
Abstract
We present a new approach for the analysis of water in nominally anhydrous minerals using transmission Raman spectroscopy. Using this approach, the laser was shone through thin, nearly transparent samples of clinopyroxene, garnet and synthetic rhyolite glass. To remove mineral-induced background and to improve the quality of the OH spectral region, specifically for clinopyroxene, a reference spectrum of a dehydrated crystal was measured and subtracted. Water contents of all clinopyroxene samples were previously determined by Fourier transformed infrared spectroscopy (FTIR). The application of transmission Raman spectroscopy and a reference spectrum of a dry sample revealed a noticeable improvement in the quality of spectra and thus the detection limit, compared to the standard backscattering configurations. We show that the quality of transmission spectra and the detection limit depend on the sample thickness, and that the thickness has to be taken into account when measuring and comparing OH-integrated intensity and water content if the results are used for OH quantification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During growth from hydrous magma or metamorphic fluids, nominally anhydrous minerals (NAMs) such as clinopyroxene, olivine and garnet include trace amounts of hydrogen in structural defects such as cation vacancies (2H+ vs. Mg2+) or charge deficiencies (Al3+ + H+ vs. Si4+), where it is bonded to oxygen and can be regarded as an oxide component. From this viewpoint, the presence of this hydrogen can be expressed as a water concentration. The amount of water in NAMs was shown to serve as a proxy to estimate magmatic water contents (e.g., Weis et al. 2015). Similarly the hydrous component in NAMs provides information about the deep water cycle and the storage of water in the mantle of planetary bodies (e.g., Bolfan-Casanova 2005; Peslier 2010). Common methods for analysis of water in NAMs are Fourier transformed infrared spectroscopy (FTIR) and secondary ion mass spectrometry (SIMS) which, however, may require orientation of the crystals, suffer from a limited spatial resolution or result in the loss of sample material. An alternative method may be provided by Raman spectroscopy (e.g., Thomas et al. 2008a, b; Bolfan-Casanova et al. 2014). One of the most prominent advantages of this method is the very small analysis spot size (typically on the order of a micrometer) and its non-destructive nature. Yet backscattering Raman spectroscopy may be affected by strong fluorescence background, or heating of dark crystals which both might interfere with the bands of weakly Raman-active molecules such as water. In addition, the obtained Raman spectrum is only representative for a single spot within the sample instead of, similar to transmission FTIR, the whole thickness of the sample. An alternative approach is the application of transmission Raman spectroscopy. Up to date, most Raman measurements in geoscience have been performed in the backscattering configuration whereas transmission Raman spectroscopy found most of its application in the field of biology, medicine and pharmacy (e.g., Matousek and Parker 2007; Matousek et al. 2011; Griffen et al. 2015). Specifically for the analysis of pharmaceutical tablets and capsules, transmission Raman spectroscopy has shown to be better than the conventional backscattering configuration. The main advantage of transmission over the conventional backscattered Raman spectroscopy is that it provides a more ‘in depth’ analysis of the bulk sample instead of a single sample point. As a result, the signal to noise ratio can be improved significantly as more molecules, structural groups or bonds of interest are activated and background such as fluorescence arising from, for example, surficial contaminants becomes less significant in the spectrum. Another problem arising during Raman measurements is the effect of sample heating if dark-colored samples are analyzed, specifically if the laser beam is tightly focused onto the sample as in the case for backscattered configuration or when high laser powers are applied to compensate for low concentrations of the chemical bonds in question. To remedy this sort of problem, the preparation of thin, almost transparent samples may reduce the heat emission background. In this study, we tested the analysis of water and silica bands in orientated, very thin and nearly transparent crystals of clinopyroxene and garnet, as well as a synthetic rhyolite glass standard by the technique of transmission Raman spectroscopy. Prior to Raman analysis, the water contents of clinopyroxene had been measured by FTIR spectroscopy. By measuring in a forward scattering configuration, i.e. transmission, the quality of the Raman spectra was improved. Moreover, Raman signals originating from the OH-species, which are more IR- and less Raman-active, were increased and spectra became more representative of the whole sample, rather than a single point within the crystal or glass. The improved spectra, particularly for clinopyroxene, can be used for a mineral-specific internal calibration using water and silica bands, in other words, for development of a method for quantification of water in NAMs using Raman spectroscopy.
Samples
The majority of clinopyroxene crystals in this study come from the Canary Islands. The included rock samples are El Hierro island ankaramite lava bombs from Tanganasoga volcano (Carracedo et al. 2001; Longpré et al. 2009; Weis et al. 2016). To get a comparison for different crystal chemistries, a clinopyroxene crystal from a gabbro from the well-known Rum igneous intrusion was added (e.g., Emeleus and Troll 2014). All studied clinopyroxene crystals are diopsides (cf. Weis et al. 2016). One garnet crystal, a grossular from Ala valley in Piedmont in Italy, was included in the study (Arredondo and Rossman 2002). Furthermore, a synthetic rhyolite glass with known water content (3.25 wt%) was added.
Methods
Rock samples (n = 6) were crushed to obtain loose clinopyroxene crystals of a size suitable for the analysis (≥ 300 µm). These were hand-picked under a binocular microscope and individual clinopyroxene crystals were then mounted in a thermoplastic resin for further processing. With the help of crystal morphology and optical microscopy (extinction angles), the selected crystals (n = 22) were oriented along their crystallographic c-axis and their (100) and (010) crystal faces, on which the directions of the main refractive indices (α, β and γ) occur. A detailed procedure of the crystal alignment is described in Stalder and Ludwig (2007). Various particle size-grades of Al2O3-grinding paper were used to thin and polish the oriented crystals to a thickness of a few hundred micrometers for FTIR analysis. After the FTIR analysis, crystals were polished to near complete transparency at thicknesses between 15 and 50 microns on selected crystal faces for the transmission Raman analysis. Two reference crystals were dehydrated by heating them in a glass-tube furnace at 700 °C for 32 h. One selected clinopyroxene crystal was split into three pieces and each piece was polished to a different thickness to test the effect of varying sample thickness on the analysis. Similarly, four pieces of different thickness (36–957 µm) were prepared for the rhyolite glass. To investigate the influence of varying sample composition on transmission Raman spectroscopy, a thin, nearly transparent clinopyroxene crystal was partly fixed on top of parts of the thicker (~ 100 µm) Ala garnet crystal. All crystals or glass pieces were then mounted on 1 mm apertures.
FTIR spectroscopy
Polarized FTIR spectra in the range 2000–5000 cm−1 were measured on the oriented clinopyroxene crystals along the directions of the main refractive indices (α, β and γ) to obtain the total absorbance: Atotal = A α + A β + A γ for the OH-bands. Measurements for A α and A γ were done on the (010) crystal face and A β on (100). The polished crystals were analyzed with a Bruker Vertex 70 spectrometer equipped with a NIR source (halogen lamp), a CaF2 beamsplitter, a wiregrid polarizer (KRS-5) and an InSb detector. The crystal thicknesses varied between 150 and 500 µm, with most crystals being between 200 and 400 µm thick for both the (100) and (010) orientations. Small apertures (100 to 400 µm) were applied for masking during analysis to avoid cracks and inclusions in the crystals. On occasion, impurities were present in the beam path (e.g. opaque phases), but these appeared not to have imposed any significant effect on the OH region of the spectra. For each individual spectrum, 128 scans were taken and averaged. Subsequently, the obtained spectra were baseline corrected by a polynomial function and the OH-bands fitted with the software PeakFit. The corresponding water contents were then calculated using both the wavenumber-dependent calibration function established by Libowitzky and Rossman (1997) and the mineral-specific (augite) calibration of Bell et al. (1995).
Raman spectroscopy
Experiments were carried out using a Raman spectroscopic system designed and built at the Department of Earth Sciences, Uppsala University. The key system components include a high-throughput, single-stage imaging spectrometer (HoloSpec, Kaiser Optical Systems, Inc.) equipped with a holographic transmission grating and thermoelectrically cooled CCD detector (Newton, Andor Technology) with a spectral pixel resolution of about 4 cm−1, two Argon ion lasers (487.99 nm, 40 mW, linearly polarized and 514.53 nm, 10 mW, linearly polarized), a DPSS laser (532.42 nm, 150 mW, linearly polarized) and an optical imaging system (magnification 20×, spatial resolution ∼ 1 µm). The system was set up in both a 0° forward scattering (transmission) as well as a 180° backscatter configuration, by introducing a movable mirror. Two holographic notch filters (Ondax, Inc.) blocked the Rayleigh line. To enhance the weak Raman signal of the OH region, the confocal pinhole aperture was removed. The spectrometer was calibrated by fluorescence lines of a neon lamp. The focal spot size of the laser beam on the sample was ca. 5–10 µm. No analyzer was used. The majority of the measurements were performed applying a laser excitation of 514 nm. Spectra for clinopyroxene were taken at room temperature in both geometries for 30 min using an output laser power of 5 mW (corresponding to 3.2 mW on the sample). Measurements for rhyolite glass were done in forward scattering configuration with the 488 nm excitation to remove the strong fluorescence bands interfering with the OH region. To avoid saturation of the detector, measurements for the different pieces of water-rich rhyolite glass lasted 30 s each, applying 35 mW of input laser power, corresponding to 28.6 mW on the sample. To test the effect of varying sample composition on transmission Raman spectroscopy, spectra for clinopyroxene and garnet were taken with a laser excitation of 532 nm at 22 mW (corresponding to 20 mW on the sample) for 15 min in forward scattering configuration. Subsequently, one measurement was taken on the combined sample with clinopyroxene attached on garnet. To keep the consistency, Raman signal intensity of the strongest silica band was optimized by fine tuning the laser focus. As in the case of the FTIR analysis, the obtained spectra were baseline corrected with the software PeakFit. To improve this process, especially for clinopyroxene which showed a more complicated background, the spectrum collected under the same experimental conditions from a dehydrated reference crystal was subtracted from the obtained clinopyroxene spectra to remove any background (e.g., fluorescence) arising from the crystals. In the course of the fitting process, the area (integrated intensity) under the OH-bands was determined for each sample. The used Raman system is normally operated with excitation lasers lasing at 514 and 532 nm. Application of the non-standard laser (488 nm) and the static grating in this study resulted in a high-wavenumber range from 3419 to 5562 cm−1 and the low-wavenumber range from 940 to 3398 cm−1 which had an effect on the spectra measured for rhyolite glass.
Due to the cut-off at 3419 cm−1 from the static grating when using the non-standard laser (488 nm), we measured a reference spectrum for the rhyolite glass with a commercial system to investigate potential peak cut-off or loss. The measurement was done in normal backscattering configuration using a Horiba Jobin Yvon LabRAM-HR Evolution confocal micro-Raman spectrometer, with 473 nm diode laser excitation at the Department of Mineralogy and Crystallography at Vienna University. The system is equipped with a Peltier-cooled CCD detector, z-focus, and a software-controlled x–y sample stage. For the 473 nm excitation, the exposure time was four minutes, and the on-sample laser power was 8 mW using a 1800 grooves/mm grating.
Results
FTIR spectroscopy
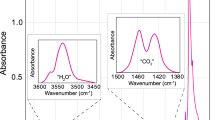
All clinopyroxene crystals showed absorption maxima at 3630, 3530, and 3460 cm−1 in the IR spectra (Fig. 1), which corresponds to the vibrational bands expected for OH in diopside (e.g., Skogby 2006). The OH-band at around 3630 cm−1 is prominent for Eǀǀα and Eǀǀβ, while the two bands around 3530 and 3460 cm−1 are dominant for Eǀǀγ. This infrared-pleochroic behavior is typical for OH-bands in clinopyroxene (e.g., Beran 1976) and thus the influence of possible OH-bearing impurities (e.g. hydrous minerals, melt/fluid inclusions) on the spectra can be excluded. Overall water contents of the analyzed crystals ranged between 149 and 820 ppm H2O for the Libowitzky and Rossman (1997) calibration (Table 1). Water contents corresponding to the spectra differed for the calibrations of Bell et al. (1995) and Libowitzky and Rossman (1997). With the mineral-specific calibration by Bell et al. (1995), values were about 15% higher, but similar to previous studies we prefer to use water contents derived through the wavenumber-dependent calibration (e.g., Stalder 2004; Stalder and Ludwig 2007; Sundvall and Stalder 2011; Mosenfelder and Rossman 2013; Weis et al. 2015, 2016). No OH-bands were observed in the two annealed reference crystals implying water contents below the detection limit or their complete dehydration.

a Representative IR-spectra of clinopyroxene crystal T2R1 analyzed in this study. Polarized measurements with Eǀǀα and Eǀǀγ were done on the (010) crystal face while Eǀǀβ was measured on (100). Absorbances have been normalized to 1 mm thickness. The spectra show the three main vibrational bands of water at 3630, 3530, 3460 cm−1, which are expected for diopside (Skogby 2006) and relate to different OH-dipole orientations (see text for details). b Representative Raman spectra for measurements done in forward scattering configuration on clinopyroxene crystals in different crystal orientations. For Raman measurements, it can be seen that the OH-bands were strongest for Eǀǀα and weakest for Eǀǀγ. On occasion plasma lines from the laser were seen on the spectrum. These were, however, accounted for or removed during further data processing. Similar as to FTIR analysis, the typical OH-bands at 3630 and 3530 cm−1 could be observed. Missing from the Raman spectra, however, was a clear band at 3460 cm−1
Potential uncertainties for calculated water contents can arise from baseline correction and measurements of the crystal thickness. However, due to the quality of the spectra and the relatively large thickness of the crystals a maximum error of ± 10% is assumed for the precision of the calculated clinopyroxene water contents. In addition, the uncertainty regarding the accuracy of the values due to the calibration for absorption coefficients is another ± 10% (cf. Bell et al. 1995; Libowitzky and Rossman 1997) resulting in an overall uncertainty of ± 20% for the calculated clinopyroxene water contents.
Transmission Raman spectroscopy
The obtained spectra showed peaks similar in shape and position to the FTIR analysis (Fig. 1). For measurements in the forward scattering configuration plasma lines from the laser could occasionally be observed on the spectra. These were, however, removed during the later data processing. Further, the quality of the clinopyroxene spectra was improved after the subtraction of the reference spectrum which visualized the extent of sample-induced background (Fig. 2). After baseline correction, the curves for clinopyroxene could be fitted with bands between 3500 and 3630 cm−1, similar to the bands obtained with the FTIR spectroscopy. The band at 3630 cm−1 was visible for all three orientations but strongest for Eǀǀα and weakest for Eǀǀγ (Fig. 1). A weak band at 3530 cm−1 was also visible for all orientations. The band at 3460 cm−1 as observed for FTIR spectroscopy was not clearly observed on the Raman spectra for clinopyroxene. No difference in the peak positions was seen between the crystals from Tanganasoga and the crystal from Rum. The peak intensity and the area under the OH-bands increased with increasing water content in the crystals. Low-wavenumber spectra measured with the 514 and 532 nm laser excitations corresponded to Raman spectra previously observed for diopside and grossular (e.g., Kolesov and Geiger 1998; Huang et al. 2000; Arredondo and Rossman 2002; Prencipe et al. 2012). The two dominant features for diopside are the bands at 667 and 1013 cm− 1 associated with a Si–O–Si bending mode and a Si-O stretching mode, respectively. The intensity of the Si-region of the spectrum varied quite strongly with orientation with Eǀǀα having the strongest and Eǀǀγ the lowest Raman activity (Fig. 3). No clear correlation for the integrated intensity of the silicate bands and the water content of the crystals as seen in prior studies for olivine (Bolfan-Casanova et al. 2014) was observed. Glass spectra showed a typical asymmetric OH-band at 3550 cm− 1, which reflects stretching vibrational contributions from molecular water as well as hydroxyl groups (e.g., Chabiron et al. 2004) (Fig. 4). For separately tested crystal and glass pieces, the integrated intensity of the OH- and silicate bands increased with increasing thickness. Due to the good quality of the spectra, for clinopyroxene resulting also from the background subtraction using a reference spectrum, as well as from repeated fitting of the same spectra showing similar results, a maximum uncertainty of 20% is assumed for the integrated intensities. Measurements on the part where clinopyroxene and garnet were attached on top of each other revealed Raman bands from both minerals on the spectrum (Fig. 4).

Representative Raman spectrum of clinopyroxene crystal TR7 analyzed in forward scattering configuration. Also shown is a reference spectrum taken of a dehydrated crystal. The reference spectrum was then subtracted from each obtained Raman spectrum to remove any background arising from clinopyroxene. The result was a clearer spectrum with the OH-peaks clearly visible which then could be further processed with the software PeakFit

Low-wavenumber spectra taken in forward scattering configuration and different crystal orientations for clinopyroxene. Crystal orientation has a big effect on the peak intensity of the Si-bands. The Raman scattering effect was again strongest for Eǀǀα and weakest for Eǀǀβ

a Raman spectrum of the OH region of the synthetic rhyolite glass taken in forward scattering geometry with a 488 nm excitation energy. The spectrum is cut below 3419 cm−1 as the applied Raman system was initially designed for a 514 nm laser. b Transmission Raman spectra of the low-wavenumber region of clinopyroxene and garnet individually and the two crystals overlapping taken in forward scattering configuration
Backscattered Raman spectroscopy
The Raman spectra taken for the clinopyroxene crystals with the 514 nm wavelength in the backscatter configuration revealed the same peaks as observed for the forward scattering configuration, however, the bands were of lower intensity, and for example the band at 3530 cm−1 not as clearly visible as for the transmission configuration (Fig. 5). Other than sample-induced background, clearly visible through the reference spectrum of the dehydrated crystal, again no OH-bands were observed at 3460 cm− 1 (Fig. 5). The observed and integrated intensity of the Raman bands was lower than for the forward scattering configuration.

Difference between Raman spectra of clinopyroxene taken in forward (black) and backscatter (blue) configuration with a 514 nm excitation. For both configurations, the same OH-bands could be observed; however, the intensity was higher for spectra taken in transmission configuration. In addition, the band at 3530 cm−1 was better visible for the transmission configuration than the backscatter. This also made a clear identification of all the OH-bands easier
The reference spectrum taken for the rhyolite glass with the 473-nm excitation wavelength revealed that parts of the Raman peak lie below 3419 cm−1 extending down to ~ 3000 cm−1 resulting in a partial loss of the OH-band for measurements in the forward scattering configuration. After assigning bands to the spectrum taken in backscatter mode and integrating the total area, we estimate that with the bands missing for the transmission configuration, about 25% of the total integrated area underneath the OH region are missing with the cut-off at 3419 cm−1. Yet, since most of the OH-band in the rhyolite glass is visible, specifically the main band at ~ 3550 cm−1, the effect of the cut-off is believed to be small when testing the effect of varying thickness on transmission Raman spectroscopy with the different glass pieces, as the cut-off and the subsequent peak fitting process are the same for each spectrum.
Discussion
Because for clinopyroxene the water band was strongest for Eǀǀα, only spectra taken in this configuration are considered from now onwards in the study. Previous studies testing the use of Raman spectroscopy to determine the water content in nominally anhydrous minerals concentrated on garnet and enstatite (Thomas et al. 2008b, 2015; Bolfan-Casanova et al. 2014), whereas this study will focus mainly on the results obtained for clinopyroxene, more precisely diopside. Two methods have been presented for the purpose of OH quantification in NAMs using Raman spectroscopy. The first one, referred to as the “comparator technique”, uses an external calibration standard, such as volcanic or synthetic glass, where the water content, refractive index and density are known (e.g., Thomas et al. 2008a, b). The second method employs an internal calibration using the normalization of integrated OH-intensities with the area of one or several peaks, potentially from the silica region of a mineral’s spectrum (e.g., Arredondo and Rossman 2002; Bolfan-Casanova et al. 2014). A disadvantage of the comparator technique is that the exact density and the refractive index of the reference glass or the studied samples may be unknown which affects the correction of integrated Raman intensities. Further, the method does not fully account for other potential matrix effects such as reflection/refraction or self-absorption (Bolfan-Casanova et al. 2014). Thus, a comparison of the comparator standard to a pre-established mineral-specific calibration may be needed (see Bolfan-Casanova et al. 2014). A mineral-specific calibration for clinopyroxene has until now, however, not been established. Our data show a linear correlation of OH-integrated Raman intensity and water contents (Fig. 6). Yet an internal calibration should be done to account for random fluctuation in intensity by normalizing the integrated OH-intensities with, for example, the integrated intensity of the low-wavenumber Si-range (Fig. 2) as done in previous studies (e.g. Arredondo and Rossman 2002; Bolfan-Casanova et al. 2014). The number of peaks for normalization can differ (e.g., Thomas 2000; Thomas et al. 2008a; Arredondo and Rossman 2002; Bolfan-Casanova et al. 2014) but should be kept constant for application of the calibration curve. One critical issue with such a normalization can be the effect of crystal orientation. The intensity of the OH and the Si-O stretching modes are strongly affected by crystal orientation which, in turn, will affect the ratio between the two peaks for similar water contents analyzed with a slight difference in crystal orientation. An internal normalization of the integrated OH-intensities of the diopside crystals by their entire Si-region crystals results in a linear trend between water content and integrated peak intensity (Fig. 6b). Some outliers occur which probably represent the negative effect of a slight misorientation of the crystal during analysis as the intensity of Si-bands seems easily affected by this (Fig. 3). Previously, calibration curves were established for olivine and enstatite on the basis of synthetic crystals (Bolfan-Casanova et al. 2014), and a new data and sample set for a calibration specifically for common volcanic and plutonic diopside are presented here.

Relation between a the integrated intensity of OH-bands and b Si-band normalized integrated OH Raman intensity vs. the water contents of the clinopyroxene crystals determined by FTIR and wavenumber-dependent absorption coefficients by Libowitzky and Rossman (1997). In both cases a positive linear trend can be observed. For the normalization some outliers may result from effects of crystal orientation
Spectral quality and detection limit
A concise detection limit for water quantification in NAMs using Raman spectroscopy has not been established and is probably very dependent on factors such as laser wavelength and power, general system alignment and also the sample properties such as color and contaminants which are the cause for strong background noise (e.g. Arredondo and Rossman 2002). In previous works, the detection limits were occasionally rather high (~ 1000 ppm), however, recent studies showed that detection of water contents down to ~ 3 ppm is possible (Arredondo and Rossman 2002; Thomas et al. 2008b, 2009, 2015; Bolfan-Casanova et al. 2014). The detection of very low water contents in garnets (3 to 50 ppm H2O), however, required a high laser power (1 W, 140 mW on the sample; see Thomas et al. 2008b). Water contents as low as 75 ppm have been measured in olivines with an output laser power of only 70 mW (Bolfan-Casanova et al. 2014). In this study, the lowest detected water content was 150 ppm H2O (see Table 1) and was achieved with an on-sample laser power of only 3.2 mW. Considering the analytical uncertainty and the quality of the OH-bands obtained for the crystal with 150 ppm H2O, the detection limit may even be lower (~ 100 ppm) and remains to be tested properly. Of course, applying a higher laser power as in previous studies would decrease the detection limit even further. The application of forward scattering configuration over the backscatter configuration led to an improvement of spectral quality as it enhanced certain peaks on the spectrum. For the spectra taken in the transmission mode, the band at 3530 cm−1 was enhanced and clearly visible while this was not the case on the spectra taken in backscatter configuration (Fig. 5). Overall for measurements taken with the same laser power and time interval, the OH-bands stood out more clearly on the spectrum for the forward scattering geometry. One major aspect that further helped in achieving high-quality spectra for low water contents may also be the analysis of a dehydrated reference sample and the subsequent subtraction of the reference spectrum from all other measurements (Fig. 2). This reduced sample induced background significantly and made especially the identification of weak water bands in crystals with low H2O contents easier. Yet, despite the improved spectral quality from background reduction and increased peak intensities no band at 3460 cm−1 was observed. Either the band was yet too weak to be seen or it simply is not Raman-active. Another important factor improving the detection limit may come from an improved signal to noise ratio due to the application of transmission rather than backscattered Raman spectroscopy, thus sampling more volume, and the fact that samples were almost transparent thus reducing the effect of heat emission. Similarly, the improvement of spectral quality and the low detection limit may come from measurements on orientated crystals to obtain the strongest Raman scattering activity of the sample (e.g. Eǀǀα). The factors influencing the detection limit are of course many and may be difficult to compare for different Raman spectrometers. Yet, we provide some aspects that can and should be taken into account to improve the detection limit when analyzing low OH-concentrations by Raman spectroscopy.
Effect of thickness on transmission Raman spectroscopy
The overall observed trend between increasing water content and increasing integrated OH-intensities shows several outliers, for which a larger difference in sample thickness to the other crystals is distinctive (Fig. 6a). The test done on both clinopyroxene and glass samples on how crystal thickness affects Raman intensity of OH-bands during forward scattering shows a positive correlation that can be fitted to a logarithmic trend (Fig. 7a, b). This shows that for a certain range of thicknesses there is a rapid increase in peak intensity and for progressively thicker specimens (> 200 µm) the magnitude of the integrated intensity levels off (Fig. 7a). This is expected since with increasing thickness, at some point, the maximally possible Raman-probed volume will be reached. Beyond this point, a portion of the sample will start to lie outside the depth of focus and the focal cylinder of the laser beam (about 200–250 µm) and thus the scattering activity will be much less. Yet this imposes a problem when analyzing crystals of different water content and thickness as it can lead to over- or underestimation of H2O contents during quantification. The effect of thickness may be taken away by an internal normalization with the integrated intensities of the Si-range as shown earlier. However, the internal normalization may be difficult if quantification methods such as the comparator technique are applied, since such a normalization can be more difficult for glass samples. In this case, the samples need a normalization to account for the thickness. Since the thickness has a logarithmic effect on the intensity of OH-bands, the integrated areas can be normalized by the natural logarithm of the crystals’ thickness. Testing this on our sample set reveals an overall positive linear trend between water content and integrated intensities similar to the internal normalization applying Si-peaks (Figs. 6b, 8a). Further, testing the normalization, for example, on the three crystal sections of different thickness brings their normalized integrated intensities very close to the same value (Table 1). The same is observed for the different-thick rhyolite glass pieces, with the thinnest sample being an outlier. The reason for this is not fully known yet most likely arises from an error during the measurement (e.g., imperfect focus).

Relation between integrated Raman intensities and thickness. a, b For differently thick rhyolite glass and clinopyroxene sections, a logarithmic trend between increasing thickness and integrated intensity of OH-bands can be seen. c Enlargement of the 0 to 50 µm part of (b). It can be seen that the logarithmic trend becomes near-linear for very short intervals of thickness (~ 15 µm) such as for the clinopyroxene crystals used in this study
Another aspect which is revealed by the thickness–intensity relation is that for small intervals of thickness (e.g. ~ 15 µm) there is a near-linear progression (Fig. 7c). Thus for small thickness-intervals a simple normalization by thickness can be done. When the integrated OH-intensities of our clinopyroxene crystals are normalized by their thickness, the data plot along two distinct linear correlations for two distinct intervals of crystal thickness (Fig. 8b). As soon as the sample thickness becomes too large, a normalization by thickness may lead to an underestimation of the integrated peak intensity for the same water content, which is again indicated by the three analyzed diopside crystal segments. Given the observed correlations between sample thickness and intensity, ideally, all analyzed samples should have either the same thickness or be within a small thickness interval when applying the forward scattering configuration.

Relation between clinopyroxene water content and thickness-normalized integrated OH-intensities. a Normalizing the integrated OH-intensities by the natural logarithm of the crystal thickness results in a positive linear trend similar to the internal normalization done with the Si-bands. b For small differences in crystal thickness integrated OH-intensities can be normalized simply by thickness. This results in linear-trends for small intervals of crystal thickness (e.g., 30–50 µm). Crystal thicknesses should thus be kept very close or simply uniform when analyzing in the forward scattering geometry to obtain best results
It is important to note that the thickness also has a great effect on the detection limit since with increasing thickness the chances are increased to detect low water contents as peak intensities increase. This is further supported by our spectra as those taken in forward scattering geometry show certain OH-bands better than spectra taken in backscatter configuration. In addition, the transmission configuration can affect the measurement time, as less time is needed to obtain spectra of better quality. One critical aspect though is the change in transparency of the crystal accompanied with crystal thickness which, in turn, can cause sample heating and thus increase background from the thermal emission, or risk of sample damage and dehydration.
Effect of sample composition on transmission Raman spectroscopy
Since with the transmission configuration basically a spectrum over the whole sample thickness is taken, influences of heterogeneous sample composition or inclusions can be expected if they are in the beam path. This was revealed when measuring in forward scattering configuration on the overlapping clinopyroxene and garnet crystals where peaks of both minerals appeared on the spectrum (Fig. 4b). Due to the smaller thickness of the clinopyroxene (30 µm vs. 95 µm) bands of the garnet crystal are stronger, yet clinopyroxene bands are visible and interference of bands from both minerals occurs (Fig. 4b). This shows that transmission Raman spectroscopy is an effective way, similar to FTIR, to obtain and measure the composition of a sample as a whole rather than selected spots. By such it may be possible to detect changes in crystal composition or to analyze inclusions together with their host crystal. In this way, the forward scattering configuration can be applied for two-dimensional mapping including variations within the sample along depth. However, the more “bulk-type” sample analysis also has to be taken into account when evaluating the spectra as peaks of two different scattering mediums in the sample may overlap.
Summary and conclusions
In this study, we have tested transmission Raman spectroscopy as an alternative method to the more conventional backscattered Raman measurements and other analytical methods measuring water content in NAMs. It was shown that Raman spectroscopy can be used for qualitative determination of OH in minerals with transmission Raman spectroscopy providing certain advantages over backscatter Raman spectroscopy. This provides further opportunities for quantification of OH in NAMs using Raman spectroscopy. One major conclusion of this work is that for transmission Raman spectroscopy, besides sample orientation, the sample thickness is very important for a low detection limit but also for a potential quantification of water contents especially when applying the comparator technique. Thicknesses should ideally be kept very close or the same for all analyzed mediums and integrated intensities require a normalization. Altogether, the combination of nearly transparent samples, laser transmission and background reduction by applying a reference spectrum of a dehydrated crystal increased the quality of obtained Raman spectra and made the detection of even low water contents (~ 140 ppm) with low laser power (5 mW) possible. One limitation with transmission Raman spectroscopy is that it may be difficult to perform on most commercial Raman spectrometers as these systems may not be easily tuned to a 0° scattering configuration or a specific instrument for the transmission configuration is required (e.g., Cobalt Light Systems TRS100). Due to the big variation in water contents, yet relatively homogenous crystal chemistry, the set of clinopyroxene crystals presented here has great potential to serve as a standard material for an internal, mineral-specific calibration for OH quantification using Raman spectroscopy in general.
References
Arredondo EH, Rossman GR (2002) Feasibility of determining the quantitative OH content of garnets with Raman spectroscopy. Am Mineral 87:307–311
Bell DR, Ihinger PD, Rossman GR (1995) Quantitative analysis of trace OH in garnet and pyroxenes. Am Mineral 80:465–474
Beran A (1976) Messung des Ultrarot-Pleochroismus von Mineralen XIV Der Pleochroismus der OH-Streckfrequenz in Diopsid. Tscherm Mineral Petrog Mitt 23:79–85
Bolfan-Casanova N (2005) Water in the Earth’s mantle. Mineral Mag 69:229–257
Bolfan-Casanova N, Montagnac G, Reynard B (2014) Measurement of water contents in olivine using Raman spectroscopy. Am Mineral 99:149–156
Carracedo JC, Badiola ER, Guillou H, de La Nuez J, Pérez Torrado FJ (2001) Geology and volcanology of La Palma and El Hierro. Western Canaries Estud Geol 57:175–273
Chabiron A, Pironon J, Massare D (2004) Characterization of water in synthetic rhyolitic glasses and natural melt inclusions by Raman spectroscopy. Contrib Mineral Petrol 146:485–492
Emeleus CH, Troll VR (2014) The Rum Igneous Centre, Scotland. Mineral Mag 78:805–839
Griffen JA, Owen AW, Matousek P (2015) Development of Transmission Raman Spectroscopy towards the in line, high throughput and non-destructive quantitative analysis of pharmaceutical solid oral dose. Analyst 140:107–112
Huang E, Chen CH, Huang T, Lin EH, Xu J (2000) Raman spectroscopic characteristics of Mg–Fe–Ca pyroxenes. Am Mineral 85:473–479
Kolesov BA, Geiger CA (1998) Raman spectra of silicate garnets. Phys Chem Miner 25:142–151
Libowitzky E, Rossman GR (1997) An IR absorption calibration for water in minerals. Am Mineral 82:1111–1115
Longpré MA, Troll VR, Hansteen TH, Anderson E (2009) Ankaramitic lavas and clinopyroxene megacrysts from the Tanganasoga Volcano, El Hierro Island (Canary Archipelago) Eos Transactions AGU, Fall Meet Suppl, Abstract V51A-1662, vol 90, no 52
Matousek P, Parker AW (2007) Non-invasive probing of pharmaceutical capsules using transmission Raman spectroscopy. J Raman Spectrosc 38:563–567
Matousek P, Everall N, Littlejohn D, Nordon A, Bloomfield M (2011) Dependence of signal on depth Transmission Raman spectroscopy. Appl Spectrosc 65:724–733
Mosenfelder J, Rossman GR (2013) Analysis of hydrogen and fluorine in pyroxenes: II Clinopyroxene. Am Mineral 98:1042–1054
Peslier AH (2010) A review of water contents of nominally anhydrous natural minerals in the mantles of Earth, Mars and the Moon. J Volcanol Geotherm Res 197:239–258
Prencipe M, Mantovani L, Tribaudino M, Bersani D, Lottici PP (2012) The Raman spectrum of diopside: a comparison between ab initio calculated and experimentally measured frequencies. Eur J Mineral 24:457–464
Skogby H (2006) Water in natural mantle minerals I: pyroxenes. Rev Mineral Geochem 62:155–168
Stalder R (2004) Influence of Fe, Cr and Al on hydrogen incorporation in orthopyroxene. Eur J Mineral 16:703–711
Stalder R, Ludwig T (2007) OH incorporation in synthetic diopside. Eur J Mineral 19:373–380
Sundvall R, Stalder R (2011) Water in upper mantle pyroxene megacrysts and xenocrysts: a survey study. Am Mineral 96:1215–1227
Thomas R (2000) Determination of water contents of granite melt inclusions by confocal laser Raman microprobe spectroscopy. Am Mineral 85:868–872. https://doi.org/10.2138/am-2000-5-631
Thomas R, Metrich N, Scaillet B, Kamenetsky VS, Davidson P (2008a) Determination of water in Fe-rich basalt glasses with confocal micro-Raman spectroscopy. Z geol Wiss 36:31–37
Thomas S-M, Thomas R, Davidson P, Reichart P, Koch-Müller M, Dollinger G (2008b) Application of Raman spectroscopy to quantify trace water concentrations in glasses and garnets. Am Mineral 93:1550–1557
Thomas S-M, Koch-Müller M, Reichart P, Rhede D, Thomas R, Wirth R, Matsyuk S (2009) IR calibrations for water determination in olivine, r-GeO2, and SiO2 polymorphs. Phys Chem Minerals 36:489–509
Thomas SM, Wilson K, Koch-Müller M et al (2015) Quantification of water in majoritic garnet. Am Mineral 100:1084–1092. https://doi.org/10.2138/am-2015-5136
Weis FA, Skogby H, Troll VR, Deegan FM, Darehn B (2015) Magmatic water contents determined through clinopyroxene: Examples from the Western Canary Islands, Spain. Geochem Geophys 16:2127–2146
Weis FA, Stalder R, Skogby H (2016) Experimental hydration of natural volcanic clinopyroxene phenocrysts under hydrothermal pressures (05-3 kbar). Am Mineral 101:2233–2247
Acknowledgements
We thank Valentin Troll, Frances Deegan, Kirsten Zaczek, Juan Carlos Carracedo and Tobias Mattsson for providing the samples from the Canary Islands and the Rum igneous intrusion. Lisa Samrock, Soophie Omidian and Per-Olof Persson are thanked for their help with sample preparation. We thank Ulf Hålenius for the help determining the crystal thicknesses of thin samples. We thank Prof. Lutz Nasdala and Dr. Chutimun Chanmuang at the Institute of Mineralogy and Crystallography at Vienna University for giving us the opportunity to use their Raman spectrometer. Nathalie Bolfan-Casanova and Federica Schiavi are being thanked for providing the rhyolite glass sample. Financial support for this project was provided by the Swedish Research Council. The data for this paper are available in the text, tables, and references therein and from the corresponding author on request. The two unknown reviewers are thanked for their comments that helped to improve this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Weis, F.A., Lazor, P. & Skogby, H. Hydrogen analysis in nominally anhydrous minerals by transmission Raman spectroscopy. Phys Chem Minerals 45, 597–607 (2018). https://doi.org/10.1007/s00269-018-0945-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-018-0945-2