
Abstract
Diffuse large B cell lymphoma (DLBCL) comprises specific subtypes, disease entities, and other not otherwise specified (NOS) lymphomas. This review will focus on DLBCL NOS because of their prevalence and their heterogeneity with respect to morphology, clinical presentation, biology, and response to treatment. Gene expression profiling of DLBCL NOS has identified molecular subgroups that correlate with prognosis and may have relevance for treatment based on signaling pathways. New technologies have revealed that the “activated B cell” subgroup is linked to activation of the nuclear factor kB (NF-kB) pathway, with mutations found in CD79A/B, CARD11, and MYD88, and loss of function mutations in TNFAIP3. The “germinal center B cell-like” subgroup is linked to mutational changes in EZH2 and CREBBP. Biomarkers that are related to pathways promoting tumor cell growth and survival in DLBCL have been recognized, although their predictive role requires clinical validation. Immunohistochemistry for detecting the expression of these biomarkers is a practical technique that could provide a rational for clinical trial design.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The application of advanced immunologic, biochemical, and genetic techniques in the classification of tumors of hematopoietic and lymphoid tissues has led to the identification of many distinct disease entities as proposed by the World Health Organization (WHO) working group [1]. Among tumors of lymphoid tissues, diffuse large B cell lymphoma (DLBCL) is the most common lymphoma, accounting for about 30 % of the cases, and comprises specific subtypes or disease entities. However, most cases are still classified as DLBCL, not otherwise specified (NOS) [2]. DLBCL NOS is a heterogeneous category with respect to morphology, clinical presentation, biology, and response to treatment. DLBCL can be subclassified based on cytologic appearance (e.g., centroblastic or immunoblastic morphology) and the site of primary involvement (nodal or extranodal) and according to the clinical background from which they arise (e.g., normal or compromised immunity) [3]. Importantly, these disparate features are reflected by the wide spectrum of clinical outcomes and treatment response, so that the development of new therapeutic strategies is urgently needed to address this heterogeneity in DLBCL [4, 5].
In recent years, gene expression profiling (GEP) studies identified gene expression signatures that can define the “cell of origin” of DLBCL cases. Subsequently, immunohistochemical signatures were developed to capture the distinction between the germinal center (GC) B-like DLBCL (GCB DLBCL) subgroup and the activated B-like DLBCL (ABC DLBCL) subgroup. Among the many advantages, GEP provides new information about the underlying molecular mechanisms of DLBCL and enables the grouping of more homogeneous subsets of patients with poorer prognosis who may benefit from improved treatments [6]. GEP also helps to select tumors for which next generation sequencing (NGS) technology may recognize specific oncogenic pathways, thus selecting patients who may benefit from identifiable therapeutic agents [6–9] (Fig. 1). However, an effective use of this information still requires reliable tests and treatment strategies that exploit these data.

Evolution in the study of diffuse large B cell lymphoma focusing on prognosis and biomarkers detection
This paper will focus on DLBCL, NOS, because of their prevalence and their heterogeneity. This is a synthetic review of the different biological abnormalities found in DLBCL, NOS, that reveal diagnostic or prognostic biomarkers. This review also describes discoveries that help to identify DLBCL subgroups and to recognize related oncogenic pathways, thereby providing a rationale for a more individualized approach in the treatment of this group of neoplasms.
DLBCL classification
In 1994 the International Lymphoma study group [10] unified within a single “diffuse large B cell lymphoma” category, three high-grade lymphomas originally defined as “histiocytic” [11] and then, with updated terminology, named centroblastic lymphoma and its variants [12], immunoblastic B cell lymphoma [12, 13], and large B cell anaplastic Ki1+ lymphoma [14]. Notably, the DLBCL category included lymphoma subtypes that in a previous international classification (working formulation for clinical usage) [15], were split into two different prognostic groups (intermediate and high grade) [16]. Therefore, it was not surprising that important clinical studies showed that DLBCL NOS was heterogeneous with respect to clinical outcome [4]. Interestingly, the recent WHO classification proposal, in addition to specific DLBCL subtypes and disease entities, formally acknowledges the remaining DLBCL as DLBCL NOS. This category includes lymphomas with centroblastic, immunoblastic, and anaplastic morphology as common morphological variants [1]. The clinical relevance of these DLBCL variants remain debatable, although tumors composed predominantly of centroblasts have a better prognosis than those composed of immunoblasts [17]. Coherently, other studies have shown that immunoblastic morphology is highly significant in predicting an adverse outcome [18]. However, conclusions about the prognostic significance of these morphologic variants are hampered by poor reproducibility and a lack of consensus among pathologists [1–3].
DLBCL subgrouping by cell of origin
GEP of DLBCLs has identified molecular subgroups, which correlate with prognosis, and may have relevance for treatment based on signaling pathways (see below) [6–9, 19]. The original GEP studies have shown that at least two major subgroups of DLBCL could be identified. They resemble either germinal center B cells (GCBs) or activated B cells (ABCs), establishing a putative cell of origin [19]. GEP studies have demonstrated a significantly worse prognosis for the ABC subtype. The prognostic value of GEP has been independently validated by examining other selected gene sets [20, 21]. A six-gene model (favorable LMO2, BCL6, FN1; unfavorable CCND2, SCYA3, BCL2) was reported to identify approximately one third of DLBCL patients whose 5-year survivals were less than 27 % [21]. Furthermore, expressions of LMO2 and TNFRSF9 have been used to develop a two-gene signature based on tumor and microenvironment [22] (Fig. 2). In a large series of DLBCL patients, this simple model added prognostic value to the clinical International Prognostic Index (IPI) [22]. Very recently, a 20-gene gene expression assay in formalin fixed paraffin-embedded tissues has been proposed for the determination of cell of origin subgroups of DLBCL [23]. By this, assay identification of ABC versus GCB subgroups from paraffin-embedded tissue is now possible. In the validation cohort, these assays proved to be accurate and robust with a rapid turnaround time (Fig. 1).

Diffuse large B cell lymphoma not otherwise specified. Subgroups with prognostic significance. DLBCL diffuse large B cell lymphoma, NOS not otherwise specified, GC B-like germinal center B cell like, Non GCB-like germinal center B cell like, ABC activated B cell
Regarding the correlation between the morphologic variants of DLBCL NOS and GEP, a single study demonstrated that the immunoblastic subtype was enriched for cases with an ABC profile, whereas purely centroblastic neoplasms were more often GCB [24]. Consistently, an immunohistochemical study, performed in HIV-associated lymphomas, showed that the expression of BCL6 (a GC marker) and MUM1 (a post-GC marker) were mutually exclusive. BCL6 was generally restricted to the centroblastic and MUM1 to the immunoblastic variants of DLBCL [25]. However, cases co-expressing BCL6 and MUM1 were observed. They were considered as non-GC-related lymphomas (reviewed in [26]).
Because it is impractical to perform GEP involving mRNA expression in every case, various immunohistochemical profiles have been tested as surrogates [9, 27–31]. Although the correspondence was not exact, prognostic correlations were drawn with immunohistochemically defined groups. The first algorithm using CD10, MUM1, and BCL6 [27] did not appear to distinguish groups of significantly differing prognosis in almost all the series examined, when patients were treated with rituximab-containing regimens. Refinements of the approach have led to the incorporation of further markers including stromal response markers and microvessel density [32]. However, the concordance rate between the immunohistochemically defined and GEP-defined subgroups has been a variable [29, 30, 33, 34]. These observations raise the question whether the core of the original GEP and immunohistochemical algorithms still needs to be maintained. The question is still open, but it remains that MUM1 should be retained within the immunohistochemical profile together with BCL6, since it is a marker of transition from BCL6 positivity (GC B cells) to CD138 expression (immunoblasts and plasma cells) [35].
Conflicting results have been found in HIV-associated DLBCL [36, 37]. In particular, the role of molecular subgroups in predicting outcome in HIV-associated DLBCL is still unclear. A recent study aiming at understanding the role of oncogenic pathway-related biomarkers has shown that the molecular pathogenesis of immunodeficiency-associated lymphomas differs from that of lymphomas of the immunocompetent host with similar histology [38]. Immune deregulation, viral infections, and chronic antigenic stimulation may provide alternative survival signals that render neoplastic B cells less dependent on genetic lesions otherwise important for lymphomagenesis in immunocompetent host [39].
In conclusion, regarding DLBCL subgrouping by cell of origin, the concordance between GEP and immunohistochemistry data has been poor [29], and the reproducibility of the classification in ABC and GCB subtypes using different immunohistochemical algorithms was also very poor [40, 41]. Therefore, it is important to stress that immunohistochemistry cannot yet be used as a reliable technique for DLBCL classification. Technical standardization on immunohistochemical markers for formalin fixed paraffin-embedded tissue is advisable.
Genetic landscape of DLBCL NOS
Owing to the application of new technologies (Fig. 2), the ABC and GCB DLBCL subgroups, originally formulated on a cell-of-origin model, have more recently been shown to harbor different pathways of cellular transformation and oncogenesis [42, 43]. In the first study [44] that examined DBLCL with NGS technology, using a combination of whole genome sequencing (WGS), exome sequencing, and RNA sequencing, a recurrent and much targeted somatic mutation affecting the polycomb repressor-2 complex gene EZH2 was identified. EZH2 gene mutation was found in 22 % of DLBCL NOS, all of which were confined to the GCB subgroup. Regarding the ABC subgroup, the major signaling alteration appeared to be the constitutive activation of the nuclear factor kB (NF-kB) pathway through chronic stimulation of the B cell receptor (BCR) pathway [44].
Table 1 summarizes the major discoveries in the genetic landscape of DLBCL NOS using NGS technology [24, 42, 44–60] (reviewed in [7–9, 61–63]). A role for the CBM complex, CARD11, BCL10, and MAL1, downstream of BCR in NF-kB activation, has been demonstrated. Mutations in CARD11 are observed in approximately 10 % of ABC DLBCLs [42]. A majority of other ABC DLBCLs has been shown to have chronic activation of the BCR pathway through various mechanisms including activating mutations of CD79A and CD79B and recruitment of Bruton’s tyrosine kinase, which is required for CARD11 signaling [60]. CREBBP mutations are observed in 22 % of all DLBCL, with enrichment in the GCB subtype, whereas E300 mutations are observed in 10 % of all DLBCL [47]. These mutations might be functionally significant in that tumor cells harboring mutant genes could be deficient in acetylating BCL6 and p53, leading to constitutive activation of the BLC6 oncoprotein and to decreased p53 tumor suppressor activity [47]. Importantly, recurrent mutations in several genes affecting histone modification have been identified [48]. A recent study [45] confirmed the above-reported findings [44, 46, 47] identifying EZH2, MYD88, CREBBP, MLL2, MEF2B, and CD58, in addition to several other genes, as targets of recurrent mutation in DLBCL [45]. In summary, the ABC subgroup is particularly linked to activation of the NF-kB pathway. The GCB subgroup of DLBCL is less clearly dependent upon deregulation of a particular pathway.
The identification and functional characterization of the molecular bases of deregulated signaling in DLBCL NOS subgroups is providing the preclinical rationale for therapeutic inhibition of the involved pathways [6, 64, 65]. In the light of these discoveries, the next steps should include the recognition of biomarkers related to oncogenic pathways that are deregulated by these gene mutations and the validation of their immunohistochemical detection [66] (Fig. 3 and Table 2). At present, the expression of these biomarkers can be detected by immunohistochemistry, but its performance must increase in some of the protein targets for which antibodies are not ideal yet [67].

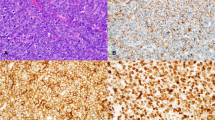
This composite figure shows some examples of immunohistochemical detection of prognostic/potentially predictive biomarker expression in diffuse large B cell lymphoma not otherwise specified (DLBCL NOS). Most tumor cells are immunostained for the various biomarkers tested. Immunostaining for pAKT and pERK1/2 is nuclear and cytoplasmic; similar immunostaining is also observed in breast cancer tumor cells (positive controls, inset). Immunostaining for pSYK, p53, and MYC is nuclear, whereas immunostaining for BCL2 is cytoplasmic. Type of specimen: lymph nodes involved by DLBCL NOS. Type of stabilization of specimen: formalin-fixed paraffin-embedded samples. Antibodies: suitable for paraffin-embedded tissues. Images acquired with the Olympus Dot.Slide Virtual microscopy system using an Olympus BX51 microscopy equipped with PLAN APO × 2/0.08 and UPLAN SApo × 40/0.95 objectives
Prognostic markers in DLBCL
p53 expression, MYC deregulation, BCL2 rearrangement, and protein expression in DLBCL (Table 3)
TP53
The TP53 (tumor protein 53) gene encodes the tumor suppressor p53 protein, which plays a crucial part in maintaining genomic stability. p53 exerts transcriptional control on multiple genes involved in cell cycle regulation, DNA repair, and gene transcription [58]. It also interacts with numerous cytosolic proteins associated with the intrinsic mitochondrial apoptosis pathway and autophagy [58]. In p53-deficient mice, malignant lymphoma is the predominant malignancy, occurring in about two thirds of animals [68]. TP53 dysfunction occurs mostly commonly via mutation in the coding sequence, but aberrations at the gene promoter and gene polymorphisms may also contribute [69]. TP53 mutation occurs in about 20 % of DLBCL. However, disruption of p53-dependent apoptosis appears to be essential in lymphomagenesis, acting through overexpression of anti-apoptotic proteins including BCL2 and BCL-XL and surviving [58]. Recent data also indicate that a subset of DLBCL harbors a complementary set of alterations of p53 and its downstream cell cycle proteins, collaborating to perturb p53 function leading to lymphomagenesis [70].
In general, TP53 aberrations in DLBCL are associated with poor outcomes, although the exact impact varies somewhat in different studies, owing probably to the adoption of different detection methods, which include immunohistochemical staining for p53, fluorescence in situ hybridization (FISH) for 17p13.1 (which contains TP53) deletion, and direct gene mutation analysis. Of these methods, mutational analysis gives the most consistent results. FISH analysis showing loss of heterozygosity (LOH) is less predictive, as LOH needs to collaborate with TP53 mutation in order to abrogate p53 function. Immunohistochemical analysis is observer dependent, and a pattern of p53 + p21− has been proposed to be more closely associated with TP53 gene mutation [71].
In the pre-rituximab era, TP53 mutation had been shown to be a poor prognostic indicator in CHOP-treated cases [72]. The poor prognostic impact still remained in patients treated with ritixumab-CHOP, both for the GCB and ABC subtypes [73]. In addition to prognostic significance, TP53 perturbations may be a potential molecular marker of DLBCL that are amenable to targeted treatment, particularly with CDK inhibitors [70].
MYC
MYC was originally identified as a putative oncogene involved in the t(8;14)(q24;q32) translocation in Burkitt lymphoma (BL) [74]. MYC rearrangements have subsequently been found in other subtypes of aggressive lymphomas [75]. It is a transcription factor with diverse biologic functions. In oncogenesis, MYC regulates numerous genes involved in cellular proliferation, growth, and DNA replication. By activating CCND2 and other CDKs and suppressing cell cycle inhibitors, MYC promotes transition into S phase. It also regulates many micro-RNAs with oncogenic or tumor suppressor function [74, 75].
MYC rearrangements occur in 9–14 % of DLBCL [55, 76]. In contrast to BL, MYC rearrangement in DLBCL is often associated with multiple karyotyic aberrations and other genetic lesions [75]. Clinically, patients with MYC-rearranged DLBCL were usually >60 years old, presenting with higher IPI scores and more advanced-stage disease, often with extranodal involvement [55, 76]. Accordingly, MYC rearrangements portend a poor prognosis in DLBCL treated with standard rituximab-CHOP. In one study, the 5-year overall survival of patients with MYC rearrangement was significantly inferior at 33 %, as compared with 72 % in patients without MYC rearrangement [55]. In another study, the survival at 2 years of MYC-rearranged cases was 35 %, as compared with 61 % in non-rearranged cases [76].
Determination of MYC rearrangement with FISH is costly, time-consuming, and not routinely available. Moreover, MYC overexpression may also be due to MYC amplification and other cellular mechanisms. The advent of a sensitive and specific monoclonal anti-MYC antibody recognizing the N-terminus of MYC has allowed immunohistochemical detection of nuclear MYC with high accuracies in paraffin-embedded sections [77]. Cases with MYC rearrangement show strong nuclear MYC staining (>70 % of cells) [77]. However, MYC overexpressing cases may not always harbor MYC rearrangement, suggesting alternative mechanisms of upregulation of MYC.
BCL2
BCL2 is an oncogene originally identified from the t(14;18)(q32;q21) translocation found in follicular lymphoma [78]. Overexpression of BCL2 leads to extended B cell survival and follicular lymphoproliferation in transgenic mice, recapitulating the human disease [78]. It is an anti-apoptosis protein important in normal B cell development and differentiation. BCL2 overexpression provides a survival advantage to neoplastic B cells and may play a part in resistance to chemotherapy [79].
BCL2 overexpression can be related to BCL2 rearrangement or other cellular mechanisms. Early immunohistochemical studies coupled with FISH analysis had shown BCL2 overexpression in about 24–55 % of cases of DLBCL [80–82]. BCL2 overexpression tended to be associated with advanced stage and inferior survivals. However, the results were not always consistent. These conflicting findings were partly resolved, when later studies showed that the prognostic significance of BCL2 might vary depending on the cellular context. With immunohistochemical analysis, it was observed that BCL2 expression in GCB DLBCL was associated with t(14;18) and did not correlate with prognosis. However, BCL2 expression in ABC DLBCL was not associated with t(14;18), but often with BCL2 gene amplification and activation of the NFKB pathway, and portended inferior survivals [83]. These findings were also confirmed by FISH analysis of BCL2 rearrangement, which was found to correlate strongly with GCB phenotype, but did not significantly impact on survivals [84].
Complex genetic alterations involving MYC, BCL2, and BCL6
The prognostic impact of MYC and BCL2 rearrangement and overexpression taken individually appears significant in DLBCL, but is not always unequivocal. Emerging data indicate that rearrangement and overexpression of MYC and BCL2 may collaborate to negative-effect survivals.
In earlier studies, where FISH was used to analyze MYC and BCL2 rearrangements, DLBCL with concomitant MYC/BCL2 rearrangements had dismal survivals, with about 60 % of patients dying within 6 months, and a 5-year survival of less than 10 % [54]. Notably, about two thirds of these “double-hit” lymphomas were classified as B cell lymphoma unclassifiable, with features intermediate between DLBCL and BL. These observations were further extended with routine immunohistochemical analysis of MYC and BCL2 overexpression. In one study, MYC+/BCL2+ cases were found to constitute 21 % of 167 cases of DLBCL [85]. MYC protein overexpression (in >40 % of neoplastic cells) predicted inferior survivals with rituximab-CHOP only when BCL2 protein was concomitantly overexpressed (>50 % of neoplastic cells). Although MYC protein overexpression correlated strongly with MYC rearrangement, actual concomitant MYC/BCL2 rearrangement occurred in 5 % of cases, which was associated also with a dismal prognosis. In another study, 29 % of 193 cases of DLBCL were MYC+/BCL2+ on immunohistochemical evaluation. These patients had inferior response rates, overall survival, and disease-free survivals, which were independent of IPI scores and GBC/ABC origins. Consistently, on FISH analysis, the negative prognostic impact of MYC rearrangement was only evident when BCL2 was also rearranged [86]. Recent data on a large cohort of DLBCL treated uniformly with rituximab-CHOP also confirmed the negative prognostic relevance of the MYC+/BCL2+ phenotype, with concomitant TP53 mutation conferring an even worse prognosis in such cases [63]. These studies have shown some salient features. The poor outcome of MYC + DLBCL is largely the result of concurrent BCL2 overexpression, and it is the concomitant MYC+/BCL2+ phenotype that predicts outcome. Immunohistochemistry is significantly more sensitive than FISH in defining MYC and BCL2 overexpression. The MYC+/BCL2+ phenotype occurs both in GCB and ABC DLBCL, suggesting that molecular pathways accounting for MYC and BCL2 overexpression might be heterogeneous. Although some of the cases are morphologically B cell lymphoma with features intermediate between DLBCL and BL, histopathologic evaluation is inadequate in predicting the MYC+/BCL2+ phenotype. Finally, patients have a high median age of nearly 70 years, suggesting that MYC+/BCL2+ cases may be a disease predominantly of the elderly population.
Recent data have indicated a possible third player BCL6 in impacting on prognosis in DLBCL. BCL6 was originally identified from chromosomal breakpoints at 3q27 [87]. BCL6 is expressed predominantly in GCB cells and is essential for GC formation. In DLBCL, BCL6 is deregulated by gene translocation or somatic hypermutation. Early studies had shown that high BCL6 gene expression was associated with a better outcome [88]. BCL6 expression occurred predominantly in GCB DLBCL [49], which might explain the more favorable outcome. In a recent study of elderly patients treated with rituximab-CHOP, low BCL6 expression (<25 % of neoplastic cells) as defined by immunohistochemistry was shown in concert with high BCL2 expression to impart an unfavorable outcome to cases with MYC rearrangement or overexpression, independent of the IPI scores [89]. Hence, a “triple-hit” phenotype of MYChigh, BCL2high, and BCL6low has been proposed to confer an even worse prognosis in DLBCL than MYC+/BCL2+ cases.
While these results have shown that immunohistochemical staining for MYC, BCL2, and BCL6 is robust, can be performed routinely, and preferred over FISH analysis, the reproducibility of the defined cutoff has to be defined. However, the double-hit or triple-hit status of DLBCL obviously has to be determined in future trials of DLBCL, particularly when novel agents are tested. Outside clinical trials, it is still unclear how information on MYC, BCL2, and BCL6 may be used in the treatment of individual patients in order to improve outcome. Rituximab-CHOP does not appear to be satisfactory treatment for these cases. As many of these patients are elderly, aggressive chemotherapy or hematopoietic stem cell transplantation may not be feasible. It remains to be defined if therapy targeted against MYC or BCL2 may be valid therapeutic options.
Microenvironmental, viral, and host’s factors influencing DLBCL prognosis
Retrospective studies have confirmed the worse prognosis for patients with DLBCL ABC subgroup. However, when multivariate analyses are used, it is clear that some of the differences may well be related to older age at diagnosis and other adverse presentation features [34]. In fact, in addition to cytomorphology, immunophenotype, and molecular characteristics derived from newer tools, DLBCL prognosis may be influenced by diverse factors such as the “stromal signature” of the background, viral infections, and host’s factors (age and site) (reviewed in [26]). Moreover, CD5-positive DLBCL has been suggested in some studies, mainly from Japan, to have distinct clinical features [90]. These patients are often older and present with bulky retroperitoneal disease. These cases arise de novo and have no relation to other lymphomas which express CD5, i.e., chronic lymphocytic leukemia or mantle cell lymphoma. Ki-67 index has been reported to be a prognostic marker [91]. A high proliferation rate, as assessed by immunohistochemistry with anti-Ki67 antibody, has been associated with adverse outcome [2]. Coherently, the proliferation signature by one gene expression proved to be a strong predictor of poor survival [24]. In the pre-rituximab era, the IPI, which is based on clinical parameters, such as age, stage, serum lactate deydrogenases level, and performance status, an extent of extranodal involvement proved to be highly valuable for the prediction of prognosis in patients with DLBCL [92]. However, the IPI seems to have lost some of its high predictive value in the rituximab era [93]. Very recently, the so-called enhanced IPI, an updated version of the IPI for patients with DLBCL treated in the rituximab era, has been proposed [94]. Table 4 highlights highly aggressive DLBCL subgrouped according to adverse prognostic factors: lack of CD20 expression, special phenotypes linked to cell of origin and CD5 expression, EBV infection, and complex karyotypes [95, 96].
The microenvironment in DLBCL
The microenvironment and the inflammatory response may provide clues to the behavior of DLBCL according to two patterns of stromal signature predictive of good survival, “stromal 1” (including genes encoding for extracellular matrix proteins), and poor outcome, “stromal 2” (including angiogenetic switch-related genes) [97]. A recent study on the tumor microenvironment and viral components has shown that DLBCL occurring in HIV-infected patients is highly angiogenic with markedly higher blood-vessel density than sporadic DLBCL cases. Importantly, the investigation has also highlighted the role of Epstein Barr virus (EBV) in angiogenesis [98].
DLBCL associated with immune dysfunction and infectious agents
DLBCL associated with infectious agents include the spectrum of HIV- and gamma herpesviruses-associated lymphomas and are highly aggressive tumors (Table 5) [39, 99, 100]. EBV-associated lymphomas that are related to chronic inflammation or senescence of the immune system represent distinct disease entities occurring in non AIDS-related settings.
DLBCL associated with chronic inflammation
DLBCL associated with chronic inflammation most commonly involves body cavities. The prototype for this category is pyothorax-associated lymphoma (PAL)[101, 102]. Other cases of DLBCL occurring in the setting of chronic inflammation (such as chronic skin ulcers or osteomyelitis) are also frequently positive for EBV (reviewed in [26]).
EBV-positive DLBCL of the elderly
EBV-positive DLBCL of the elderly, also known as age-related or senile EBV-associated lymphoproliferative disorders [103], is diagnosed in patients older than 50 years with no known cause of immunodeficiency or prior lymphoma [103]. Seventy percent of these patients present with extranodal involvement and >50 % have advanced disease with poor prognosis and a short survival rate.
DLBCL based on an anatomic site
Some peculiar DLBCL subtypes are specifically related to their sites of presentation. Examples include primary cutaneous DLBCL, “leg type” [104], primary mediastinal (thymic) large B cell lymphoma [105–107], DLBCL of the central nervous system [108, 109], and primary large B cell lymphomas of bones [110, 111]. These DLBCL subtypes express BCL2 and MUM1/IRF4 but not CD10, are frequently related to an ABC phenotype, and are distinct entities with an aggressive behavior [112].
Biomarkers enrichment strategies to guide therapy
At the molecular level, DLBCL is a heterogeneous disease. Both GEP studies and DNA sequencing studies have demonstrated that DLBCL can be further subdivided into smaller, more homogeneous groups [6–9]. These findings not only explain why patients respond differently to a specific therapy but also provide an opportunity for designing tailored treatments based on tumor characteristics. Such characteristic biomarkers will need to be reproducible across different laboratories, and preferentially, should guide therapeutic options.
In recent years, GEP methods were used to stratify patients based on the cell of origin, into GCB or ABC DLBCL. While this approach helped advancing the field and patient selection for clinical trials, it continues to lack mechanistic precision. Both genetic and protein biomarkers can be used to more precisely stratify patients for evaluating novel treatment regimens based on molecular mechanisms that support tumor growth and survival. However, at the present time, the ideal biomarker or biomarker set remains elusive. For this reason, it is advised to include several biomarkers as part of correlative discovery biomarker analysis. As shown in Fig. 4, a biopsy can be analyzed for a set of biomarkers: genetic mutations, cell surface proteins, and intracellular proteins. Other biomarkers can also be included, such as gene expression status. Regardless, it would be more efficient to screen patient tumors for several biomarkers at the same time using an umbrella protocol. For example, a lymphoma specimen can be examined for the presence of genetic alterations using targeted sequencing strategies. At the same time, the biopsy can be examined for several immunohistochemistry-based biomarkers, such as the cell of origin, MYC and BCL2 expression, and the expression of phosphoproteins that are associated with activated oncogenic signaling pathways, such as pSTAT3, pERK, and pAKT. Based on this analysis, patients can be offered targeted agents that match and are more suitable for their tumors. Thus, in an “umbrella” clinical trial, several targeted agents can be simultaneously tested in biomarker-defined populations. However, when a tumor expresses several biomarkers at the same time, a prioritization algorithm should be implemented. For example, if a lymphoma specimen is shown to have EZH2 mutation and pSTAT3 expression, experimental treatment with an EZH2 inhibitor or a JAK2 inhibitor can be offered, but which treatment should be considered first? Eventually, the coexpression of more than one targeted biomarker may offer an opportunity to guide new treatment strategies [64, 113]. For example, a lymphoma specimen that coexpresses high levels of MYC and BCL2 proteins may respond better to a combination regimen that targets both proteins. The selection of such combination regimens should be based on synergistic effects in preclinical studies and should first be evaluated in phase 1 trials to establish its safety in the clinical setting.

Genetic and protein biomarkers can be used to stratify patients for new treatment strategies. The figure lists a set of biomarkers including genetic mutations, cell surface proteins, and intracellular proteins.
Because many genetic mutations in DLBCL occur at a very low frequency, it would be more efficient to group several mutations into one mechanistic category, such as activation of well-defined oncogenic signaling pathways [65]. Such a strategy will require linking signaling pathways with a unifying set of biomarkers that can be detected by immunohistochemistry. For example, mutations in PI3K, AKT, TSC, mTOR, deletion of PTEN, and phosphorylate PRAS40 protein may be grouped into one bucket of “activated PI3K pathway.” Mechanism-based treatment strategies can then be evaluated to inhibit different components in this pathway, rather than targeting each genetic mutation separately. This strategy can be more efficient for enriching patients for specific trials.
Concluding remarks
The era of treating all patients with DLBCL with the same regimen is fading away. New strategies of “divide and concur” are gaining momentum as it divided patients into several groups based on their tumor characteristics. Identifying these patients through clinical biomarkers is now feasible and may allow in the future the administration of more precise therapy for different patients. These strategies are currently being evaluated in clinical trials. For example, bortezomib and ibrutinib are in trials for patients affected by ABC DLBCL, while EZH2 and BCL6 inhibitors may be used for other patients affected for GCB DLBCL. Patient participation in these clinical trials is critical for expediting our progress in improving the cure rate of patients with DLBCL.
References
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC, Lyon
Stein H, Warnke RA, Chan WC, Jaffe ES, Chan JKC, Gatter KC, Campo E (2008) Diffuse large B-cell lymphoma, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds) WHO classification of tumours of haematopietic and lymphoid tissues, 4th edn. IARC press, Lyon, pp 233–237
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES (2011) The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117(19):5019–5032. doi:10.1182/blood-2011-01-293050
Shankland KR, Armitage JO, Hancock BW (2012) Non-Hodgkin lymphoma. Lancet 380(9844):848–857. doi:10.1016/S0140-6736(12)60605-9
Cheson BD (2013) Therapy for diffuse large B-cell lymphoma: getting personal. Lancet 381(9880):1793–1794. doi:10.1016/S0140-6736(13)60849-1
Campo E (2013) Whole genome profiling and other high throughput technologies in lymphoid neoplasms—current contributions and future hopes. Mod Pathol 26(Suppl):1S97–S110. doi:10.1038/modpathol.2012.179
Staudt LM (2013) Therapy of DLBCL based on genomics. Hematol Oncol 31(Suppl):126–128. doi:10.1002/hon.2062
Slack GW, Gascoyne RD (2013) Next-generation sequencing discoveries in lymphoma. Adv Anat Pathol 20(2):110–116. doi:10.1097/PAP.0b013e3182862724
Menon MP, Pittaluga S, Jaffe ES (2012) The histological and biological spectrum of diffuse large B-cell lymphoma in the World Health Organization classification. Cancer J 18(5):411–420. doi:10.1097/PPO.0b013e31826aee97
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC (1994) A revised european-american classification of lymphoid neoplasms: A proposal from the international lymphoma study group. Blood 84(5):1361–1392
Rappaport H (1966) Tumors of the hematopoietic system. Series I ed. Armed Forces Institute of Pathology, Washington
Gérard-Marchant R, Hamlin I, Lennert K, Rilke F, Stansfeld A, van Unnik J (1974) Classification of non-Hodgkin’s lymphomas. Lancet 406-408
Lukes RJ, Collins RD (1974) Immunologic characterization of human malignant lymphomas. Cancer 34(4 Supp):suppl:1488–suppl:1503
Stansfeld AG, Diebold J, Noel H, Kapanci Y, Rilke F, Kelenyi G, Sundstrom C, Lennert K, van Unnik JA, Mioduszewska O (1988) Updated kiel classification for lymphomas. Lancet 1(8580):292–293
[Anonymous] (1982) National cancer institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. The non-Hodgkin’s lymphoma pathologic classification project. Cancer 49(10):2112–2135
Rosenberg SA (1994) Classification of lymphoid neoplasms. Blood 84(5):1359–1360
Mounier N, Briere J, Gisselbrecht C, Emile JF, Lederlin P, Sebban C, Berger F, Bosly A, Morel P, Tilly H, Bouabdallah R, Reyes F, Gaulard P, Coiffier B (2003) Rituximab plus CHOP (R-CHOP) overcomes bcl-2-associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood 101(11):4279–4284. doi:10.1182/blood-2002-11-3442
Ott G, Ziepert M, Klapper W, Horn H, Szczepanowski M, Bernd HW, Thorns C, Feller AC, Lenze D, Hummel M, Stein H, Muller-Hermelink HK, Frank M, Hansmann ML, Barth TF, Moller P, Cogliatti S, Pfreundschuh M, Schmitz N, Trumper L, Loeffler M, Rosenwald A (2010) Immunoblastic morphology but not the immunohistochemical GCB/nonGCB classifier predicts outcome in diffuse large B-cell lymphoma in the RICOVER-60 trial of the DSHNHL. Blood 116(23):4916–4925. doi:10.1182/blood-2010-03-276766
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403(6769):503–511. doi:10.1038/35000501
Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR (2002) Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med 8(1):68–74. doi:10.1038/nm0102-68
Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R (2004) Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med 350(18):1828–1837. doi:10.1056/NEJMoa032520
Alizadeh AA, Gentles AJ, Alencar AJ, Liu CL, Kohrt HE, Houot R, Goldstein MJ, Zhao S, Natkunam Y, Advani RH, Gascoyne RD, Briones J, Tibshirani RJ, Myklebust JH, Plevritis SK, Lossos IS, Levy R (2011) Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood 118(5):1350–1358. doi:10.1182/blood-2011-03-345272
Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, Rosenwald A, Campo E, Chan WC, Connors JM, Smeland EB, Mottok A, Braziel RM, Ott G, Delabie J, Tubbs RR, Cook JR, Weisenburger DD, Greiner TC, Glinsmann-Gibson BJ, Fu K, Staudt LM, Gascoyne RD, Rimsza LM (2014) Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 123(8):1214–1217. doi:10.1182/blood-2013-11-536433
Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, Lopez-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM, Lymphoma/Leukemia Molecular Profiling Project (2002) The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346(25):1937–1947. doi:10.1056/NEJMoa012914
Carbone A, Gloghini A, Larocca LM, Capello D, Pierconti F, Canzonieri V, Tirelli U, Dalla-Favera R, Gaidano G (2001) Expression profile of MUM1/IRF4, BCL-6, and CD138/syndecan-1 defines novel histogenetic subsets of human immunodeficiency virus-related lymphomas. Blood 97(3):744–751
Said JW (2013) Aggressive B-cell lymphomas: how many categories do we need? Mod Pathol 26(Suppl):1S42–1S56. doi:10.1038/modpathol.2012.178
Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, Staudt LM, Connors JM, Armitage JO, Chan WC (2004) Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103(1):275–282. doi:10.1182/blood-2003-05-1545
Salles G, de Jong D, Xie W, Rosenwald A, Chhanabhai M, Gaulard P, Klapper W, Calaminici M, Sander B, Thorns C, Campo E, Molina T, Lee A, Pfreundschuh M, Horning S, Lister A, Sehn LH, Raemaekers J, Hagenbeek A, Gascoyne RD, Weller E (2011) Prognostic significance of immunohistochemical biomarkers in diffuse large B-cell lymphoma: A study from the lunenburg lymphoma biomarker consortium. Blood 117(26):7070–7078. doi:10.1182/blood-2011-04-345256
Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD, Ott G, Rosenwald A, Braziel RM, Campo E, Vose JM, Lenz G, Staudt LM, Chan WC, Weisenburger DD (2011) Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol 29(2):200–207. doi:10.1200/JCO.2010.30.0368
Choi WW, Weisenburger DD, Greiner TC, Piris MA, Banham AH, Delabie J, Braziel RM, Geng H, Iqbal J, Lenz G, Vose JM, Hans CP, Fu K, Smith LM, Li M, Liu Z, Gascoyne RD, Rosenwald A, Ott G, Rimsza LM, Campo E, Jaffe ES, Jaye DL, Staudt LM, Chan WC (2009) A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res 15(17):5494–5502. doi:10.1158/1078-0432.CCR-09-0113
Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, Wilson WH (2009) Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 113(24):6069–6076. doi:10.1182/blood-2009-01-199679
Perry AM, Cardesa-Salzmann TM, Meyer PN, Colomo L, Smith LM, Fu K, Greiner TC, Delabie J, Gascoyne RD, Rimsza L, Jaffe ES, Ott G, Rosenwald A, Braziel RM, Tubbs R, Cook JR, Staudt LM, Connors JM, Sehn LH, Vose JM, Lopez-Guillermo A, Campo E, Chan WC, Weisenburger DD (2012) A new biologic prognostic model based on immunohistochemistry predicts survival in patients with diffuse large B-cell lymphoma. Blood 120(11):2290–2296. doi:10.1182/blood-2012-05-430389
Nyman H, Jerkeman M, Karjalainen-Lindsberg ML, Banham AH, Leppa S (2009) Prognostic impact of activated B-cell focused classification in diffuse large B-cell lymphoma patients treated with R-CHOP. Mod Pathol 22(8):1094–1101. doi:10.1038/modpathol.2009.73
Barrans SL, Crouch S, Care MA, Worrillow L, Smith A, Patmore R, Westhead DR, Tooze R, Roman E, Jack AS (2012) Whole genome expression profiling based on paraffin embedded tissue can be used to classify diffuse large B-cell lymphoma and predict clinical outcome. Br J Haematol 159(4):441–453. doi:10.1111/bjh.12045
Gaidano G, Carbone A (2000) MUM1: a step ahead toward the understanding of lymphoma histogenesis. Leukemia 14(4):563–566
Chadburn A, Chiu A, Lee JY, Chen X, Hyjek E, Banham AH, Noy A, Kaplan LD, Sparano JA, Bhatia K, Cesarman E (2009) Immunophenotypic analysis of AIDS-related diffuse large B-cell lymphoma and clinical implications in patients from AIDS malignancies consortium clinical trials 010 and 034. J Clin Oncol 27(30):5039–5048. doi:10.1200/JCO.2008.20.5450
Dunleavy K, Little RF, Pittaluga S, Grant N, Wayne AS, Carrasquillo JA, Steinberg SM, Yarchoan R, Jaffe ES, Wilson WH (2010) The role of tumor histogenesis, FDG-PET, and short-course EPOCH with dose-dense rituximab (SC-EPOCH-RR) in HIV-associated diffuse large B-cell lymphoma. Blood 115(15):3017–3024. doi:10.1182/blood-2009-11-253039
Capello D, Gloghini A, Martini M, Spina M, Tirelli U, Bertoni F, Rinaldi A, Morra E, Rambaldi A, Sinigaglia F, Larocca LM, Carbone A (2011) Mutations of CD79A, CD79B and EZH2 genes in immunodeficiency-related non-Hodgkin lymphomas. Br J Haematol 152(6):777–780. doi:10.1111/j.1365-2141.2010.08445.x
IARC (2012) IARC monograph on the evaluation of carcinogenic risk to humans. vol. 100. A review of human carcinogens. part B: biological agents. IARC, Lyon
Coutinho R, Clear AJ, Owen A, Wilson A, Matthews J, Lee A, Alvarez R, da Silva MG, Cabecadas J, Calaminici M, Gribben JG (2013) Poor concordance among nine immunohistochemistry classifiers of cell-of-origin for diffuse large B-cell lymphoma: implications for therapeutic strategies. Clin Cancer Res 19(24):6686–6695. doi:10.1158/1078-0432.CCR-13-1482
Culpin RE, Sieniawski M, Angus B, Menon GK, Proctor SJ, Milne P, McCabe K, Mainou-Fowler T (2013) Prognostic significance of immunohistochemistry-based markers and algorithms in immunochemotherapy-treated diffuse large B cell lymphoma patients. Histopathology 63(6):788–801. doi:10.1111/his.12223
Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM (2008) Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319(5870):1676–1679. doi:10.1126/science.1153629
Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG (2010) MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood 116(9):1515–1523. doi:10.1182/blood-2010-03-272567
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42(2):181–185. doi:10.1038/ng.518
Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, Saksena G, Stransky N, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Hernandez-Lemus E, Schwarz-Cruz y Celis A, Imaz-Rosshandler I, Ojesina AI, Jung J, Pedamallu CS, Lander ES, Habermann TM, Cerhan JR, Shipp MA, Getz G, Golub TR (2012) Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A 109(10):3879–3884. doi:10.1073/pnas.1121343109
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470(7332):115–119. doi:10.1038/nature09671
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, Mullighan CG, Gaidano G, Rabadan R, Brindle PK, Dalla-Favera R (2011) Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471(7337):189–195. doi:10.1038/nature09730
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA (2011) Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476(7360):298–303. doi:10.1038/nature10351
Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, Wright G, Sanger WG, Pickering DL, Jain S, Horsman DE, Shen Y, Fu K, Weisenburger DD, Hans CP, Campo E, Gascoyne RD, Rosenwald A, Jaffe ES, Delabie J, Rimsza L, Ott G, Muller-Hermelink HK, Connors JM, Vose JM, McKeithan T, Staudt LM, Chan WC, Leukemia/Lymphoma Molecular Profiling Project (2007) Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia 21(11):2332–2343. doi:10.1038/sj.leu.2404856
Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Campo E, Jaffe ES, Delabie J, Smeland EB, Rimsza LM, Fisher RI, Weisenburger DD, Chan WC, Staudt LM (2008) Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A 105(36):13520–13525. doi:10.1073/pnas.0804295105
Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, Aster JC, Murty VV, Shipp MA, Dalla-Favera R (2006) Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 203(2):311–317
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L (2009) Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459(7247):717–721. doi:10.1038/nature07968
Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R (2011) Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 43(9):830–837. doi:10.1038/ng.892
Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJ, Siebert R, Kuruvilla J, Klasa R, Connors JM, Gascoyne RD, Horsman DE (2009) Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 114(11):2273–2279. doi:10.1182/blood-2009-03-212191
Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, Gascoyne RD (2009) MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 114(17):3533–3537. doi:10.1182/blood-2009-05-220095
Scott DW, Mungall KL, Ben-Neriah S, Rogic S, Morin RD, Slack GW, Tan KL, Chan FC, Lim RS, Connors JM, Marra MA, Mungall AJ, Steidl C, Gascoyne RD (2012) TBL1XR1/TP63: a novel recurrent gene fusion in B-cell non-Hodgkin lymphoma. Blood 119(21):4949–4952. doi:10.1182/blood-2012-02-414441
Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, Izutsu K, Sakata-Yanagimoto M, Hangaishi A, Yagita H, Fukayama M, Seto M, Kurokawa M, Ogawa S, Chiba S (2009) Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci 100(5):920–926
Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, Greiner TC, McDonnell TJ, Young KH (2012) Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 119(16):3668–3683. doi:10.1182/blood-2011-11-366062
Davis RE, Brown KD, Siebenlist U, Staudt LM (2001) Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 194(12):1861–1874
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, Xiao W, Powell J, Jiang JK, Thomas CJ, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Johnson NA, Rimsza LM, Campo E, Jaffe ES, Wilson WH, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pierce SK, Staudt LM (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463(7277):88–92. doi:10.1038/nature08638
Barton S, Hawkes EA, Wotherspoon A, Cunningham D (2012) Are we ready to stratify treatment for diffuse large B-cell lymphoma using molecular hallmarks? Oncologist 17(12):1562–1573. doi:10.1634/theoncologist.2012-0218
Rossi D, Ciardullo C, Gaidano G (2013) Genetic aberrations of signaling pathways in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. doi:10.1016/j.semcancer.2013.04.002
Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, Liu WM, Visco C, Li Y, Miranda RN, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Choi WW, Zhao X, van Krieken JH, Huang Q, Huh J, Ai W, Ponzoni M, Ferreri AJ, Zhou F, Slack GW, Gascoyne RD, Tu M, Variakojis D, Chen W, Go RS, Piris MA, Moller MB, Medeiros LJ, Young KH (2013) MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from the international DLBCL rituximab-CHOP consortium program. Blood 121(20):4021–4031. doi:10.1182/blood-2012-10-460063, quiz 4250
Younes A (2011) Beyond chemotherapy: new agents for targeted treatment of lymphoma. Nat Rev, Clin Oncol 8(2):85–96. doi:10.1038/nrclinonc.2010.189
Younes A, Berry DA (2012) From drug discovery to biomarker-driven clinical trials in lymphoma. Nat Rev, Clin Oncol 9(11):643–653. doi:10.1038/nrclinonc.2012.156
Chau CH, Rixe O, McLeod H, Figg WD (2008) Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res 14(19):5967–5976. doi:10.1158/1078-0432.CCR-07-4535
Dancey JE, Dobbin KK, Groshen S, Jessup JM, Hruszkewycz AH, Koehler M, Parchment R, Ratain MJ, Shankar LK, Stadler WM, True LD, Gravell A, Grever MR, Biomarkers Task Force of the NCI Investigational Drug Steering Committee (2010) Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res 16(6):1745–1755. doi:10.1158/1078-0432.CCR-09-2167
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356(6366):215–221. doi:10.1038/356215a0
Jardin F, Coiffier B (2013) TP53 and outcome in DLBCL: not only the coding region. Blood 121(22):4433–4434. doi:10.1182/blood-2013-04-493486
Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, Inguilizian H, Mermel C, Currie T, Dogan A, Kutok JL, Beroukhim R, Neuberg D, Habermann TM, Getz G, Kung AL, Golub TR, Shipp MA (2012) Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 22(3):359–372. doi:10.1016/j.ccr.2012.07.014
Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, Campo E, Delabie J, Gascoyne RD, Ott G, Rimsza L, Muller-Hermelink HK, Jaffe ES, Rosenwald A, Staudt LM, Chan WC, Greiner TC (2007) Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 110(13):4396–4405. doi:10.1182/blood-2007-02-072082
Leroy K, Haioun C, Lepage E, Le Metayer N, Berger F, Labouyrie E, Meignin V, Petit B, Bastard C, Salles G, Gisselbrecht C, Reyes F, Gaulard P, Groupe d’Etude des Lymphomes de l’Adulte (2002) p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Ann Oncol 13(7):1108–1115
Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Zhao XF, Choi WW, Zhao X, van Krieken JH, Huang Q, Huh J, Ai W, Ponzoni M, Ferreri AJ, Zhou F, Kahl BS, Winter JN, Xu W, Li J, Go RS, Li Y, Piris MA, Moller MB, Miranda RN, Abruzzo LV, Medeiros LJ, Young KH (2012) Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an international DLBCL rituximab-CHOP consortium program study. Blood 120(19):3986–3996. doi:10.1182/blood-2012-05-433334
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8(12):976–990. doi:10.1038/nrc2231
Ott G, Rosenwald A, Campo E (2013) Understanding MYC driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. doi:10.1182/blood-2013-05-498329
Barrans S, Crouch S, Smith A, Turner K, Owen R, Patmore R, Roman E, Jack A (2010) Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 28(20):3360–3365. doi:10.1200/JCO.2009.26.3947
Green TM, Nielsen O, de Stricker K, Xu-Monette ZY, Young KH, Moller MB (2012) High levels of nuclear MYC protein predict the presence of MYC rearrangement in diffuse large B-cell lymphoma. Am J Surg Pathol 36(4):612–619. doi:10.1097/PAS.0b013e318244e2ba
McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ (1989) Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57(1):79–88
Reed JC (1997) Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol 34(4 Suppl 5):9–19
Hermine O, Haioun C, Lepage E, d’Agay MF, Briere J, Lavignac C, Fillet G, Salles G, Marolleau JP, Diebold J, Reyas F, Gaulard P (1996) Prognostic significance of bcl-2 protein expression in aggressive non-Hodgkin’s lymphoma. groupe d’etude des lymphomes de l’adulte (GELA). Blood 87(1):265–272
Gascoyne RD, Adomat SA, Krajewski S, Krajewska M, Horsman DE, Tolcher AW, O’Reilly SE, Hoskins P, Coldman AJ, Reed JC, Connors JM (1997) Prognostic significance of bcl-2 protein expression and bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 90(1):244–251
Hill ME, MacLennan KA, Cunningham DC, Vaughan Hudson B, Burke M, Clarke P, Di Stefano F, Anderson L, Vaughan Hudson G, Mason D, Selby P, Linch DC (1996) Prognostic significance of BCL-2 expression and bcl-2 major breakpoint region rearrangement in diffuse large cell non-Hodgkin’s lymphoma: a British national lymphoma investigation study. Blood 88(3):1046–1051
Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, Lynch J, Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Campo E, Ott G, Muller-Hermelink HK, Delabie J, Jaffe ES, Grogan TM, Connors JM, Vose JM, Armitage JO, Staudt LM, Chan WC (2006) BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol 24(6):961–968. doi:10.1200/JCO.2005.03.4264
Copie-Bergman C, Gaulard P, Leroy K, Briere J, Baia M, Jais JP, Salles GA, Berger F, Haioun C, Tilly H, Emile JF, Banham AH, Mounier N, Gisselbrecht C, Feugier P, Coiffier B, Molina TJ (2009) Immuno-fluorescence in situ hybridization index predicts survival in patients with diffuse large B-cell lymphoma treated with R-CHOP: a GELA study. J Clin Oncol 27(33):5573–5579. doi:10.1200/JCO.2009.22.7058
Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, Chan WC, Iqbal J, Meyer PN, Lenz G, Wright G, Rimsza LM, Valentino C, Brunhoeber P, Grogan TM, Braziel RM, Cook JR, Tubbs RR, Weisenburger DD, Campo E, Rosenwald A, Ott G, Delabie J, Holcroft C, Jaffe ES, Staudt LM, Gascoyne RD (2012) Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30(28):3452–3459. doi:10.1200/JCO.2011.41.0985
Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, Nielsen O, Gadeberg OV, Mourits-Andersen T, Frederiksen M, Pedersen LM, Moller MB (2012) Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30(28):3460–3467. doi:10.1200/JCO.2011.41.4342
Ye BH, Lista F, Lo Coco F, Knowles DM, Offit K, Chaganti RS, Dalla-Favera R (1993) Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 262(5134):747–750
Lossos IS, Jones CD, Warnke R, Natkunam Y, Kaizer H, Zehnder JL, Tibshirani R, Levy R (2001) Expression of a single gene, BCL-6, strongly predicts survival in patients with diffuse large B-cell lymphoma. Blood 98(4):945–951
Horn H, Ziepert M, Becher C, Barth TF, Bernd HW, Feller AC, Klapper W, Hummel M, Stein H, Hansmann ML, Schmelter C, Moller P, Cogliatti S, Pfreundschuh M, Schmitz N, Trumper L, Siebert R, Loeffler M, Rosenwald A, Ott G, German High-Grade Non-Hodgkin Lymphoma Study Group (2013) MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121(12):2253–2263. doi:10.1182/blood-2012-06-435842
Yamaguchi M, Nakamura N, Suzuki R, Kagami Y, Okamoto M, Ichinohasama R, Yoshino T, Suzumiya J, Murase T, Miura I, Ohshima K, Nishikori M, Tamaru J, Taniwaki M, Hirano M, Morishima Y, Ueda R, Shiku H, Nakamura S (2008) De novo CD5+ diffuse large B-cell lymphoma: results of a detailed clinicopathological review in 120 patients. Haematologica 93(8):1195–1202. doi:10.3324/haematol.12810
Miller TP, Grogan TM, Dahlberg S, Spier CM, Braziel RM, Banks PM, Foucar K, Kjeldsberg CR, Levy N, Nathwani BN (1994) Prognostic significance of the ki-67-associated proliferative antigen in aggressive non-Hodgkin’s lymphomas: a prospective southwest oncology group trial. Blood 83(6):1460–1466
Armitage JO, Weisenburger DD (1998) New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. non-Hodgkin’s lymphoma classification project. J Clin Oncol 16(8):2780–2795
Sehn LH, Berry B, Chhanabhai M, Fitzgerald C, Gill K, Hoskins P, Klasa R, Savage KJ, Shenkier T, Sutherland J, Gascoyne RD, Connors JM (2007) The revised international prognostic index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 109(5):1857–1861
Zhou Z, Sehn LH, Rademaker AW, Gordon LI, Lacasce AS, Crosby-Thompson A, Vanderplas A, Zelenetz AD, Abel GA, Rodriguez MA, Nademanee A, Kaminski MS, Czuczman MS, Millenson M, Niland J, Gascoyne RD, Connors JM, Friedberg JW, Winter JN (2014) An enhanced international prognostic index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123(6):837–842. doi:10.1182/blood-2013-09-524108
Dolcetti R, Dal Col J, Martorelli D, Carbone A, Klein E (2013) Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin Cancer Biol. doi:10.1016/j.semcancer.2013.07.005
Carbone A, Tripodo C, Carlo-Stella C, Santoro A, Gloghini A (2013) Inflammation and lymphoma. In: Gupta SC, Sung B, Aggarwal BB (eds) Inflammation and cancer. Springer, Basel
Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, Vose J, Bast M, Fu K, Weisenburger DD, Greiner TC, Armitage JO, Kyle A, May L, Gascoyne RD, Connors JM, Troen G, Holte H, Kvaloy S, Dierickx D, Verhoef G, Delabie J, Smeland EB, Jares P, Martinez A, Lopez-Guillermo A, Montserrat E, Campo E, Braziel RM, Miller TP, Rimsza LM, Cook JR, Pohlman B, Sweetenham J, Tubbs RR, Fisher RI, Hartmann E, Rosenwald A, Ott G, Muller-Hermelink HK, Wrench D, Lister TA, Jaffe ES, Wilson WH, Chan WC, Staudt LM, Lymphoma/Leukemia Molecular Profiling Project (2008) Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 359(22):2313–2323. doi:10.1056/NEJMoa0802885
Liapis K, Clear A, Owen A, Coutinho R, Greaves P, Lee AM, Montoto S, Calaminici M, Gribben JG (2013) The microenvironment of AIDS-related diffuse large B-cell lymphoma provides insight into the pathophysiology and indicates possible therapeutic strategies. Blood 122(3):424–433. doi:10.1182/blood-2013-03-488171
Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, WHO International Agency for Research on Cancer Monograph Working Group (2009) A review of human carcinogens—part B: biological agents. Lancet Oncol 10(4):321–322
Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF (2009) HIV-associated lymphomas and gamma-herpesviruses. Blood 113(6):1213–1224. doi:10.1182/blood-2008-09-180315
Aozasa K (2006) Pyothorax-associated lymphoma. J Clin Exp Hematop 46(1):5–10
Fukayama M, Ibuka T, Hayashi Y, Ooba T, Koike M, Mizutani S (1993) Epstein-barr virus in pyothorax-associated pleural lymphoma. Am J Pathol 143(4):1044–1049
Chan JKC, Aozasa K, Gaulard P (2008) DLBCL associated with chronic inflammation. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC press, Lyon, pp 245–246
Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, Kerl H, Kurrer M, Knobler R, Pimpinelli N, Sander C, Santucci M, Sterry W, Vermeer MH, Wechsler J, Whittaker S, Meijer CJ (2005) WHO-EORTC classification for cutaneous lymphomas. Blood 105(10):3768–3785
Savage KJ, Monti S, Kutok JL, Cattoretti G, Neuberg D, De Leval L, Kurtin P, Dal Cin P, Ladd C, Feuerhake F, Aguiar RC, Li S, Salles G, Berger F, Jing W, Pinkus GS, Habermann T, Dalla-Favera R, Harris NL, Aster JC, Golub TR, Shipp MA (2003) The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 102(12):3871–3879. doi:10.1182/blood-2003-06-1841
Traverse-Glehen A, Pittaluga S, Gaulard P, Sorbara L, Alonso MA, Raffeld M, Jaffe ES (2005) Mediastinal gray zone lymphoma: the missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol 29(11):1411–1421
Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Campo E, Montserrat E, Lopez-Guillermo A, Ott G, Muller-Hermelink HK, Connors JM, Braziel R, Grogan TM, Fisher RI, Miller TP, LeBlanc M, Chiorazzi M, Zhao H, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Staudt LM (2003) Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 198(6):851–862. doi:10.1084/jem.20031074
Ferreri AJ (2011) How I treat primary CNS lymphoma. Blood 118(3):510–522. doi:10.1182/blood-2011-03-321349
Sung CO, Kim SC, Karnan S, Karube K, Shin HJ, Nam DH, Suh YL, Kim SH, Kim JY, Kim SJ, Kim WS, Seto M, Ko YH (2011) Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood 117(4):1291–1300. doi:10.1182/blood-2010-07-297861
Martinez A, Ponzoni M, Agostinelli C, Hebeda KM, Matutes E, Peccatori J, Campidelli C, Espinet B, Perea G, Acevedo A, Mehrjardi AZ, Martinez-Bernal M, Gelemur M, Zucca E, Pileri S, Campo E, Lopez-Guillermo A, Rozman M, International Extranodal Lymphoma Study Group (2012) Primary bone marrow lymphoma: an uncommon extranodal presentation of aggressive non-Hodgkin lymphomas. Am J Surg Pathol 36(2):296–304. doi:10.1097/PAS.0b013e31823ea106
Bruno Ventre M, Ferreri AJ, Gospodarowicz M, Govi S, Messina C, Porter D, Radford J, Heo DS, Park Y, Martinelli G, Taylor E, Lucraft H, Hong A, Scarfo L, Zucca E, Christie D, International Extranodal Lymphoma Study Group (2014) Clinical features, management, and prognosis of an international series of 161 patients with limited-stage diffuse large B-cell lymphoma of the bone (the IELSG-14 study). Oncologist 19(3):291–298. doi:10.1634/theoncologist.2013-0249
Gloghini A, De Leval L (2013) Immunophenotyping of non-Hodgkin lymphomas. In: Carbone A, Younes A (eds) Non-Hodgkin lymphomas: advanced diagnostics and personalized therapies, Future Medicine, pp 32–55. doi:10.2217/9781780841717
Younes A (2013) XIV. the rationale for combining targeted and biological anti-lymphoma drugs. Hematol Oncol 31(Suppl):181–183. doi:10.1002/hon.2072
Review criteria
PubMed and MEDLINE were used for database searches to identify articles published from 1989 to July 2013. Only articles or abstracts in English were considered for this review. Search terms included the following: “DLBCL,” “Biomarker*,” “Prognosis,” “Diagnosis,” and “Treatment.” The authors used their own judgment about which papers to include from the literature search based on the relevance of the article to the clinical scenario. This review also includes a summary of the authors’ work and knowledge based on reading the oncology literature. This review is not a systematic review; it includes a selection of references.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in part by an Institutional grant from Centro di Riferimento Oncologico Aviano for an intramural project “Molecular Medicine” (A.C.).
A. Carbone is Chairman of the Italian TNM Staging Committee UICC
A. Carbone is member of the WHO IARC Monograph Working Group on Biological Agents, Lyon, 2009
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Carbone, A., Gloghini, A., Kwong, YL. et al. Diffuse large B cell lymphoma: using pathologic and molecular biomarkers to define subgroups for novel therapy. Ann Hematol 93, 1263–1277 (2014). https://doi.org/10.1007/s00277-014-2116-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-014-2116-y