
Abstract
The interactions of chronic lymphocytic leukemia cells with the microenvironment in secondary lymphoid tissues and the bone marrow are known to promote CLL cell survival and proliferation. CD38 and CD49d are both independent prognostic risk parameters in CLL with important roles in shaping these interactions. Both are reported to influence CLL cell trafficking between blood and lymphoid organs as well as their survival and proliferation within the lymphoid organs, thereby impacting the pathophysiology of the disease. The expression of CD38 and CD49d is associated in the majority of cases, and they exist as part of macromolecular complexes. Here, we review the current evidence for the individual and associated contributions of these molecules to CLL pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The heterogeneity of CLL in regard to the tumor microenvironment
Chronic lymphocytic leukemia (CLL), a B cell non-Hodgkin’s lymphoma with a leukemic appearance, is a remarkably heterogeneous disease that can follow a wide variety of courses. Patients with an indolent course survive for many years. Others, however, show a rapidly fatal disease despite aggressive therapy and die within 2–3 years of diagnosis (reviewed in [1]). Mere staging by Rai [2] or Binet [3] is not sufficient to predict at an early stage of the disease which clinical course a patient will experience. Several more recently suggested prognostic markers, apparently involved in the cellular processes underlying CLL pathogenesis, may aid to classify patients according to clinical risk. These markers include chromosomal aberrations such as deletion of 17p13, 11q22-23, or 13q1, and trisomy 12 [4] that directly influence cell fate or transformation, as well as molecular markers for CLL cell interactions with the tumor microenvironment [5]. Among the molecular prognostic factors, the mutational status of immunoglobulin variable region (IGHV) genes [6, 7], the expression of CD38 on the surface of CLL cells [6], and the intracellular expression of zeta-associated protein 70 [8, 9] are the best-established ones. A more recently discovered marker is CD49d, the alpha4 subunit of the VLA-4 integrin (alpha4beta1). High CD49d expression predicts reduced overall survival and time to first treatment in CLL patients [10, 11].
The pathogenic significance of CLL cell interactions with the lymphoid microenvironment has become increasingly acknowledged in recent years. CLL cell proliferation is supposed to take place in lymph nodes and, to a lesser extent in bone marrow, with up to 2 % of the entire clone being newly generated per day [12], and is most likely supported by activated T lymphocytes that express CD40 ligand [13–15]. Signals from T lymphocytes and from other accessory cells in this environment, such as stromal or nurse-like cells, also provide pro-survival support to the malignant cells [16–19]. Not only does the microenvironment influence CLL cells, but CLL cells alter their microenvironment to their advantage by priming T cells towards an immune suppressive phenotype [20] or inducing stromal cells to provide pro-survival signals [21–24], which contributes to chemoresistance and treatment failure. Minimal residual disease after therapy is attributed to supportive microenvironmental signals and prognostically associated with shortened progression-free and overall survival rates of CLL patients [25–27]. Eradicating residual CLL cells within their protective niches in secondary lymphoid tissues and bone marrow is thus considered a major therapeutic goal for achieving permanent remission.
As dissected in the following chapters, the prognostic markers CD49d and CD38 have been reported to be involved in various cellular functions relevant to CLL pathogenesis: CLL cell homing to lymphoid organs, survival, and proliferation. However, CD49d and CD38 expression is associated in about 80 % of CLL patients, and these molecules are reported to physically interact within multi-protein complexes. Because of this, it is difficult to gauge the individual contribution of each molecule to key pathogenic functions in CLL. In light of the high heterogeneity of reports dealing with either molecule and the fact that CD49d is still a newcomer among the prognostic factors, we review the current evidence for the individual and associated contributions of these molecules to CLL pathophysiology.
CD38 and VLA-4 in general
CD38
CD38 is a highly conserved 45 kDa transmembrane type II glycoprotein with a short cytoplasmatic tail, a single-spanning transmembrane domain, and a large extracellular domain (257 aa) [28, 29]. CD38 can be localized on the plasma membrane, in the cytoplasm, and in the inner nuclear membrane of cells [30, 31]. It is expressed in numerous cells types of the hematopoietic system, such as lymphocytes, myeloid cells, natural killer (NK) cells, platelets, and erythrocytes, as well as in solid tissues, including various cell types of the brain, the eye, in pancreatic islet cells, smooth muscle cells, and osteoclasts and osteoblasts [31]. CD38 is an important enzyme for the regulation of calcium signaling and the cell’s energy transfer homeostasis [29]. The products of the enzymatic reactions catalyzed by CD38 are all involved in the release of different intracellular calcium stores, mostly independent of the traditional inositol triphosphate (IP3) pathway [32–37]. As such, CD38 has been shown to play a critical role in diverse immune functions: in T cell activation [34], neutrophil chemotaxis [38], dendritic cell migration [39], and monocyte chemokine production [40]. Furthermore, CD38-mediated calcium control has also been implicated in various other functions of different cell types: in the insulin secretion of pancreatic beta cells [41, 42], in the oxytocin production of neurons [43–45], in bone resorption of osteoclasts [46], as well as the responsiveness of airway smooth muscle cells [47].
In addition to these enzymatic functions, CD38 is a well-known lymphocyte differentiation antigen with proposed receptor and adhesion molecule functions. This versatility of CD38 and its characteristic to function as dimers, multimers, or as part of multi-protein complexes makes it difficult to fully comprehend its biology. The proposed functions of CD38 as a receptor vary greatly and depend on its association with other surface molecules. CD38 is laterally associated with the main signaling complexes of lymphocytes that are organized in lipid rafts. In T cells, CD38 is capable of interacting with the T cell receptor (TCR)/CD3 complex; in B cells, with the BCR/CD19 complex; and in NK cells, with the CD16/CD81 complex [31]. CD31 (PECAM-1), expressed by, e.g., endothelial cells has been described as a CD38 ligand [48]. CD38+ lymphocytes show a weak, selectin-like adhesion to endothelial cells [49], which appears to be mediated by CD38–CD31 interactions [48].
In B cells, ligation of CD38 by agonistic antibodies triggers different in vitro responses depending on the differentiation stage of the cells. In immature B cell precursors, CD38 ligation inhibited DNA synthesis and induced apoptosis, thereby blocking B cell hematopoiesis [50]. In tonsillar germinal center B cells, CD38 mediated pro-survival signaling [51]. In mature circulating B cells, CD38 ligation induced proliferation by promoting the expression of CD25, MHC-II, and certain cytokines [31, 52].
In summary, CD38 is a widely expressed enzyme and receptor, involved in various cellular functions, making it difficult to pinpoint one particular function that would most critically impact CLL pathobiology.
CD49d
CD49d belongs to the family of integrin alpha subunits. Integrins are heterodimers of non-covalently linked alpha and beta subunits. The human CD49d (alpha4 integrin) subunit can associate with either CD29 (beta1 integrin) or with beta7 integrin [53, 54]. The combination of CD49d with beta7 mediates lymphocyte binding to mucosal addressin cell adhesion molecule-1, and its expression defines lymphocytes capable of trafficking through the intestines and the intestinal lymphoid tissues [55, 56]. In contrast, very late antigen-4 (VLA-4) is formed by the combination CD49d/CD29 [53] and is expressed on leukocytes, including B and T cells, and on CD34+ hematopoietic stem/progenitor cells (HSPCs). VLA-4 is the major CD49d-containing combination found in resting CLL cells [57]. VLA-4 has two major ligands: VCAM-1 [58], expressed on endothelial cells and bone marrow stromal cells [59], and the extracellular matrix molecule fibronectin [60].
The use of mouse models has proved fundamental in revealing the essential role of VLA-4 in fetal and adult hematopoiesis (reviewed in [56, 61]). Germline deletion of either VLA-4 subunit, CD49d or CD29, resulted in embryonic lethality in mice. Chimeric mouse models generated with CD29 integrin-expressing or CD29 integrin-deficient embryonic stem (ES) cells indicated that loss of CD29 does not impact haematopoietic stem cell formation or their differentiation into different lineages but severely compromises their ability to colonize the fetal liver [62, 63]. Adult murine hematopoiesis, however, did not seem to rely on CD29 [64]. Similar chimeric models generated with CD49d-positive or CD49d-negative ES cells demonstrated that this integrin subunit is critical for proper lineage differentiation and maturation of the hematopoietic system [65, 66]. Later, more refined inducible knockout models revealed that CD49d is essential for bone marrow homing and retention of progenitor cells [67, 68]. Moreover, absence of CD49d in HSPCs hindered both their self-renewal capacity and their ability to reconstitute hematopoiesis [69].
Homing is a rapid process that describes the active migration of cells from the blood, through the vascular endothelium, into lymphoid organs. It differs from engraftment, for which cell proliferation within the lymphoid tissue environment is essential [70]. Using adoptive transfers of human cells into immune-deficient mice, VLA-4 was identified as a key molecule for both the bone marrow homing and the engraftment of normal and leukemic human HSPCs [71, 72]. VLA-4 exists in multiple conformational states, including high-affinity-activated but also low-affinity-extended states [73]. These specific conformations allow VLA-4, in contrast to other integrins, to support not only firm adhesion but also rolling of lymphocytes on VCAM-1 displaying endothelial cells. Chemokine-induced inside-out activation of VLA-4, e.g., by the bone marrow chemokine CXCL12, induces an upregulation of its adhesive properties to VCAM-1. This mediates the arrest of HSPCs on the BM vessels, which is a prerequisite for their BM homing [72]. Functional VLA-4 expression is also indispensable for retention of normal HSPCs as well as leukemic blasts in bone marrow [74, 75]. Consequently, targeted disruption of VLA-4 function by anti-CD49d antibodies or small-molecular-weight VLA-4 antagonists is known to result in rapid release of HSPCs into the peripheral circulation and to act synergistically or additive with conventional mobilization regimes (for review, see [61]).
Thus, in contrast to CD38, the biological functions of CD49d and the CD49d/CD29 integrin combination VLA-4 are well defined, with a principal involvement in bone marrow homing and retention of hematopoietic cells, processes important to CLL pathophysiology.
CD38 and CD49d as prognostic markers in CLL
The prognostic role of CD38 in CLL was first proposed on the basis of an immunophenotypic study of CLL cases with known IGHV sequences [6]. CD38 predicted shorter overall survival rates when expressed on 30 % or more CLL cells [6]. Since this first report in 1999, CD38 expression has been well established as an independent prognostic factor in CLL by numerous reports, however, with various cut-off levels. While Hamblin et al. [76] and Del Poeta et al. [77] concur with 30 % as the best cut-off, others proposed 20 % [78, 79] or even 7 % [80, 81]. Further studies are still necessary to define a common cut-off level (reviewed in [82, 83]).
In 2008, two studies concluded that high expression of CD49d is a robust adverse prognostic marker in CLL [10, 11]. When analyzed retrospectively, CLL patients with ≥30 % CD49d-positive tumor cells revealed significantly shorter treatment-free and overall survival than patients with <30 % CD49d positivity [10]. A prospective analysis indicated that an alternative cut-off level of 45 % CD49d expression might be superior to the 30 % level [11]. Following these first reports, the prognostic relevance of increased CD49d expression was rapidly and unequivocally confirmed by several groups, using the 30 % cut-off level [84–88] (Table 1). Comparative analyses of CD49d mRNA and protein levels demonstrated its transcriptional–translational consistency [57, 84] which allows its determination by flow cytometry as well as PCR-based assays for risk categorization. As with CD38, high CD49d expression acts as an independent prognostic marker but is highly associated with other risk parameters such as IGHV, ZAP70, CD38, and the presence of chromosomal aberrations [10, 11].
Remarkably, the finding of differential CD49d expression in CLL is an older discovery than anticipated. In 1996, it had already been demonstrated that CD49d expression in CLL is variable, with higher expression of CLL samples of advanced (Rai III, IV) than early stages [89]. Zucchetto and colleagues were the first in 2006 that reported the strong association of CD38 and CD49d expression on CLL cells using both parameters as categorical variables [90]. Comparing overall survival rates, a combined CD38 low/CD49d low phenotype was attributed to the best prognosis. Out of the 115 investigated samples, 27 cases (23 %), however, displayed a discordant CD38 low/CD49d high or CD38 high/CD49d low phenotype. Patients with this discordant phenotype showed better overall survival rates compared with the combined CD38 high/CD49d high phenotype. Recently, we found a comparable 21.5 % rate of discordant cases when analyzing 144 samples [86]. In our analysis, both a CD38 high or CD49d high phenotype were sufficient to predict shortened time to first treatment, even when the presence of the second marker was low. This implies that a relevant proportion of our patients would be misclassified with regards to risk if we were to base our stratification solely on CD38 expression. Our data thus support the previous suggestion of a scoring system based on several antigens, including CD38 and CD49d, as an additional tool for accurate risk categorization in CLL [91].
CD38 and CD49d in CLL cell migration and homing
In light of the current evidence that suggests that CLL localization within supportive lymphoid niches is critical to disease progression, it seems logical that at least a part of the peripheral blood CLL pool constantly recirculates into bone marrow and secondary lymphoid organs (Scheme 1). Nevertheless, compared with healthy B lymphocytes, CLL cells display an impaired in vitro transendothelial migratory (TEM) capacity over human umbilical vein endothelial cells [92], a widely accepted endothelial model. These data are in line with the early in vivo observation that 51Cr-labeled CLL cells of a CLL patient left the circulation at a dramatically diminished degree compared with healthy lymphocytes [93].

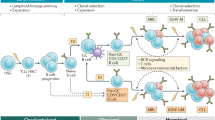
Hypothetical model of CD49d (VLA-4) and CD38 implications in the distinct steps of the CLL life cycle. CD49d is a key molecule for homing of CLL cells with a major mechanistical role in (1) rolling and (2) arrest of CLL cells on the bone marrow and lymph node endothelia. CD38 may contribute to rolling of lymphocytes on the endothelium cells and to (3) transendothelial migration. A macromolecular complex (MMC), involving both CD38 and CD49d, is relevant for (4) invasion within the lymphoid tissue and (5) survival of CLL cells, with additional individual anti-apoptotic contributions of the molecules. (6) CLL proliferation is strongly associated with CD38 expression, with a presumable but yet-to-be-described role of CD49d. Mobilization of CLL cells from the lymphoid organs likely requires downregulation of CD49d expression or function. CD49d stands for the functional CD49d/CD29 (VLA-4). MMC: macromolecular complex including CD49d/CD29/CD38/MMP9/CD44v
This transmigratory defect was firstly attributed to low L-selection expression [94], which is a major mediator of lymphocyte tethering on endothelial cells. Subsequently, it became clear that the transmigratory capacity of CLL cells varied between patients, with CLL cells from patients with advanced disease and bearing lymph node enlargement displaying increased TEM rates [95]. CCR7 and VLA-4 were identified as key factors in this process and a correlation between high CD49d expression and lymphadenopathy was demonstrated [95]. In these patients, high CD49d levels appear to overcome some of the migratory defects of the CLL cells [96]. While entry of normal B lymphocytes into lymph nodes is dependent on LFA-1, CLL cells with reduced LFA-1 levels (compared with normal B lymphocytes) were able to cross human umbilical vein endothelial cells in vitro by a mechanism that required an interplay between VLA-4 and LFA-1 [57, 96]. CD49d expression was decisive for in vivo homing of human CLL cells into the bone marrow of immune-deficient mice [57, 86]. Moreover, analyzing human bone marrow CLL specimens, we also observed an association of CD49d expression and high leukemic BM infiltration [86]. The association of CD49d and MMP9 (see in more detail in chapters below) may further contribute to invasiveness of CLL cells in the dense BM environment.
CD38 expression was also described to define CLL cells with an increased migratory potential as tested by transwell chemotaxis assays [97]. In addition, lentiviral transfection of CLL cells with CD38 resulted in their enhanced motility to CXCL12 [98]. By using an inhibitory anti-CD38 antibody (clone: SUN-4B7), the authors uncovered a CD38 contribution to BM homing of CLL cells. In light of the expression of CD31, reported to be a ligand of CD38, in the endothelium, a role of CD38 in CLL cell extravasation appears logical. However, when comparing the integrin expression on CD38 high or low CLL cells, increased VLA-4 (CD49d subunit) and LFA-1 (CD18 subunit) expression was found in the CD38 high subtype [99], making it difficult to ascribe enhanced migration functions solely to CD38. In short-term homing assays wherein CLL cells from discordant CD38+/VLA-4− and CD38-/VLA-4+ cases were transplanted into NOD/SCID mice, only cells expressing VLA-4 were capable of entering the BM [86]. Importantly, the enhanced engraftment of CD38 positive samples in NOD/SCID mice and the higher proliferation rates in the spleens of these xenogeneic animals have been attributed to CD38-dependent proliferation rather than homing of the CLL cells [100].
Thus, the evidence indisputably demonstrates that CLL cells require a functional VLA-4 to enter the bone marrow. Additionally, VLA-4 potentially compensates for CLL-associated LFA-1 defects during lymph node entry. While the role for CD38 in homing is still ambiguous, CD38-associated proliferation (discussed in more detail below) may play a role in the successful engraftment of the tumor cells within these lymphoid tissues.
CD38 and CD49d in CLL cell survival
Both, CD38 and CD49d have been described to contribute to CLL survival, in a direct or indirect manner. A direct contribution would involve the induction of anti-apoptotic signaling cascades upon ligand binding, whereas an indirect contribution would be to ensure that the tumor cells are in the right place at the right time in order to avail of the favorable signals emanating from the microenvironment.
Ligation of CD38 in IL-2-treated CLL cells led to an increase in long-term survival [101]. Whether engagement of CD38 by its supposed ligand CD31 induces direct pro-survival signals in CLL is still controversially discussed. Co-culture of CD38 expressing CLL cells with CD31-transfected murine cell lines or CD31+ nurse like cells enhanced survival of CLL cells and induced their proliferation [102, 103]. The CD31-specificity of these results could be demonstrated by addition of an antagonistic anti-CD31 mAb that abrogated the effects [102]. Other authors, however, have reported that they found no increase in cell survival or proliferation of either CD38 high or low expressing CLL cells co-cultured with endothelial cells or CD31-transfected fibroblasts in the presence or absence of either anti-CD31 or anti-CD38 blocking mAbs [104]. Discordant results were also obtained when studying the modulation of apoptosis regulators upon incubation of CLL cells from CD38 high-risk patients with CD31-transfected fibroblasts [104, 105]. Whether these findings reflect the in vivo situation remains to be confirmed.
Similarly, a direct ligand-triggered survival function of VLA-4 was suggested in CLL, as tumor cells could be rescued from spontaneous or drug-induced in vitro apoptosis by direct cell–cell contact with stromal cells [19, 106]. These studies suggested that beta1 integrins contribute to this protective adhesion [106]. Furthermore, another series of studies reported that culturing CLL cells on fibronectin- or VCAM-1-coated plates increased their in vitro viability and chemoresistance, which was accompanied by an increased Bcl-2/Bax ratio and elevated Bcl-xL levels [107, 108]. The authors also suggested that a general chemoresistant phenotype is related to high VLA-4 expression of CLL cells. However, Majid and colleagues [85] did not find a correlation between CD49d expression and in vitro resistance to fludarabine in liquid cultures albeit they still observed a protective adhesion of CD49d high cells to fibronectin-coated plates. Similarly, we confirmed higher adhesion rates of VLA-4+ CLL cells to protective stromal cells than of VLA-4− CLL cells [86]. Nevertheless, VLA-4 low CLL cells were still protected from spontaneous apoptosis to a similar extent by the presence of stroma, suggesting that this CLL subgroup uses alternative viability signals. In line with these observations, cell adhesion-mediated drug resistance, induced by culturing CLL cells on a follicular dendritic cell line, was shown to be independent of VCAM-1 [109] but dependent on other signals, e.g., CD44. One possible explanation for these divergent findings is that different groups investigated the protective effects of VLA-4-mediated cell adhesion over different lengths of time. Of note, Zucchetto et al. [110] found that protective VLA-4/VCAM-1 interactions between CLL cells and VCAM-1-transfected fibroblasts first become apparent after 7 days of co-culture and continuously increased with the most dramatic effects being observed after 21 days, a much longer time period than studied in most previous reports.
As we found VLA-4/CD38 low-risk samples to be less sensitive towards spontaneous apoptosis ex vivo [86], they appear less dependent on continuous external stimulation. Hypothetically, they are in a more quiescent mitotic state, based on their lower basal Ki-67 expression. Our data are consistent with the report by Coscia and colleagues [111] who observed that high-risk CLL cells with unmutated IGHV genes were extremely vulnerable when removed from the microenvironmental signals that induce supportive NF–kB signaling in these cells. Furthermore, NF–kB signaling via its transcriptional target TAp63 has been shown to result in increased VLA-4 expression on high-risk samples [112], thus potentially creating a positive feedback loop leading to the accumulation of survival signals in the tumor cells. Of note, other intercellular interactions leading to the activation of CLL cells, such as their interaction with vascular endothelium or CD40L-transfected fibroblasts, also result in the upregulation of VLA-4 expression on the tumor cells [103, 113]. Whether these interactions result in the concomitant increase of the ligands VCAM-1 and fibronectin has not been investigated yet. However, the implication of these findings is that VLA-4-mediated adhesion may help to strengthen the intercellular contacts, thereby allowing stable and long-term bi-directional signaling between the tumor cells and the microenvironment. VLA-4 may also support survival of CLL cells as part of a larger protein complex including MMP9 and CD44v [114, 115]. In this setting, VLA-4 supports the adhesion of CLL cells to proMMP9, which results in pro-survival signals towards the CLL cells via the hemopexin domain of MMP9 [115]. Intriguingly, proMMP9 hereby acts as a non-canonical ligand for VLA-4 inducing a Lyn-Stat3-Mcl-1 pro-survival signaling cascade distinct from VCAM-1 induced survival signals.
CD38 and CD49d in CLL cell proliferation
An enormous amount of effort has been put into defining the link between CD38 and CLL cell proliferation and determining whether CD38 plays an active or passive role in this process. The most recent findings suggest that CD38 expression marks an activated and recently born CLL cell subset [116–118]. The proliferation marker Ki-67 that characterizes cell cycle entry and the mitosis regulating anaphase-promoting complex/cyclosome (APC/C) are significantly increased in CLL cells of CD38 high-risk patients compared with low-risk samples [86, 118] [99]. Moreover, CD38+ CLL subclones within individual patient samples express increased expression of the early activation marker CD69, the B cell activation marker CD27, and of Ki-67 [118–120]. Elegant in vivo labeling studies verified that the CD38+ peripheral blood CLL cell pool comprises more newly proliferated CLL cells than the CD38- pool [116]. Consistently, Ki-67 positive cells were frequently positive for CD38 in CLL proliferation centers in lymph nodes [15]. In vitro, CLL cell proliferation can be induced by activation of T lymphocytes or CD40 ligand stimulation, and this is accompanied by an increase in CD38 expression on the CLL cell [15, 121]. These observations, however, are correlative rather than proof of a direct functional role for CD38 in this cellular function. A single study implicated an active role for CD38 in CLL proliferation: CD38 ligation on CLL cells by an agonistic antibody in the presence of IL-2 provoked intracellular calcium signals and proliferation of the tumor cells [101]. What is clear, however, is that CD38 expression can be regulated by the microenvironment and can serve as a marker for an activated or recently activated CLL phenotype [15, 99, 101, 122]. It is likely that CD38 expression within the CLL clone is transient and CD38-positive tumor cells eventually become CD38-negative, as proposed by Calissano et al. [116]. This is supported by the fact that CD38+ and CD38− subclones do not show any difference in telomere length [118, 119] and that there is no clonal evolution of the CD38+ subclone [119]. This would also imply that a CD38− subclone could become CD38+ given the proper stimuli. Whether the upregulation of CD38 is a prerequisite to proliferation remains to be determined and is hindered by the lack of small molecule specific inhibitors of CD38, as well as difficulties in achieving stable and long-term knockdowns in CLL cells.
In contrast to CD38, there is much less data linking VLA-4 to CLL proliferation. We recently demonstrated that, similar to CD38, VLA-4 expression of bone marrow-derived CLL cells is higher than that of peripheral blood CLL cells and that the proliferating CLL cell fraction was enriched in the VLA-4+ subclone [86]. Notably, VLA-4 high-risk CLL cells also displayed increased in vitro proliferation rates upon co-culture with CD40L-transfected fibroblasts [121]. How VLA-4 impacts CLL cell proliferation remains to be elucidated.
Challenges in separating individual and shared contributions of VLA-4 and CD38 in CLL pathophysiology
A possible molecular basis for the high correlation of CD49d and CD38 expression in CLL could be their physical association in multi-protein-complexes. Recent reports suggest a variety of possible protein combinations [98, 110, 114, 115, 123, 124]. Two recent reports demonstrate a colocalization and physical association of CD38 and CD49d/CD29 by a combination of immunofluorescence and immunoprecipitation approaches [110, 123]. Whether this interaction occurs via the CD49d or the CD29d subunit of the VLA-4 integrin could not unequivocally be clarified [110, 123]. Interpatient variability, which is usually high in CLL, adds further complexity: Buggins et al. reported [123] a multimer-complex involving CD38, CD49d, MMP9, and CD44 and observed a co-immunoprecipitation of CD38 with CD49d in the majority, but of CD38 with MMP9 in only about half of the investigated samples. Redondo-Munoz et al. reported the association of CD49d and MMP9 particularly with CD44 variant forms instead of pan CD44 [124]. All the reported complex structures may represent novel CLL high-risk-specific therapeutic targets as they do not appear to form in normal B cells or in low-risk cases. Notably, besides direct interactions, CD38 and CD49d may also indirectly influence each other as parts of a consecutive chain of events [125].
Given this complexity, it is difficult to separate association-intrinsic from molecule-specific functions, particularly when interpreting correlative analyses. In addition, the use of blocking antibodies in functional studies bears the risk of co-capping, crosslinking, or steric hindrance of the partner molecule, which could be overcome by the use of small molecule inhibitors, which are increasingly being developed. Furthermore, a genetic modulation of CD38 and VLA-4 expression could help to correctly define their individual contributions. The successful lentiviral introduction of CD38 in CD38-negative CLL cells has only recently been achieved [126] and contributes to a better understanding of the molecule. Furthermore, introduction of CD38 into cells of the CLL-derived prolymphocytic leukemia cell line MEC1 increased their adhesion to VLA-4 ligands, indicating functional CD38-VLA-4 interaction [110]. Conversely, lentiviral transfer of short hairpin RNA (shRNA) could be used for stable and specific reduction of CD38 expression in high-risk CLL. However, genetic manipulations of the cell cycle-arrested primary peripheral blood CLL cells are still a challenge. To achieve an efficient knockdown, it would likely be necessary to combine shRNA approaches with long-term culture and cell cycle induction of CLL cells. To this end, co-culture techniques that mimic the proliferative and supportive microenvironment in CLL are continuously being improved [121].
An alternative approach to separate the functions of CD38 and VLA-4, feasible in the absence of efficient knockdown techniques, is the analysis of discordant cases. Notably, the correlation of risk factors in CLL samples is not absolute with a considerable rate of discordancy. Functional analyses using these discordant samples are useful to define the dispensability or compensation of a specific molecule in a cellular function. Taking this road, we have been able to demonstrate that CD38 is not required for BM homing of CLL cells while VLA-4 is indispensable. However, in light of the potential enzymatic function of CD38, it is still conceivable that in CD38/VLA-4 double-positive cases CD38 can exert a supportive function in energy-dependent VLA-4 activation. The basis of this crosstalk remains to be elucidated.
Therapeutic implications
Collectively, the data demonstrate that CLL cells with high-risk features are in fact those that are most exquisitely dependent on microenvironmental stimuli for their survival and proliferation. Notably, the pathophysiological and prognostic importance of this crosstalk bears therapeutical consequences. Finding a therapeutic means of interfering with the bi-directional communication between CLL cells and the supportive microenvironment would go a long way to finding a definite cure for this disease. Besides single targeting of CD38 or VLA-4, disrupting the macromolecular complexes housing these proteins, or inhibiting downstream signaling, are all conceivable strategies. Additionally, immunomodulatory drugs such as lenalidomide, whose molecular mechanism of action is still unclear, may indirectly impact CD38 and VLA-4 expression and function, and this should be further investigated.
The ubiquitous expression of CD38 in many different cell types and tissues obviously raises concerns regarding the safety of widespread inhibition of CD38 function. Currently, there are three different anti-CD38 antibodies under evaluation for safety in clinical trials (Table 2). Two monoclonal antibodies are being tested in multiple myeloma (daratumumab, identifier: NCT00574288 and MOR03087, identifier: NCT01421186; http://clinicaltrials.gov) and a third in selected CD38+ hematological malignancies including CLL (SAR650984, identifier: NCT01084252; http://clinicaltrials.gov). These antibodies are supposed to bind to CD38+ tumor cells and trigger antibody-dependent cellular cytotoxicity rather than inhibit the biological CD38 functions.
Treatment strategies using VLA-4 inhibitors supposedly interfere with the recirculation of CLL cells into bone marrow and lymph nodes. Recently, the recombinant anti-VLA-4 antibody natalizumab demonstrated the potential to overcome stromal cell-induced resistance of B cell lymphoma cells against cytotoxic drugs and rituximab in vitro [127]. Natalizumab is already approved as an anti-inflammatory drug, and a number of small molecule inhibitors for VLA-4 have been developed [61], primarily for use in multiple sclerosis or asthma. However, most of the clinical trials using these small-molecular-weight antagonists for VLA-4 have been terminated due to low efficacy or side effects of the substances. A new generation of currently developed VLA-4 inhibitors might overcome the previous problems and widen the therapeutic spectrum of VLA-4 antagonism towards tumor therapy. In fact, VLA-4 antagonizing nanoparticles recently demonstrated adhesion-inhibitory and cytotoxic effects that resulted in reduced tumor growth in a multiple myeloma mouse model [128].
Moreover, targeting of VLA-4 downstream signals might provide an alternative approach. VLA-4 antagonism is known to mobilize stem and progenitor cells from bone marrow [61], and its ligand VCAM-1 is highly expressed in both CLL BM and lymph nodes (unpublished observation). It is therefore expected that VLA-4 targeting will not only impede CLL cell recirculation to these lymphoid niches, but additionally mobilize tumor cells from lymphoid organs, similar to the effects seen with novel small molecule inhibitors. Notably, ibrutinib, the clinically active BTK inhibitor PCI-32765, was recently shown to impair VLA-4-mediated adhesion of CLL cells [129]. This is consistent with the clinical observation of a transient lymphocytosis of ibrutinib-treated patients due to mobilization of CLL cells from lymphoid organs into the peripheral blood [130]. Other small molecule antagonists used in clinical trials for treatment of CLL, e.g., the phosphatidylinositol 3-kinase inhibitor CAL-101 [131], might also affect VLA-4-mediated cellular functions in CLL.
Conclusively, it is evident that CD38 and VLA-4 are more than just markers of an aggressive CLL cell type and that they play functional roles in the pathobiology of the disease. As such, they represent therapeutic targets that may be exploited in addition to, or in combination with, the currently developed novel approaches of interfering with CLL cell–tumor host interactions. Targeting these molecules should also be tested for its potential in avoiding the frequent relapses and development of chemoresistance in CLL.
References
Chiorazzi N, Rai KR, Ferrarini M (2005) Chronic lymphocytic leukemia. N Engl J Med 352(8):804–815. doi:10.1056/NEJMra041720
Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS (1975) Clinical staging of chronic lymphocytic leukemia. Blood 46(2):219–234
Binet JL, Auquier A, Dighiero G, Chastang C, Piguet H, Goasguen J, Vaugier G, Potron G, Colona P, Oberling F, Thomas M, Tchernia G, Jacquillat C, Boivin P, Lesty C, Duault MT, Monconduit M, Belabbes S, Gremy F (1981) A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48(1):198–206
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, Dohner K, Bentz M, Lichter P (2000) Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343(26):1910–1916. doi:10.1056/NEJM200012283432602
Dal-Bo M, Bertoni F, Forconi F, Zucchetto A, Bomben R, Marasca R, Deaglio S, Laurenti L, Efremov DG, Gaidano G, Del Poeta G, Gattei V (2009) Intrinsic and extrinsic factors influencing the clinical course of B-cell chronic lymphocytic leukemia: prognostic markers with pathogenetic relevance. J Transl Med 7:76. doi:10.1186/1479-5876-7-76
Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, Lichtman SM, Schulman P, Vinciguerra VP, Rai KR, Ferrarini M, Chiorazzi N (1999) Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94(6):1840–1847
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK (1999) Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94(6):1848–1854
Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, Kipps TJ (2002) Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 100(13):4609–4614. doi:10.1182/blood-2002-06-1683
Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, Zhao H, Ibbotson RE, Orchard JA, Davis Z, Stetler-Stevenson M, Raffeld M, Arthur DC, Marti GE, Wilson WH, Hamblin TJ, Oscier DG, Staudt LM (2003) ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 101(12):4944–4951. doi:10.1182/blood-2002-10-3306
Gattei V, Bulian P, Del Principe MI, Zucchetto A, Maurillo L, Buccisano F, Bomben R, Dal-Bo M, Luciano F, Rossi FM, Degan M, Amadori S, Del Poeta G (2008) Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood 111(2):865–873. doi:10.1182/blood-2007-05-092486
Shanafelt TD, Geyer SM, Bone ND, Tschumper RC, Witzig TE, Nowakowski GS, Zent CS, Call TG, Laplant B, Dewald GW, Jelinek DF, Kay NE (2008) CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: a prognostic parameter with therapeutic potential. Br J Haematol 140(5):537–546. doi:10.1111/j.1365-2141.2007.06965.x
Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, Murphy EJ, Koduru P, Ferrarini M, Zupo S, Cutrona G, Damle RN, Wasil T, Rai KR, Hellerstein MK, Chiorazzi N (2005) In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 115(3):755–764. doi:10.1172/JCI23409
Carbone A, Gloghini A, Gruss HJ, Pinto A (1995) CD40 ligand is constitutively expressed in a subset of T cell lymphomas and on the microenvironmental reactive T cells of follicular lymphomas and Hodgkin’s disease. Am J Pathol 147(4):912–922
Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, Ruffing N, Montagna L, Piccoli P, Chilosi M, Caligaris-Cappio F (2002) Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L + T cells by producing CCL22. Eur J Immunol 32(5):1403–1413. doi:10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y
Patten PE, Buggins AG, Richards J, Wotherspoon A, Salisbury J, Mufti GJ, Hamblin TJ, Devereux S (2008) CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood 111(10):5173–5181. doi:10.1182/blood-2007-08-108605
Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ (2000) Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 96(8):2655–2663
Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A (2007) Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an "angiogenic switch". Leuk Res 31(7):899–906. doi:10.1016/j.leukres.2006.11.024
Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV (1996) Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol 92(1):97–103
Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, Sivina M, Wierda WG, Estrov Z, Keating MJ, Shehata M, Jager U, Gandhi V, Kay NE, Plunkett W, Burger JA (2009) Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 114(20):4441–4450. doi:10.1182/blood-2009-07-233718
Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, Byrd JC, Gribben JG (2008) Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest 118(7):2427–2437. doi:10.1172/JCI35017
Cols M, Barra CM, He B, Puga I, Xu W, Chiu A, Tam W, Knowles DM, Dillon SR, Leonard JP, Furman RR, Chen K, Cerutti A (2012) Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL, and CD40L. J Immunol 188(12):6071–6083. doi:10.4049/jimmunol.1102066
Ding W, Knox TR, Tschumper RC, Wu W, Schwager SM, Boysen JC, Jelinek DF, Kay NE (2010) Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood 116(16):2984–2993. doi:10.1182/blood-2010-02-269894
Ding W, Nowakowski GS, Knox TR, Boysen JC, Maas ML, Schwager SM, Wu W, Wellik LE, Dietz AB, Ghosh AK, Secreto CR, Medina KL, Shanafelt TD, Zent CS, Call TG, Kay NE (2009) Bi-directional activation between mesenchymal stem cells and CLL B-cells: implication for CLL disease progression. Br J Haematol 147(4):471–483. doi:10.1111/j.1365-2141.2009.07868.x
Lutzny G, Kocher T, Schmidt-Supprian M, Rudelius M, Klein-Hitpass L, Finch AJ, Durig J, Wagner M, Haferlach C, Kohlmann A, Schnittger S, Seifert M, Wanninger S, Zaborsky N, Oostendorp R, Ruland J, Leitges M, Kuhnt T, Schafer Y, Lampl B, Peschel C, Egle A, Ringshausen I (2013) Protein kinase c-beta-dependent activation of NF-kappaB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell 23(1):77–92. doi:10.1016/j.ccr.2012.12.003
Bottcher S, Ritgen M, Fischer K, Stilgenbauer S, Busch RM, Fingerle-Rowson G, Fink AM, Buhler A, Zenz T, Wenger MK, Mendila M, Wendtner CM, Eichhorst BF, Dohner H, Hallek MJ, Kneba M (2012) Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol Off J Am Soc Clin Oncol 30(9):980–988. doi:10.1200/JCO.2011.36.9348
Kwok M, Rawstron A, Varghese A, Hillmen P (2009) Minimal residual disease is a predictor for progression-free and overall survival in chronic lymphocytic leukemia (CLL) that is independent of the type or line of therapy. Blood 114 (22):Abstract 540
Moreton P, Kennedy B, Lucas G, Leach M, Rassam SM, Haynes A, Tighe J, Oscier D, Fegan C, Rawstron A, Hillmen P (2005) Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol Off J Am Soc Clin Oncol 23(13):2971–2979. doi:10.1200/JCO.2005.04.021
Alessio M, Roggero S, Funaro A, De Monte LB, Peruzzi L, Geuna M, Malavasi F (1990) CD38 molecule: structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J Immunol 145(3):878–884
Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q (2005) Crystal structure of human CD38 extracellular domain. Structure 13(9):1331–1339. doi:10.1016/j.str.2005.05.012
Adebanjo OA, Anandatheerthavarada HK, Koval AP, Moonga BS, Biswas G, Sun L, Sodam BR, Bevis PJ, Huang CL, Epstein S, Lai FA, Avadhani NG, Zaidi M (1999) A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol 1(7):409–414. doi:10.1038/15640
Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 88(3):841–886. doi:10.1152/physrev.00035.2007
Guse AH (1999) Cyclic ADP-ribose: a novel Ca2 +-mobilising second messenger. Cell Signal 11(5):309–316
Gustafsson AJ, Muraro L, Dahlberg C, Migaud M, Chevallier O, Khanh HN, Krishnan K, Li N, Islam MS (2011) ADP ribose is an endogenous ligand for the purinergic P2Y1 receptor. Mol Cell Endocrinol 333(1):8–19. doi:10.1016/j.mce.2010.11.004
Magnone M, Bauer I, Poggi A, Mannino E, Sturla L, Brini M, Zocchi E, De Flora A, Nencioni A, Bruzzone S (2012) NAD + levels control Ca2+ stores replenishment and mitogen-induced increase of cytosolic Ca2+ by ADPR-dependent TRPM2 gating in human T lymphocytes. J Biol Chem. doi:10.1074/jbc.M111.324269
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411(6837):595–599. doi:10.1038/35079100
Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293(5533):1327–1330. doi:10.1126/science.1062473
Sumoza-Toledo A, Penner R (2011) TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589(Pt 7):1515–1525. doi:10.1113/jphysiol.2010.201855
Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE (2001) Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7(11):1209–1216. doi:10.1038/nm1101-1209
Partida-Sanchez S, Goodrich S, Kusser K, Oppenheimer N, Randall TD, Lund FE (2004) Regulation of dendritic cell trafficking by the ADP-ribosyl cyclase CD38: impact on the development of humoral immunity. Immun 20(3):279–291
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2 + influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14(7):738–747. doi:10.1038/nm1758
Johnson JD, Ford EL, Bernal-Mizrachi E, Kusser KL, Luciani DS, Han Z, Tran H, Randall TD, Lund FE, Polonsky KS (2006) Suppressed insulin signaling and increased apoptosis in CD38-null islets. Diabetes 55(10):2737–2746. doi:10.2337/db05-1455
Kato I, Yamamoto Y, Fujimura M, Noguchi N, Takasawa S, Okamoto H (1999) CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J Biol Chem 274(4):1869–1872
Higashida H, Yokoyama S, Munesue T, Kikuchi M, Minabe Y, Lopatina O (2011) CD38 gene knockout juvenile mice: a model of oxytocin signal defects in autism. Biol Pharm Bull 34(9):1369–1372
Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, Yamada N, Hayashi K, Noguchi N, Kato I, Okamoto H, Matsushima A, Salmina A, Munesue T, Shimizu N, Mochida S, Asano M, Higashida H (2007) CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446(7131):41–45. doi:10.1038/nature05526
Salmina AB, Lopatina O, Ekimova MV, Mikhutkina SV, Higashida H (2010) CD38/cyclic ADP-ribose system: a new player for oxytocin secretion and regulation of social behaviour. J Neuroendocrinol 22(5):380–392. doi:10.1111/j.1365-2826.2010.01970.x
Sun L, Iqbal J, Dolgilevich S, Yuen T, Wu XB, Moonga BS, Adebanjo OA, Bevis PJ, Lund F, Huang CL, Blair HC, Abe E, Zaidi M (2003) Disordered osteoclast formation and function in a CD38 (ADP-ribosyl cyclase)-deficient mouse establishes an essential role for CD38 in bone resorption. FASEB J: Off Publ Fed Am Soc Exp Biol 17(3):369–375. doi:10.1096/fj.02-0205com
Deshpande DA, White TA, Guedes AG, Milla C, Walseth TF, Lund FE, Kannan MS (2005) Altered airway responsiveness in CD38-deficient mice. Am J Respir Cell Mol Biol 32(2):149–156. doi:10.1165/rcmb.2004-0243OC
Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, Dianzani U, Stockinger H, Malavasi F (1998) Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol 160(1):395–402
Dianzani U, Funaro A, DiFranco D, Garbarino G, Bragardo M, Redoglia V, Buonfiglio D, De Monte LB, Pileri A, Malavasi F (1994) Interaction between endothelium and CD4 + CD45RA + lymphocytes. Role of the human CD38 molecule. J Immunol 153(3):952–959
Kumagai M, Coustan-Smith E, Murray DJ, Silvennoinen O, Murti KG, Evans WE, Malavasi F, Campana D (1995) Ligation of CD38 suppresses human B lymphopoiesis. J Exp Med 181(3):1101–1110
Zupo S, Rugari E, Dono M, Taborelli G, Malavasi F, Ferrarini M (1994) CD38 signaling by agonistic monoclonal antibody prevents apoptosis of human germinal center B cells. Eur J Immunol 24(5):1218–1222. doi:10.1002/eji.1830240532
Funaro A, Morra M, Calosso L, Zini MG, Ausiello CM, Malavasi F (1997) Role of the human CD38 molecule in B cell activation and proliferation. Tissue Antigens 49(1):7–15
Hemler ME, Elices MJ, Parker C, Takada Y (1990) Structure of the integrin VLA-4 and its cell-cell and cell-matrix adhesion functions. Immunol Rev 114:45–65
Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, Weissman IL, Hamann A, Butcher EC (1993) Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74(1):185–195
Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC (1995) Alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell 80(3):413–422
Rose DM, Han J, Ginsberg MH (2002) Alpha4 integrins and the immune response. Immunol Rev 186:118–124
Hartmann TN, Grabovsky V, Wang W, Desch P, Rubenzer G, Wollner S, Binsky I, Vallon-Eberhard A, Sapoznikov A, Burger M, Shachar I, Haran M, Honczarenko M, Greil R, Alon R (2009) Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res 69(7):3121–3130. doi:10.1158/0008-5472.CAN-08-4136
Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR (1990) VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 60(4):577–584
Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, Lobb R (1989) Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell 59(6):1203–1211
Guan JL, Hynes RO (1990) Lymphoid cells recognize an alternatively spliced segment of fibronectin via the integrin receptor alpha 4 beta 1. Cell 60(1):53–61
Rettig MP, Ansstas G, DiPersio JF (2012) Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leuk Off J Leuk Soc Am Leuk Res Fund UK 26(1):34–53. doi:10.1038/leu.2011.197
Hirsch E, Iglesias A, Potocnik AJ, Hartmann U, Fassler R (1996) Impaired migration but not differentiation of haematopoietic stem cells in the absence of beta1 integrins. Nature 380(6570):171–175. doi:10.1038/380171a0
Fassler R, Meyer M (1995) Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev 9(15):1896–1908
Bungartz G, Stiller S, Bauer M, Muller W, Schippers A, Wagner N, Fassler R, Brakebusch C (2006) Adult murine hematopoiesis can proceed without beta1 and beta7 integrins. Blood 108(6):1857–1864. doi:10.1182/blood-2005-10-007658
Arroyo AG, Yang JT, Rayburn H, Hynes RO (1996) Differential requirements for alpha4 integrins during fetal and adult hematopoiesis. Cell 85(7):997–1008
Arroyo AG, Yang JT, Rayburn H, Hynes RO (1999) Alpha4 integrins regulate the proliferation/differentiation balance of multilineage hematopoietic progenitors in vivo. Immunity 11(5):555–566
Scott LM, Priestley GV, Papayannopoulou T (2003) Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol 23(24):9349–9360
Priestley GV, Ulyanova T, Papayannopoulou T (2007) Sustained alterations in biodistribution of stem/progenitor cells in Tie2Cre + alpha4(f/f) mice are hematopoietic cell autonomous. Blood 109(1):109–111. doi:10.1182/blood-2006-06-026427
Priestley GV, Scott LM, Ulyanova T, Papayannopoulou T (2006) Lack of alpha4 integrin expression in stem cells restricts competitive function and self-renewal activity. Blood 107(7):2959–2967. doi:10.1182/blood-2005-07-2670
Lapidot T, Dar A, Kollet O (2005) How do stem cells find their way home? Blood 106(6):1901–1910. doi:10.1182/blood-2005-04-1417
Kollet O, Spiegel A, Peled A, Petit I, Byk T, Hershkoviz R, Guetta E, Barkai G, Nagler A, Lapidot T (2001) Rapid and efficient homing of human CD34(+)CD38(−/low)CXCR4(+) stem and progenitor cells to the bone marrow and spleen of NOD/SCID and NOD/SCID/B2m(null) mice. Blood 97(10):3283–3291
Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, Slav MM, Nagler A, Lider O, Alon R, Zipori D, Lapidot T (2000) The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95(11):3289–3296
Chigaev A, Sklar LA (2012) Aspects of VLA-4 and LFA-1 regulation that may contribute to rolling and firm adhesion. Front Immunol 3:242. doi:10.3389/fimmu.2012.00242
Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M, Kato J, Hirayama Y, Sakamaki S, Kohda K, Miyake K, Niitsu Y (2003) Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med 9(9):1158–1165. doi:10.1038/nm909
Oostendorp RA, Dormer P (1997) VLA-4-mediated interactions between normal human hematopoietic progenitors and stromal cells. Leuk Lymphoma 24(5–6):423–435
Hamblin TJ, Orchard JA, Ibbotson RE, Davis Z, Thomas PW, Stevenson FK, Oscier DG (2002) CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 99(3):1023–1029
Del Poeta G, Maurillo L, Venditti A, Buccisano F, Epiceno AM, Capelli G, Tamburini A, Suppo G, Battaglia A, Del Principe MI, Del Moro B, Masi M, Amadori S (2001) Clinical significance of CD38 expression in chronic lymphocytic leukemia. Blood 98(9):2633–2639
Durig J, Naschar M, Schmucker U, Renzing-Kohler K, Holter T, Huttmann A, Duhrsen U (2002) CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leuk Off J Leuk Soc Am Leuk Res Fund UK 16(1):30–35. doi:10.1038/sj.leu.2402339
Ibrahim S, Keating M, Do KA, O’Brien S, Huh YO, Jilani I, Lerner S, Kantarjian HM, Albitar M (2001) CD38 expression as an important prognostic factor in B-cell chronic lymphocytic leukemia. Blood 98(1):181–186
Krober A, Seiler T, Benner A, Bullinger L, Bruckle E, Lichter P, Dohner H, Stilgenbauer S (2002) V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 100(4):1410–1416
Thornton PD, Fernandez C, Giustolisi GM, Morilla R, Atkinson S, A’Hern RP, Matutes E, Catovsky D (2004) CD38 expression as a prognostic indicator in chronic lymphocytic leukaemia. Hematol J: Off J Eur Haematol Assoc / EHA 5(2):145–151. doi:10.1038/sj.thj.6200360
Vroblova V, Smolej L, Vrbacky F, Jankovicova K, Hrudkova M, Maly J, Krejsek J (2009) Biological prognostic markers in chronic lymphocytic leukemia. Acta Med (Hradec Kralove) 52(1):3–8
Montserrat E (2006) New prognostic markers in CLL. Hematology/the Education Program of the American Society of Hematology American Society of Hematology Education Program:279–284. doi:10.1182/asheducation-2006.1.279
Nuckel H, Switala M, Collins CH, Sellmann L, Grosse-Wilde H, Duhrsen U, Rebmann V (2009) High CD49d protein and mRNA expression predicts poor outcome in chronic lymphocytic leukemia. Clin Immunol 131(3):472–480. doi:10.1016/j.clim.2009.02.004
Majid A, Lin TT, Best G, Fishlock K, Hewamana S, Pratt G, Yallop D, Buggins AG, Wagner S, Kennedy BJ, Miall F, Hills R, Devereux S, Oscier DG, Dyer MJ, Fegan C, Pepper C (2011) CD49d is an independent prognostic marker that is associated with CXCR4 expression in CLL. Leuk Res 35(6):750–756. doi:10.1016/j.leukres.2010.10.022
Brachtl G, Sahakyan K, Denk U, Girbl T, Alinger B, Hofbauer SW, Neureiter D, Hofbauer JP, Egle A, Greil R, Hartmann TN (2011) Differential bone marrow homing capacity of VLA-4 and CD38 high expressing chronic lymphocytic leukemia cells. PloS One 6(8):e23758. doi:10.1371/journal.pone.0023758
Rossi D, Bodoni CL, Zucchetto A, Rasi S, De Paoli L, Fangazio M, Rossi FM, Ladetto M, Gattei V, Gaidano G (2010) Low CD49d expression and long telomere identify a chronic lymphocytic leukemia subset with highly favourable outcome. Am J Hematol 85(8):619–622. doi:10.1002/ajh.21756
Rossi D, Zucchetto A, Rossi FM, Capello D, Cerri M, Deambrogi C, Cresta S, Rasi S, De Paoli L, Bodoni CL, Bulian P, Del Poeta G, Ladetto M, Gattei V, Gaidano G (2008) CD49d expression is an independent risk factor of progressive disease in early stage chronic lymphocytic leukemia. Haematologica 93(10):1575–1579. doi:10.3324/haematol.13103
Eksioglu-Demiralp E, Alpdogan O, Aktan M, Firatli T, Ozturk A, Budak T, Bayik M, Akoglu T (1996) Variable expression of CD49d antigen in B cell chronic lymphocytic leukemia is related to disease stages. Leuk Off J Leuk Soc Am Leuk Res Fund UK 10(8):1331–1339
Zucchetto A, Bomben R, Dal Bo M, Bulian P, Benedetti D, Nanni P, Del Poeta G, Degan M, Gattei V (2006) CD49d in B-cell chronic lymphocytic leukemia: correlated expression with CD38 and prognostic relevance. Leuk Off J Leuk Soc Am Leuk Res Fund UK 20(3):523–525. doi:10.1038/sj.leu.2404087, author reply 528–529
Zucchetto A, Bomben R, Dal Bo M, Sonego P, Nanni P, Rupolo M, Bulian P, Dal Maso L, Del Poeta G, Del Principe MI, Degan M, Gattei V (2006) A scoring system based on the expression of six surface molecules allows the identification of three prognostic risk groups in B-cell chronic lymphocytic leukemia. J Cell Physiol 207(2):354–363. doi:10.1002/jcp.20570
Chen JR, Gu BJ, Dao LP, Bradley CJ, Mulligan SP, Wiley JS (1999) Transendothelial migration of lymphocytes in chronic lymphocytic leukaemia is impaired and involved down-regulation of both L-selectin and CD23. Br J Haematol 105(1):181–189
Bazerbashi MB, Reeve J, Chanarin I (1978) Studies in chronic lymphocytic leukaemia. The kinetics of 51Cr-labelled lymphocytes. Scand J Haematol 20(1):37–51
Gu B, Dao LP, Wiley J (2001) Impaired transendothelial migration of B-CLL lymphocytes: a defect linked to low L-selectin expression. Leuk Lymphoma 42(1–2):5–12. doi:10.3109/10428190109097671
Till KJ, Lin K, Zuzel M, Cawley JC (2002) The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood 99(8):2977–2984
Till KJ, Spiller DG, Harris RJ, Chen H, Zuzel M, Cawley JC (2005) CLL, but not normal, B cells are dependent on autocrine VEGF and alpha4beta1 integrin for chemokine-induced motility on and through endothelium. Blood 105(12):4813–4819. doi:10.1182/blood-2004-10-4054
Deaglio S, Vaisitti T, Aydin S, Bergui L, D’Arena G, Bonello L, Omede P, Scatolini M, Jaksic O, Chiorino G, Efremov D, Malavasi F (2007) CD38 and ZAP-70 are functionally linked and mark CLL cells with high migratory potential. Blood 110(12):4012–4021. doi:10.1182/blood-2007-06-094029
Vaisitti T, Aydin S, Rossi D, Cottino F, Bergui L, D’Arena G, Bonello L, Horenstein AL, Brennan P, Pepper C, Gaidano G, Malavasi F, Deaglio S (2010) CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leuk Off J Leuk Soc Am Leuk Res Fund UK 24(5):958–969. doi:10.1038/leu.2010.36
Pittner BT, Shanafelt TD, Kay NE, Jelinek DF (2005) CD38 expression levels in chronic lymphocytic leukemia B cells are associated with activation marker expression and differential responses to interferon stimulation. Leuk Off J Leuk Soc Am Leuk Res Fund UK 19(12):2264–2272. doi:10.1038/sj.leu.2403975
Aydin S, Grabellus F, Eisele L, Mollmann M, Hanoun M, Ebeling P, Moritz T, Carpinteiro A, Nuckel H, Sak A, Gothert JR, Duhrsen U, Durig J (2011) Investigating the role of CD38 and functionally related molecular risk factors in the CLL NOD/SCID xenograft model. Eur J Haematol 87(1):10–19. doi:10.1111/j.1600-0609.2011.01626.x
Deaglio S, Capobianco A, Bergui L, Durig J, Morabito F, Duhrsen U, Malavasi F (2003) CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood 102(6):2146–2155. doi:10.1182/blood-2003-03-0989
Deaglio S, Vaisitti T, Bergui L, Bonello L, Horenstein AL, Tamagnone L, Boumsell L, Malavasi F (2005) CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood 105(8):3042–3050. doi:10.1182/blood-2004-10-3873
Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P, Thomas NS, Yallop D, Devereux S, Fegan C, Buggins AG, Pepper C (2012) Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol 158(5):589–599. doi:10.1111/j.1365-2141.2012.09191.x
Tonino SH, Spijker R, Luijks DM, van Oers MH, Kater AP (2008) No convincing evidence for a role of CD31-CD38 interactions in the pathogenesis of chronic lymphocytic leukemia. Blood 112(3):840–843. doi:10.1182/blood-2008-03-144576
Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D’Arena G, Chiorino G, Malavasi F (2010) CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med 16(3–4):87–91. doi:10.2119/molmed.2009.00146
Lagneaux L, Delforge A, De Bruyn C, Bernier M, Bron D (1999) Adhesion to bone marrow stroma inhibits apoptosis of chronic lymphocytic leukemia cells. Leuk Lymphoma 35(5–6):445–453. doi:10.1080/10428199909169609
de la Fuente MT, Casanova B, Garcia-Gila M, Silva A, Garcia-Pardo A (1999) Fibronectin interaction with alpha4beta1 integrin prevents apoptosis in B cell chronic lymphocytic leukemia: correlation with Bcl-2 and Bax. Leuk Off J Leuk Soc Am Leuk Res Fund UK 13(2):266–274
de la Fuente MT, Casanova B, Moyano JV, Garcia-Gila M, Sanz L, Garcia-Marco J, Silva A, Garcia-Pardo A (2002) Engagement of alpha4beta1 integrin by fibronectin induces in vitro resistance of B chronic lymphocytic leukemia cells to fludarabine. J Leukoc Biol 71(3):495–502
Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, Kipps TJ, Choi YS, Bennett F, Reed JC (2002) Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood 100(5):1795–1801
Zucchetto A, Vaisitti T, Benedetti D, Tissino E, Bertagnolo V, Rossi D, Bomben R, Dal Bo M, Del Principe MI, Gorgone A, Pozzato G, Gaidano G, Del Poeta G, Malavasi F, Deaglio S, Gattei V (2012) The CD49d/CD29 complex is physically and functionally associated with CD38 in B-cell chronic lymphocytic leukemia cells. Leuk Off J Leuk Soc Am Leuk Res Fund UK 26(6):1301–1312. doi:10.1038/leu.2011.369
Coscia M, Pantaleoni F, Riganti C, Vitale C, Rigoni M, Peola S, Castella B, Foglietta M, Griggio V, Drandi D, Ladetto M, Bosia A, Boccadoro M, Massaia M (2011) IGHV unmutated CLL B cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL B cells. Leuk Off J Leuk Soc Am Leuk Res Fund UK 25(5):828–837. doi:10.1038/leu.2011.12
Binsky I, Lantner F, Grabovsky V, Harpaz N, Shvidel L, Berrebi A, Goldenberg DM, Leng L, Bucala R, Alon R, Haran M, Shachar I (2010) TAp63 regulates VLA-4 expression and chronic lymphocytic leukemia cell migration to the bone marrow in a CD74-dependent manner. J Immunol 184(9):4761–4769. doi:10.4049/jimmunol.0904149
Buggins AG, Pepper C, Patten PE, Hewamana S, Gohil S, Moorhead J, Folarin N, Yallop D, Thomas NS, Mufti GJ, Fegan C, Devereux S (2010) Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-kappaB activation and de novo gene transcription. Cancer Res 70(19):7523–7533. doi:10.1158/0008-5472.CAN-10-1634
Redondo-Munoz J, Escobar-Diaz E, Samaniego R, Terol MJ, Garcia-Marco JA, Garcia-Pardo A (2006) MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by alpha4beta1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood 108(9):3143–3151. doi:10.1182/blood-2006-03-007294
Redondo-Munoz J, Ugarte-Berzal E, Terol MJ, Van den Steen PE, Hernandez del Cerro M, Roderfeld M, Roeb E, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A (2010) Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell 17(2):160–172. doi:10.1016/j.ccr.2009.12.044
Calissano C, Damle RN, Hayes G, Murphy EJ, Hellerstein MK, Moreno C, Sison C, Kaufman MS, Kolitz JE, Allen SL, Rai KR, Chiorazzi N (2009) In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood 114(23):4832–4842. doi:10.1182/blood-2009-05-219634
Damle RN, Calissano C, Chiorazzi N (2010) Chronic lymphocytic leukaemia: a disease of activated monoclonal B cells. Best Pract Res Clin Haematol 23(1):33–45. doi:10.1016/j.beha.2010.02.001
Damle RN, Temburni S, Calissano C, Yancopoulos S, Banapour T, Sison C, Allen SL, Rai KR, Chiorazzi N (2007) CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood 110(9):3352–3359. doi:10.1182/blood-2007-04-083832
Lin TT, Hewamana S, Ward R, Taylor H, Payne T, Pratt G, Baird D, Fegan C, Pepper C (2008) Highly purified CD38 sub-populations show no evidence of preferential clonal evolution despite having increased proliferative activity when compared with CD38 sub-populations derived from the same chronic lymphocytic leukaemia patient. Br J Haematol 142(4):595–605. doi:10.1111/j.1365-2141.2008.07236.x
Bennett F, Rawstron A, Plummer M, de Tute R, Moreton P, Jack A, Hillmen P (2007) B-cell chronic lymphocytic leukaemia cells show specific changes in membrane protein expression during different stages of cell cycle. Br J Haematol 139(4):600–604. doi:10.1111/j.1365-2141.2007.06790.x
Asslaber D, Grossinger EM, Girbl T, Hofbauer SW, Egle A, Weiss L, Greil R, Hartmann TN (2013) Mimicking the microenvironment in chronic lymphocytic leukaemia—where does the journey go? Br J Haematol 160(5):711–714. doi:10.1111/bjh.12151
Willimott S, Baou M, Huf S, Deaglio S, Wagner SD (2007) Regulation of CD38 in proliferating chronic lymphocytic leukemia cells stimulated with CD154 and interleukin-4. Haematologica 92(10):1359–1366. doi:10.3324/haematol.11340
Buggins AG, Levi A, Gohil S, Fishlock K, Patten PE, Calle Y, Yallop D, Devereux S (2011) Evidence for a macromolecular complex in poor prognosis CLL that contains CD38, CD49d, CD44 and MMP-9. Br J Haematol 154(2):216–222. doi:10.1111/j.1365-2141.2011.08725.x
Redondo-Munoz J, Ugarte-Berzal E, Garcia-Marco JA, del Cerro MH, Van den Steen PE, Opdenakker G, Terol MJ, Garcia-Pardo A (2008) Alpha4beta1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 112(1):169–178. doi:10.1182/blood-2007-08-109249
Zucchetto A, Benedetti D, Tripodo C, Bomben R, Dal Bo M, Marconi D, Bossi F, Lorenzon D, Degan M, Rossi FM, Rossi D, Bulian P, Franco V, Del Poeta G, Deaglio S, Gaidano G, Tedesco F, Malavasi F, Gattei V (2009) CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res 69(9):4001–4009. doi:10.1158/0008-5472.CAN-08-4173
Pearce L, Morgan L, Lin TT, Hewamana S, Matthews RJ, Deaglio S, Rowntree C, Fegan C, Pepper C, Brennan P (2010) Genetic modification of primary chronic lymphocytic leukemia cells with a lentivirus expressing CD38. Haematologica 95(3):514–517. doi:10.3324/haematol.2009.014381
Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, Ansell SM, Novak AJ, Kay NE, Witzig TE, Nowakowski GS (2011) Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin alpha-4-beta-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol 155(1):53–64. doi:10.1111/j.1365-2141.2011.08794.x
Kiziltepe T, Ashley JD, Stefanick JF, Qi YM, Alves NJ, Handlogten MW, Suckow MA, Navari RM, Bilgicer B (2012) Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood Cancer J 2(4):e64. doi:10.1038/bcj.2012.10
de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M (2012) The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119(11):2590–2594. doi:10.1182/blood-2011-11-390989
Brown JR (2013) Ibrutinib (PCI-32765), the first BTK (Bruton’s tyrosine kinase) inhibitor in clinical trials. Curr Hematol Malignancy Rep 8(1):1–6. doi:10.1007/s11899-012-0147-9
Castillo JJ, Furman M, Winer ES (2012) CAL-101: a phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Exp Opin Investig Drugs 21(1):15–22. doi:10.1517/13543784.2012.640318
Acknowledgments
Work of the authors is supported by the Austrian Science Fund FWF (project W1213 and SFB program P021 to R.G., project P25015-B13 to T.N.H.), the Austrian National Bank (projects 13420 to T.N.H., 14311 to R.G., the Paracelsus Medical University Salzburg (projects E-10/11/058-HAR and E-12/15/074-HAH to T.N.H), the “Klinische Malignom und Zytokinforschung Salzburg-Innsbruck GmbH”, and the province of Salzburg.
Authors
G.B., J.P.H., R.G., and T.N.H wrote the paper and approve of the submitted and final version.
Conflict of interest
The authors declare no competing financial conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Brachtl, G., Piñón Hofbauer, J., Greil, R. et al. The pathogenic relevance of the prognostic markers CD38 and CD49d in chronic lymphocytic leukemia. Ann Hematol 93, 361–374 (2014). https://doi.org/10.1007/s00277-013-1967-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-013-1967-y