
Abstract
Myelofibrosis (MF) is a myeloproliferative neoplasm characterized by mutations (most frequently in JAK2, CALR, or MPL), burdensome symptoms, splenomegaly, cytopenia, and shortened life expectancy. In addition to other clinical manifestations, patients with MF often develop anemia, which can either be directly related to MF pathogenesis or a result of MF treatment with Janus kinase (JAK) inhibitors, such as ruxolitinib and fedratinib. Although symptoms and clinical manifestations can be similar between the 2 anemia types, only MF-related anemia is prognostic of reduced survival. In this review, I detail treatment and patient management approaches for both types of anemia presentations and provide recommendations for the treatment of MF in the presence of anemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myelofibrosis (MF) is a myeloproliferative neoplasm (MPN) characterized by abnormal megakaryocyte proliferation, along with reticulin or collagen fibrosis [1]. Nearly all patients (≈90%) have activating mutations in either JAK2, CALR, or MPL, which cause abnormal signaling that promotes cell proliferation and survival, as well as activation of several inflammation pathways [2,3,4,5,6]. MF clinical manifestations typically include anemia, thrombocytopenia, splenomegaly, and hepatomegaly that when combined can lead to burdensome symptoms such as fatigue, abdominal discomfort, night sweats, bone pain, and pruritus that impact patients’ quality of life [7, 8]. In addition to these burdensome signs and symptoms, patients with MF have increased risk of thrombosis and increased risk of progression to acute leukemia, which both also contribute to reduced survival compared with healthy controls [9, 10].
Anemia, at times reaching severe levels (< 8 g/dL), can be present at MF diagnosis and worsen over time as disease progresses (MF-related anemia), or it can manifest as a result of MF treatment with Janus kinase (JAK) inhibitors (treatment-related anemia) [11,12,13,14,15]. Although symptoms and clinical manifestations can be similar, only MF-related anemia is prognostic of reduced survival [12,13,14,15]. This review provides guidance for managing patients with either type of anemia presentation.
Sample patient—part 1
A 68-year-old female patient presented with shortness of breath. During a physical examination, she was found to have an enlarged spleen of 7 cm below the costal margin and no other significant findings. She also reported fatigue, significant night sweating, and some weight loss. Laboratory results indicated hemoglobin (Hb) of 9.7 g/dL, a white blood cell (WBC) count of 22 × 109/L with 2% blasts, and a platelet count of 122 × 109/L. Lactate dehydrogenase and erythropoietin (EPO) were both elevated (1780 U/L and 35 mU/mL, respectively). Furthermore, a bone marrow biopsy was compatible with MF.
General treatment of MF
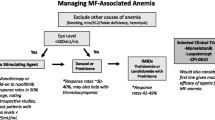
In my practice, we would first determine a prognosis of a patient by risk stratification (Fig. 1). There are several prognostic scoring systems in use, among which the Mutation and Karotype-Enhanced IPSS (MIPSS-70 + VERSION 2.0) is probably the most comprehensive, with additional options including Dynamic International Prognostic Scoring System (DIPSS)-Plus if molecular testing is not available, DIPSS if karyotyping is not available, and Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) for secondary MF. We consider a patient to be at higher risk if their risk score corresponds to high, intermediate-2, or a score in the higher intermediate range, consistent with the National Comprehensive Cancer Network guidelines [16]; such patients are typically referred to a stem cell transplant specialist for consideration of transplant procedure.

Treatment flow chart for patients with MF-associated anemia. Hematopoietic stem cell transplant should also be considered for eligible patients with high-risk disease. MF, myelofibrosis
For patients with symptomatic disease, either lower- or higher-risk MF, typical treatment choice is the oral selective JAK1/JAK2 inhibitor ruxolitinib [16]. Ruxolitinib was the first JAK inhibitor approved for the treatment of MF by the US Food and Drug Administration in 2011 [17], and treatment prolongs survival, reduces symptom burden, and reduces spleen volume, as demonstrated by multiple clinical trials [18,19,20]. Risk of death, as assessed by the prognostic scoring systems mentioned above, does not guide our decision on prescribing medications to control symptoms. Our alternative choice to ruxolitinib is the JAK2 inhibitor fedratinib, approved in the US for patients with intermediate-2 or high-risk MF, which we typically use in the second-line setting [16, 21]. For cytoreduction of high WBC or platelet counts, which are sometimes seen in patients with lower-risk, early, or prefibrotic MF, peginterferon alfa-2a or hydroxyurea are used [16]. For higher-risk patients with MF with severe thrombocytopenia (platelets < 50 × 109/L), pacritinib, an oral selective JAK2 and interleukin-1 receptor-associated kinase (IRAK1) inhibitor, is my preferred first-line treatment option [16, 22, 23]. Although both fedratinib and pacritinib provide clinical benefits, longer follow-up studies are required to determine if either provides an overall survival benefit as seen with ruxolitinib [23, 24].
Treatment options for anemia resulting from MF
Mild to moderate anemia is often present at MF diagnosis and can worsen with disease progression. Importantly, unlike JAK inhibitor treatment–related anemia discussed later in this review, MF-related anemia is associated with reduced overall survival, so proper management is essential [14]. An overview of treatment recommendations for patients with MF-related anemia in my practice is presented in Fig. 1 [16]. Patients should first be evaluated for contributing factors, including MF-related factors and exacerbating causes not directly related to MF. MF-related causes include reduced erythropoiesis, splenomegaly, and inflammatory cytokines [12, 25]. Additionally, vitamin B6, an essential element of heme synthesis, may be deficient in patients with primary or secondary MF, leading to anemia [26]. Functional iron deficiency due to inflammation is also frequently observed in patients with MF [27, 28], in which pro-inflammatory cytokine signaling upregulates hepcidin that in turn promotes storage of iron and ultimately iron-restricted anemia [29]. Functional iron deficiency is identifiable by low transferrin saturation despite normal ferritin levels [27, 28], and these patients should be treated with intravenous iron [29]. In addition, new targeted therapies are in development to modulate hepcidin signaling, including the JAK1/JAK2 and type 1 kinase activin A receptor or activin receptor-like kinase-2 (ACVR1/ALK2) inhibitor momelotinib (discussed in greater detail in the “Treatment options for anemia resulting from JAK inhibition” section) [30, 31] and the ACVR1/ALK2 inhibitor INCB000928 [32]. Although seemingly very rare, cases of patients developing primary MF and autoimmune hemolytic anemia have been reported [33]. Exacerbating causes not directly related to MF include underlying occult or gastrointestinal bleeding and deficiencies in iron folate and vitamin B12, which can lead to megaloblastic anemia [12, 25, 34, 35]. Deficiencies in iron folate and vitamin B12 are not uncommon in elderly patient populations [36] such as the MF population and are reversible via dietary or vitamin supplementation [35]. For patients with contributing factors not related to MF, the underlying cause should be treated per appropriate guidelines, and patients should be treated normally for MF, regardless of anemia presence [16, 35].
Management of patients with MF-related anemia begins with blood transfusions, with subsequent evaluation for additional anemia treatments [16]. For patients with serum EPO < 500 mU/mL, erythropoiesis-stimulating agents (ESAs) are a viable option that offers clinical benefits [16]. Up to half of the patients in this population may achieve an anemia response with ESAs, and dose escalation should be considered to achieve full benefit [37]. Importantly, ESAs can be safely added to ruxolitinib to effectively improve anemia in some patients with MF [38]. Additional treatment options are available for patients with serum EPO ≥ 500 mU/mL. The erythroid maturation agent, luspatercept, has demonstrated anemia benefits in patients with MF and myelodysplastic syndrome/MPN with ring sideroblasts who carry the SF3B1 mutation [39, 40]. It is important to note that the studies that evaluated luspatercept in MF had small patient populations, and additional investigation is warranted to further evaluate safety and efficacy. Anabolic steroid medication such as danazol can also be used for the treatment of anemia in patients with MF [16, 25]. Danazol treatment has been associated with an anemia response in these patients, including those who are transfusion-dependent [41]. Immunomodulatory imide agents (IMiDs), such as thalidomide and lenalidomide, have also demonstrated an anemia benefit in patients with MF, including those who were transfusion-dependent [42, 43]. However, this benefit was not observed in patients with myeloid metaplasia with MF who received thalidomide [44] or those with MF treated with pomalidomide, another IMiD [45]. Importantly, various treatments can be combined with ongoing ruxolitinib treatment, although the coadministration of IMiDs with steroids is currently a topic of debate. The combination of ruxolitinib with prednisone, thalidomide, and danazol has been associated with an anemia benefit in patients with MF [46]. Similarly, luspatercept combined with ruxolitinib demonstrated transfusion independence in some patients with MF [39]. Details for studies of ruxolitinib in combination with other agents, including ongoing/exploratory trials, are shown in Table 1.
MF treatment considerations in patients with MF-related anemia
In general, MF treatment is initiated as early as possible for symptomatic patients in my practice, as supported by clinical trial evidence. A pooled analysis of the COMFORT I/II trials suggested that earlier ruxolitinib initiation in patients with intermediate-2 or high-risk MF was associated with improved clinical outcomes including fewer anemia events [47]. In addition, a post hoc analysis of the phase 3 JUMP trial demonstrated that a lower IPSS score at treatment initiation was associated with better spleen response rates, suggesting that ruxolitinib treatment earlier in the disease course improves response [48]. This should be balanced by possible lead-time bias and the known relationship between lower MF disease stage and better spleen response in patients treated with ruxolitinib [49]. Nonetheless, treating MF as early as possible, before the onset of MF-related anemia, should improve outcomes, both because of the direct benefit of early intervention and indirectly due to potentially avoiding the negative outcomes associated with MF-related anemia itself.
For patients who develop MF-related anemia, anemia is not a driver for primary treatment choice and therefore is managed based on my practice’s standard MF treatment algorithm (Fig. 1). In particular, ruxolitinib is not contraindicated in patients with anemia [17]. In the COMFORT I/II trials, ruxolitinib was associated with prolonged survival in patients with MF compared with controls, regardless of baseline anemia status [14]. Regarding the choice of ruxolitinib dose, my practice follows in many patients the approach evaluated in the phase 2 REALISE trial, which established a novel ruxolitinib dosing strategy for patients with anemia based on a lower ruxolitinib starting dose (10 mg twice daily [bid] with up-titration as necessary based on platelet counts and efficacy; Fig. 2) [12]. REALISE demonstrated that patients with baseline anemia experienced improvements in spleen size and MF-related symptoms with ruxolitinib treatment, and median Hb levels remained stable throughout the study, with red blood cell (RBC) transfusion requirements decreasing or remaining stable [12].

REALISE dosing strategy for ruxolitinib in patients with MF-associated anemia. bid, twice daily; BSL, baseline spleen length; PLT, platelet count; RUX, ruxolitinib; SL, spleen length. Figure reproduced from Cervantes F, et al. Leukemia. 2021;35(12):3455–3465, under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) license (https://creativecommons.org/licenses/by/4.0/). Edits for style were made
Sample patient—Sect. 2
The patient was not interested in undergoing a hematopoietic stem cell transplant right at diagnosis and was prescribed ruxolitinib 15 mg bid, as recommended for platelet counts between 100 and 200 × 109/L. During follow-up 3 weeks after treatment initiation, the patient reported feeling better, eating more, and tolerating the treatment well. Upon examination, the spleen was smaller in size, at 2 cm below the costal margin. Laboratory results showed Hb of 8.7 g/dL, a platelet count of 67 × 109/L, and a WBC count of 14 × 109/L. Due to the decrease observed in platelet count, the patient was now prescribed a decreased dose of ruxolitinib, at 10 mg bid.
After one more month of therapy, the patient reported feeling much better than before ruxolitinib. The spleen size remained at 2 cm below the costal margin, and laboratory reports showed Hb at 7.5 g/dL, a platelet count of 82 × 109/L, and a WBC count of 17 × 109/L.
Treatment options for anemia resulting from JAK inhibition
Although MF itself can lead to anemia, JAK inhibition may also separately cause or exacerbate anemia, which often occurs early in treatment and gradually improves with long-term exposure [13, 14, 17,18,19, 50]. In the COMFORT studies, the number of patients with grade 3 or 4 anemia was higher for ruxolitinib compared with placebo; however, the lowest Hb levels were observed at weeks 8 to 12 of treatment and recovered to near-baseline levels by week 24 [14, 18, 19]. Furthermore, the number of patients with grade 3 or 4 anemia decreased over 42 months of treatment, with no patients reporting new or worsening grade 3 or 4 anemia after month 42 of treatment [13]. Importantly, new or worsening postbaseline anemia did not affect survival probability during ruxolitinib treatment in the COMFORT I/II pooled analysis [14]. In fact, patients with postbaseline anemia who received ruxolitinib had a survival advantage compared with the overall control group [14]. Likewise, transfusion dependence did not affect the survival benefit observed with ruxolitinib treatment in the COMFORT studies [20]. Similar to observations with ruxolitinib, in the JAKARTA studies of fedratinib in MF, a decrease in Hb levels was observed for 12 to 16 weeks, with a partial recovery observed afterward in the 400-mg group [50]. Taken together, these findings demonstrate that treatment-induced anemia as a result of JAK inhibition can be temporary.
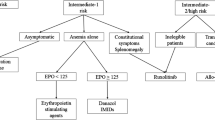
In general, management for treatment-related anemia follows the same pattern described above for MF-related anemia, where contributing factors should first be assessed and treated appropriately. In the absence of non-MF-related contributing factors, primary management includes RBC transfusion and potential addition of secondary anemia treatments (Fig. 3). If transfusions and secondary treatment options are insufficient or burdensome, JAK inhibitor dose reduction can be considered to help improve anemia [17, 21]. After recovery of anemia to acceptable levels, ruxolitinib should be continued at the given dose or with subsequent modifications if necessary. Complete blood counts should be monitored every 2 to 4 weeks until doses are stabilized [16]. I try to avoid interruptions in therapy with ruxolitinib, as it has been reported that patients may have a significant rebound in symptoms within 7 to 10 days upon sudden interruption of ruxolitinib [16].

Treatment flow chart for patients with MF and treatment-induced anemia. CBC, complete blood count; JAK, Janus kinase; MF, myelofibrosis
The JAK1/JAK2 inhibitor momelotinib, currently under investigation for patients with MF and anemia, has potent inhibitory activity against ACVR1/ALK2 and may become a second-line treatment option for patients who have to eventually stop ruxolitinib due to excessive anemia [30, 31]. This mechanism of action includes suppression of aberrant activation of hepcidin transcription in the liver and thus may improve iron homeostasis, facilitating normalized Hb levels and a decrease in transfusion requirements [30, 31]. In the phase 3 MOMENTUM trial of momelotinib versus danazol in patients with intermediate or high-risk MF previously treated with a JAK inhibitor, momelotinib provided superior clinical benefit as assessed by Myelofibrosis Symptom Assessment Form Total Symptom Score (MFSAF TSS) response and spleen response rate, as well as noninferiority for transfusion independence rate [51].
Sample patient—Sect. 3
The patient continued ruxolitinib treatment and underwent a transfusion with packed RBCs. In addition, anemia medication was provided, including an ESA as serum EPO was < 500 mU/mL. Follow-up was scheduled for every 3 to 4 weeks. After 6 months of therapy, the patient’s Hb was 8.4 g/dL, the platelet count was 77 × 109/L, and the WBC count was 12 × 109/L. The spleen was no longer palpable, no transfusions were needed, and the patient reported no symptoms.
Conclusions
Patients with MF endure burdensome symptoms and coexisting conditions as a result of their disease. In particular, patients commonly develop anemia, which can either be secondary to the disease or a result of MF treatment, further complicating disease management. Although MF treatment with JAK inhibitors can exacerbate anemia, evidence suggests that this is typically temporary and, as in the case of ruxolitinib, does not reduce survival contrary to MF-related anemia. MF treatment should be initiated as early as possible for symptomatic patients, ideally before the onset of MF-related anemia, to maximize clinical benefit. For those patients with MF who develop anemia, careful patient management, including RBC transfusions, secondary anemia treatments, JAK inhibitor dose modifications, and monitoring, can improve anemia to prevent further disease complications and improve clinical outcomes.
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405. https://doi.org/10.1182/blood-2016-03-643544
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schonegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369(25):2379–2390
Guijarro-Hernandez A, Vizmanos JL (2021) A broad overview of signaling in Ph-negative classic myeloproliferative neoplasms. Cancers (Basel) 13(5):984. https://doi.org/10.3390/cancers13050984
Geyer HL, Dueck AC, Scherber RM, Mesa RA (2015) Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflamm 2015:284706. https://doi.org/10.1155/2015/284706
Fisher DAC, Fowles JS, Zhou A, Oh ST (2021) Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol 12:683401. https://doi.org/10.3389/fimmu.2021.683401
Aguirre LEE, Jain AG, Ball S, Al Ali N, Tinsley-Vance SM, Sallman DA, Sweet K, Lancet JE, Padron E, Kuykendall AT, Komrokji RS (2021) Triple-negative myelofibrosis: disease features, response to treatment and outcomes. Blood 138(suppl 1):1494. https://doi.org/10.1182/blood-2021-151978
Tefferi A (2021) Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 96(1):145–162. https://doi.org/10.1002/ajh.26050
Mesa R, Miller CB, Thyne M, Mangan J, Goldberger S, Fazal S, Ma X, Wilson W, Paranagama DC, Dubinski DG, Boyle J, Mascarenhas JO (2016) Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer 16:167. https://doi.org/10.1186/s12885-016-2208-2
Price GL, Davis KL, Karve S, Pohl G, Walgren RA (2014) Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS One 9(3):e90299. https://doi.org/10.1371/journal.pone.0090299
Hultcrantz M, Kristinsson SY, Andersson TM, Landgren O, Eloranta S, Derolf AR, Dickman PW, Björkholm M (2012) Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 30(24):2995–3001. https://doi.org/10.1200/JCO.2012.42.1925
Badireddy M, Baradhi KM (2022) Chronic anemia. In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Cervantes F, Ross DM, Radinoff A, Palandri F, Myasnikov A, Vannucchi AM, Zachee P, Gisslinger H, Komatsu N, Foltz L, Mannelli F, Passamonti F, Gilotti G, Sadek I, Tiwari R, Zor E, Al-Ali HK (2021) Efficacy and safety of a novel dosing strategy for ruxolitinib in the treatment of patients with myelofibrosis and anemia: the REALISE phase 2 study. Leukemia 35(12):3455–3465. https://doi.org/10.1038/s41375-021-01261-x
Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, Arcasoy MO, Hexner EO, Lyons RM, Paquette R, Raza A, Jones M, Kornacki D, Sun K, Kantarjian H, Comfort-I investigators, (2017) Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 10(1):55. https://doi.org/10.1186/s13045-017-0417-z
Gupta V, Harrison C, Hexner EO, Al-Ali HK, Foltz L, Montgomery M, Sun W, Gopalakrishna P, Kantarjian H, Verstovsek S (2016) The impact of anemia on overall survival in patients with myelofibrosis treated with ruxolitinib in the COMFORT studies. Haematologica 101(12):e482–e484. https://doi.org/10.3324/haematol.2016.151449
Cervantes F, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Sirulnik A, Stalbovskaya V, McQuitty M, Hunter DS, Levy RS, Passamonti F, Barbui T, Barosi G, Harrison CN, Knoops L, Gisslinger H, COMFORT-II investigators (2013) Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 122(25):4047–4053. https://doi.org/10.1182/blood-2013-02-485888
National Comprehensive Cancer Network (2022) NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Myeloproliferative Neoplasms, version 3.2022. https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf. Accessed 9 Feb 2023
JAKAFI® (ruxolitinib). Full Prescribing Information, Incyte Corporation, Wilmington, DE, USA, 2021
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM (2012) A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 366(9):799–807. https://doi.org/10.1056/NEJMoa1110557
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G (2012) JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 366(9):787–798. https://doi.org/10.1056/NEJMoa1110556
Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, Harrison CN, Paquette R, Sun W, Naim A, Langmuir P, Dong T, Gopalakrishna P, Gupta V (2017) Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 10(1):156. https://doi.org/10.1186/s13045-017-0527-7
Inrebic® (fedratinib). Full Prescribing Information, Celgene Corporation, Summit, NJ, USA, 2019
VONJO® (pacritinib). Full Prescribing Information, CTI BioPharma Corporation, Seattle, WA, 2022
Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, Szoke A, Drummond M, Pristupa A, Granston T, Daly R, Al-Fayoumi S, Callahan JA, Singer JW, Gotlib J, Jamieson C, Harrison C, Mesa R, Verstovsek S (2018) Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol 4(5):652–659. https://doi.org/10.1001/jamaoncol.2017.5818
Waksal JA, Tremblay D, Mascarenhas J (2021) Clinical utility of fedratinib in myelofibrosis. Onco Targets Ther 14:4509–4521. https://doi.org/10.2147/ott.S267001
Stein BL (2021) Management of myelofibrosis-associated anemia: focus on standard agents and novel therapeutics in phase 3 clinical trials. Curr Hematol Malig Rep 16(5):483–489. https://doi.org/10.1007/s11899-021-00651-3
Yasuda H, Tsutsui M, Ando J, Inano T, Noguchi M, Yahata Y, Tanaka M, Tsukune Y, Masuda A, Shirane S, Misawa K, Gotoh A, Sato E, Aritaka N, Sekiguchi Y, Sugimoto K, Komatsu N (2019) Vitamin B6 deficiency is prevalent in primary and secondary myelofibrosis patients. Int J Hematol 110(5):543–549. https://doi.org/10.1007/s12185-019-02717-8
Birgegard G, Samuelsson J, Ahlstrand E, Ejerblad E, Enevold C, Ghanima W, Hasselbalch H, Nielsen CH, Knutsen H, Pedersen OB, Sørensen A, Andreasson B (2019) Inflammatory functional iron deficiency common in myelofibrosis, contributes to anaemia and impairs quality of life. From the Nordic MPN Study Group. Eur J Haematol 102(3):235–240. https://doi.org/10.1111/ejh.13198
Lucijanic M, Prka Z, Pejsa V, Stoos-Veic T, Lucijanic J, Kusec R (2018) Prognostic implications of low transferrin saturation in patients with primary myelofibrosis. Leuk Res 66:89–95. https://doi.org/10.1016/j.leukres.2018.01.017
Busti F, Marchi G, Ugolini S, Castagna A, Girelli D (2018) Anemia and iron deficiency in cancer patients: role of iron replacement therapy. Pharmaceuticals (Basel) 11(4):94. https://doi.org/10.3390/ph11040094
Chifotides HT, Bose P, Verstovsek S (2022) Momelotinib: an emerging treatment for myelofibrosis patients with anemia. J Hematol Oncol 15(1):7. https://doi.org/10.1186/s13045-021-01157-4
Verstovsek S, Chen CC, Egyed M, Ellis M, Fox L, Goh YT, Gupta V, Harrison C, Kiladjian JJ, Lazaroiu MC, Mead A, McLornan D, McMullin MF, Oh ST, Perkins A, Platzbecker U, Scheid C, Vannucchi A, Yoon SS, Kowalski MM, Mesa RA (2021) MOMENTUM: momelotinib vs danazol in patients with myelofibrosis previously treated with JAKi who are symptomatic and anemic. Future Oncol 17(12):1449–1459. https://doi.org/10.2217/fon-2020-1048
Mohan SR, Oh ST, Ali H, Hunter AM, Palandri F, Lamothe B, Cui Y, Seguy F, McBride A, Savona MR, Kiladjian J-J, Verstovsek S (2022) A phase 1/2 study of INCB000928 as monotherapy or combined with ruxolitinib (RUX) in patients (Pts) with anemia due to myelofibrosis (MF). Blood 140(suppl 1):3943–3944. https://doi.org/10.1182/blood-2022-169210
Kornblihtt LI, Vassalllu PS, Heller PG, Lago NR, Alvarez CL, Molinas FC (2008) Primary myelofibrosis in a patient who developed primary biliary cirrhosis, autoimmune hemolytic anemia and fibrillary glomerulonephritis. Ann Hematol 87(12):1019–1020. https://doi.org/10.1007/s00277-008-0516-6
Warner MJ, Kamran MT (2021) Iron deficiency anemia. StatPearls Publishing, Treasure Island, FL
Cervantes F, Correa JG, Hernandez-Boluda JC (2016) Alleviating anemia and thrombocytopenia in myelofibrosis patients. Expert Rev Hematol 9(5):489–496. https://doi.org/10.1586/17474086.2016.1154452
Laird EJ, O’Halloran AM, Carey D, O’Connor D, Kenny RA, Molloy AM (2018) Voluntary fortification is ineffective to maintain the vitamin B12 and folate status of older Irish adults: evidence from the Irish Longitudinal Study on Ageing (TILDA). Br J Nutr 120(1):111–120. https://doi.org/10.1017/S0007114518001356
Hernández-Boluda JC, Correa JG, García-Delgado R, Martínez-López J, Alvarez-Larrán A, Fox ML, García-Gutiérrez V, Pérez-Encinas M, Ferrer-Marín F, Mata-Vázquez MI, Raya JM, Estrada N, García S, Kerguelen A, Durán MA, Albors M, Cervantes F (2017) Predictive factors for anemia response to erythropoiesis-stimulating agents in myelofibrosis. Eur J Haematol 98(4):407–414. https://doi.org/10.1111/ejh.12846
Crisà E, Cilloni D, Elli EM, Martinelli V, Palumbo GA, Pugliese N, Beggiato E, Frairia C, Cerrano M, Lanzarone G, Marchetti M, Mezzabotta M, Boccadoro M, Ferrero D (2018) The use of erythropoiesis-stimulating agents is safe and effective in the management of anaemia in myelofibrosis patients treated with ruxolitinib. Br J Haematol 182(5):701–704. https://doi.org/10.1111/bjh.15450
Gerds AT, Vannucchi AM, Passamonti F, Kremyanskaya M, Gotlib JR, Palmer JM, McCaul K, Ribrag V, Mead AJ, Harrison CN, Mesa RA, Kiladjian J-J, Barosi G, Gale RP, Laadem A, Pariseau J, Gerike T, Zhang J, Linde PG, Reynolds JG, Verstovsek S (2019) A phase 2 study of luspatercept in patients with myelofibrosis-associated anemia. Blood 134(suppl 1):557. https://doi.org/10.1182/blood-2019-122546
Komrokji RS, Platzbecker U, Fenaux P, Garcia-Manero G, Mufti GJ, Santini V, Diez-Campelo M, Finelli C, Jurcic JG, Greenberg PL, Sekeres MA, Zeidan AM, DeZern AE, Savona MR, Shetty JK, Ito R, Zhang G, Ha X, Sinsimer D, Backstrom JT, Verma A (2020) Efficacy and safety of luspatercept treatment in patients with myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T): a retrospective analysis from the Medalist study. Blood 136(suppl 1):13–15. https://doi.org/10.1182/blood-2020-137232
Cervantes F, Isola IM, Alvarez-Larrán A, Hernández-Boluda JC, Correa JG, Pereira A (2015) Danazol therapy for the anemia of myelofibrosis: assessment of efficacy with current criteria of response and long-term results. Ann Hematol 94(11):1791–1796. https://doi.org/10.1007/s00277-015-2435-7
Castillo-Tokumori F, Talati C, Al Ali N, Sallman D, Yun S, Sweet K, Padron E, Lancet J, Komrokji R, Kuykendall AT (2020) Retrospective analysis of the clinical use and benefit of lenalidomide and thalidomide in myelofibrosis. Clin Lymphoma Myeloma Leuk 20(12):e956–e960. https://doi.org/10.1016/j.clml.2020.07.006
Marchetti M, Barosi G, Balestri F, Viarengo G, Gentili S, Barulli S, Demory JL, Ilariucci F, Volpe A, Bordessoule D, Grossi A, Le Bousse-Kerdiles MC, Caenazzo A, Pecci A, Falcone A, Broccia G, Bendotti C, Bauduer F, Buccisano F, Dupriez B (2004) Low-dose thalidomide ameliorates cytopenias and splenomegaly in myelofibrosis with myeloid metaplasia: a phase II trial. J Clin Oncol 22(3):424–431. https://doi.org/10.1200/JCO.2004.08.160
Abgrall JF, Guibaud I, Bastie JN, Flesch M, Rossi JF, Lacotte-Thierry L, Boyer F, Casassus P, Slama B, Berthou C, Rodon P, Leporrier M, Villemagne B, Himberlin C, Ghomari K, Larosa F, Rollot F, Dugay J, Allard C, Maigre M, Isnard F, Zerbib R, Cauvin JM, Groupe Ouest-Est Leucemies et Maladies du Sang (GOELAMS) (2006) Thalidomide versus placebo in myeloid metaplasia with myelofibrosis: a prospective, randomized, double-blind, multicenter study. Haematologica 91(8):1027–1032
Tefferi A, Al-Ali HK, Barosi G, Devos T, Gisslinger H, Jiang Q, Kiladjian JJ, Mesa R, Passamonti F, McMullin MF, Ribrag V, Schiller G, Vannucchi AM, Zhou D, Reiser D, Zhong J, Gale RP (2017) A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm-associated myelofibrosis and RBC-transfusion dependence. Leukemia 31(5):1252. https://doi.org/10.1038/leu.2017.2
Qu S, Xu Z, Qin T, Li B, Pan L, Chen J, Yan X, Wu J, Zhang Y, Zhang P, Gale RP, Xiao Z (2022) Ruxolitinib combined with prednisone, thalidomide and danazol in patients with myelofibrosis: results of a pilot study. Hematol Oncol 40(4):787–795. https://doi.org/10.1002/hon.3026
Verstovsek S, Kiladjian J-J, Vannucchi AM, Mesa RA, Scherber R, Hamer-Maansson JE, Harrison CN (2021) Does early intervention in myelofibrosis impact outcomes? A pooled analysis of the COMFORT I and II studies. Blood 138(suppl 1):1505. https://doi.org/10.1182/blood-2021-150894
Gupta V, Griesshammer M, Martino B, Foltz L, Tavares R, Al-Ali HK, Giraldo P, Guglielmelli P, Lomaia E, Bouard C, Paley C, Tiwari R, Zor E, Raanani P (2021) Analysis of predictors of response to ruxolitinib in patients with myelofibrosis in the phase 3b expanded-access JUMP study. Leuk Lymphoma 62(4):918–926. https://doi.org/10.1080/10428194.2020.1845334
Palandri F, Palumbo GA, Bonifacio M, Tiribelli M, Benevolo G, Martino B, Abruzzese E, D’Adda M, Polverelli N, Bergamaschi M, Tieghi A, Cavazzini F, Ibatici A, Crugnola M, Bosi C, Latagliata R, Di Veroli A, Scaffidi L, de Marchi F, Cerqui E, Anaclerico B, De Matteis G, Spinsanti M, Sabattini E, Catani L, Aversa F, Di Raimondo F, Vitolo U, Lemoli RM, Fanin R, Merli F, Russo D, Cuneo A, Bacchi Reggiani ML, Cavo M, Vianelli N, Breccia M (2017) Baseline factors associated with response to ruxolitinib: an independent study on 408 patients with myelofibrosis. Oncotarget 8(45):79073–79086. https://doi.org/10.18632/oncotarget.18674
Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, Masszi T, Mishchenko E, Jourdan E, Vannucchi AM, Drummond MW, Jurgutis M, Kuliczkowski K, Gheorghita E, Passamonti F, Neumann F, Patki A, Gao G, Tefferi A (2015) Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol 1(5):643–651. https://doi.org/10.1001/jamaoncol.2015.1590
Mesa RA, Gerds AT, Vannucchi A, Al-Ali HK, Lavie D, Kuykendall AT, Grosicki S, Iurlo A, Goh YT, Lazaroiu MC, Egyed M, Fox ML, McLornan DP, Perkins A, Yoon S-S, Gupta V, Kiladjian J-J, Donahue R, Kawashima J, Verstovsek S (2022) MOMENTUM: phase 3 randomized study of momelotinib (MMB) versus danazol (DAN) in symptomatic and anemic myelofibrosis (MF) patients previously treated with a JAK inhibitor. J Clin Oncol 40(16 suppl):7002–7002. https://doi.org/10.1200/JCO.2022.40.16_suppl.7002
Gowin K, Kosiorek H, Dueck A, Mascarenhas J, Hoffman R, Reeder C, Camoriano J, Tibes R, Gano K, Palmer J, Mesa R (2017) Multicenter phase 2 study of combination therapy with ruxolitinib and danazol in patients with myelofibrosis. Leuk Res 60:31–35
Gerds AT, Vannucchi AM, Passamonti F, Kremyanskaya M, Gotlib J, Palmer JM, McCaul K, Ribrag V, Mead AJ, Harrison C, Mesa R, Kiladjian J-J, Barosi G, Gerike TG, Shetty JK, Pariseau J, Miranda G, Schwickart M, Giuseppi AC, Zhang J, Backstrom JT, Verstovsek S (2020) Duration of response to luspatercept in patients (pts) requiring red blood cell (RBC) transfusions with myelofibrosis (MF) - updated data from the phase 2 ACE-536-MF-001 study. Blood 136(suppl 1):47–48. https://doi.org/10.1182/blood-2020-137265
Bose P, Daver N, Pemmaraju N, Jabbour EJ, Estrov Z, Pike A, Huynh-Lu J, Nguyen-Cao M, Wang X, Zhou L, Pierce S, Kantarjian HM, Verstovsek S (2017) Sotatercept (ACE-011) alone and in combination with ruxolitinib in patients (pts) with myeloproliferative neoplasm (MPN)-associated myelofibrosis (MF) and anemia. Blood 130(suppl 1):255. https://doi.org/10.1182/blood.V130.Suppl_1.255.255
Rampal RK, Verstovsek S, Devlin SM, King AC, Stein EM, Pemmaraju N, Mauro MJ, Kadia TM, Montalban-Bravo G, Alvarez K, Ard N, Goodman T, Taylor B, Bose P (2019) Safety and efficacy of combined ruxolitinib and thalidomide in patients with myelofibrosis: a phase II study. Blood 134(suppl 1):4163. https://doi.org/10.1182/blood-2019-127661
Acknowledgements
This study was funded by Incyte Corporation (Wilmington, DE, USA). Writing assistance was provided by Nicole Farra, PhD, an employee of ICON (Blue Bell, PA, USA), and was funded by Incyte.
Author information
Authors and Affiliations
Contributions
SV contributed substantially to the conception, content development, writing, and review, and approved submission.
Corresponding author
Ethics declarations
Competing interests
SV received research support from AstraZeneca, Blueprints Medicines Corp., Celgene, CTI BioPharma Corp., Genentech, Gilead, Incyte, ItalPharma, Novartis, NS Pharma, PharmaEssentia, Promedior, Protagonist Therapeutics, Roche, and Sierra Oncology; and is a paid consultant for Celgene, Incyte, Novartis, and Sierra Oncology.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verstovsek, S. How I manage anemia related to myelofibrosis and its treatment regimens. Ann Hematol 102, 689–698 (2023). https://doi.org/10.1007/s00277-023-05126-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05126-4