkevich P, Thonoor CM et al (2004) Disposition and pharmacokinetics of temozolomide in rat. Xenobiotica 34:487–500
Jen JF, Cutler DL, Pai SM et al (2000) Population pharmacokinetics of temozolomide in cancer patients. Pharm Res 17:1284–1289
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) (2003) Guidance for industry, bioavailability and bioequivalence studies for orally administered drug products: general considerations. http://www.fda.gov/cder/guidance/5356fnl.pdf. Accessed 3 November 2008
Guideline on the investigation of bioequivalence. http://www.emea.europa.eu/pdfs/human/qwp/140198enrev1.pdf
Hammond LA, Eckardt JR, Baker SD et al (1999) Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J Clin Oncol 17:2604–2613
Rudek MA, Donehower RC, Statkevich P et al (2004) Temozolomide in patients with advanced cancer: phase I and pharmacokinetic study. Pharmacotherapy 24:16–25
Bourgeois H, Vermorken J, Dark G et al (2007) Evaluation of oral versus intravenous dose of vinorelbine to achieve equivalent blood exposures in patients with solid tumours. Cancer Chemother Pharmacol 60:407–413
Marty M, Fumoleau P, Adenis A et al (2001) Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors. Ann Oncol 12:1643–1649
Horton TM, Thompson PA, Berg SL et al (2007) Phase I pharmacokinetic and pharmacodynamic study of temozolomide in pediatric patients with refractory or recurrent leukemia: a Children’s Oncology Group Study. J Clin Oncol 25:4922–4928
Aoki T, Nishikawa R, Mizutani T et al (2007) Pharmacokinetic study of temozolomide on a daily-for-5-days schedule in Japanese patients with relapsed malignant gliomas: first study in Asians. Int J Clin Oncol 12:341–349
Brock CS, Newlands ES, Wedge SR et al (1998) Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res 58:4363–4367
Stevens MF, Hickman JA, Langdon SP et al (1987) Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5, 1-d]-1, 2, 3, 5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res 47:5846–5852
Brada M, Judson I, Beale P et al (1999) Phase I dose-escalation and pharmacokinetic study of temozolomide (SCH 52365) for refractory or relapsing malignancies. Br J Cancer 81:1022–1030
Acknowledgments
We thank Dr. Waldo H. Belloso, Dr. Zenon Beguellín, Dr. Jo Ann Horowitz, Dr. Rudolf Kwan, Dr. Douglas Kramer, Dr. Diego Del Carretto, Jane Devane, Ingrid Banks, Virginia Dawney and Jenny Lewis for their support with this study. Financial support for medical editorial assistance was provided by Schering-Plough. We thank Jerome Sah PhD, ProEd Communications, Inc.®, for his medical editorial assistance with this manuscript.
Conflict of interest statement
P. Statkevich owns stock in Schering-Plough. M.A. Abutarif and D. Cutler have received remuneration from and own stock in Schering-Plough. B.D. Diez, Y. Zhu, F. Xuan, B. Kantesaria, M. Schwarz and M.G. Pallotta have nothing to disclose.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Fabio H. Ottaviano: deceased (2 February 1971–4 November 2006).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Diez, B.D., Statkevich, P., Zhu, Y. et al. Evaluation of the exposure equivalence of oral versus intravenous temozolomide. Cancer Chemother Pharmacol 65, 727–734 (2010). https://doi.org/10.1007/s00280-009-1078-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1078-6