
Abstract
Na–H3O jarosite was synthesized hydrothermally at 413 K for 8 days and investigated using single-crystal X-ray diffraction (XRD) and electron microprobe analysis (EMPA). The chemical composition of the studied crystal is [Na0.57(3) (H3O)0.36 (H2O)0.07]A Fe2.93(3) (SO4)2 (OH)5.70 (H2O)0.30, and Fe deficiency was confirmed by both EMPA and XRD analysis. The single-crystal XRD data were collected at 298 and 102 K, and crystal structures were refined in space group \( R\overline{3}m\). The room-temperature data match structural trends of the jarosite group, which vary linearly with the c axis. The low-temperature structure at 102 K shows an anisotropic decrease in the unit cell parameters, with c and a decreasing by 0.45 and 0.03 %, respectively. Structural changes are mainly confined to the A site environment. Only minor changes occur in FeO6 and SO4 polyhedra. The structure responds upon cooling by increasing bond length distortion and by decreasing quadratic elongation of the large AO12 polyhedra. The structural parameters at low temperature follow very similar patterns to structural changes that correspond to compositional variation in the jarosite group, which is characterised by the flexibility of AO12 polyhedra and rigidity of Fe(OH)4O2–SO4 layers. The most flexible areas in the jarosite structure are localized at AO12 edges that are not shared with neighbouring FeO6 octahedra. Importantly, for the application of XRD in planetary settings, the temperature-related changes in jarosite can mimic compositional change.
Similar content being viewed by others

Introduction
Jarosite group minerals are members of the alunite supergroup, which has the general formula AB 3(TO4)2(OH)6. A range of cations can be incorporated into jarosite group minerals such as A = Na, K, Ag, Tl, H3O, NH4, Pb, B = Fe3+ and T = S (e.g. Stoffregen et al. 2000; Bayliss et al. 2010). Jarosite senso stricto [K jarosite, KFe3(SO4)2(OH)6] and Na jarosite [NaFe3(SO4)2(OH)6] are the most common jarosite species observed in nature (e.g. Desborough et al. 2010). K-deficiency is required for the formation of Na jarosite and alkali deficiency for the formation of H3O jarosite [(H3O) Fe3(SO4)2 (OH)6].
Jarosite group minerals are mainly a product of chemical weathering. Low-temperature interaction of fluids with sulphide minerals in oxidizing and acidic environments are favourable formation conditions. On Earth, jarosite can be found in oxidation zones of sulphide-bearing ore deposits, in sediments and rocks associated with pyrite weathering, and in hydrothermally altered rocks (Dutrizac and Jambor 2000; Stoffregen et al. 2000). Jarosite group minerals are known to precipitate in acid mine drainage environment (e.g. Hudson-Edwards 2003; Jamieson et al. 2005).
Signatures of jarosite group minerals have been identified on Mars using in situ Mössbauer spectrometry by the Opportunity rover (Klingelhöfer et al. 2004). Compositions at Endurance Crater in Meridiani Planum were assigned to K–Na–H3O jarosite solid solutions with possible substitution of Al3+ for Fe3+ (Morris et al. 2006). Reflectance VIS–NIR spectra from the Mars Reconnaissance Orbiter of deposits south of Ius/Melas Chasma were assigned to non-stoichiometric H3O-bearing, Fe-deficient jarosite (Milliken et al. 2013).
Surface temperatures on Mars can range between 120 and 298 K, and crystal structure data at low temperatures can be relevant for in situ mineral identification. Only a limited number of single-crystal studies have investigated the jarosite group at low temperatures (H3O jarosite—Spratt et al. 2014; K jarosite—Mills et al. 2013; NH4 jarosite—Basciano and Peterson 2007). Mills et al. (2013) is the only study covering a wide low-temperature range and demonstrated that K jarosite undergoes a strongly anisotropic thermal expansion, with the c axis changing much stronger than the a axis. The anisotropic change observed for K jarosite is mainly related to a strong distortion of the AO12 polyhedron. Similar low-temperature effects could be inferred for H3O jarosite by comparing studies undertaken at room temperature (e.g. Majzlan et al. 2004; Plášil et al. 2014) and 173 K (Spratt et al. 2014). However, results from low-temperature data are still too limited to allow an extrapolation of structural mechanisms to other compositions within the jarosite group. For instance, the incorporation of smaller cations such as Na+ or Ag+ at the large A site could induce even larger structural distortions and related unit cell anisotropies. Our study focusses on Na–H3O jarosite, the crystal structure of which was investigated at room temperature and 102 K. Our observations are used to improve our knowledge of structural trends in the jarosite group and to further specify compositional and temperature induced changes of the jarosite topology.
Materials and methods
Synthesis of Na–H3O jarosite
Na–H3O jarosite was synthesised using a similar method as described by Basciano and Peterson (2008). A 75-ml Parr pressure vessel was used as the reaction chamber. Starting materials were 18 g of ferric sulphate hydrate, 0.3825 g of sodium sulphate and 45 ml of deionised water. The sealed reaction vessel was placed inside a furnace and heated at 413 K for 8 days. The synthesis time was four times longer than described in Basciano and Peterson (2008) to promote the growth of large crystals.
Electron microprobe analysis (EMPA)
A jarosite crystal was embedded in epoxy resin and polished for EMPA. The polished cross section of the crystal was 50 × 30 µm. The composition of the crystal was determined using a Cameca SX-100 electron microprobe. Column conditions used were 15 keV and 10 nA with an electron beam spot size of 10 µm. Counting times were set to 10 s for Na, 20 s for S, and 30 s for Fe. Jadeite (Na), barite (S), and hematite (Fe) were used as standards. The PAP program was used for the matrix correction.
The chemical composition was calculated on the basis of two sulphur per formula unit (p.f.u.) and the general formula for Na–H3O jarosite was [Na x (H3O)1−x−y (H2O)y]A Fe3−y (SO4)2 (OH)6−4y (H2O)4y . The calculation of the jarosite formula followed the approach of an NMR study of Nielsen et al. (2008) suggesting that potential non-stoichiometry at the B site is linked to hydronium deprotonation at the A site and protonation of four hydroxyl groups per vacant Fe site.
Single-crystal X-ray diffraction
A suitable subhedral crystal with dimensions of 100 × 80 × 50 μm was selected for single-crystal diffraction. The single-crystal measurements were carried out using an Agilent Xcalibur single-crystal diffractometer equipped with an EOS CCD area detector. Graphite-monochromated Mo Kα radiation was used and tube operation conditions were 50 kV and 40 mA. The crystal-to-detector distance was 70 mm. Data collection was performed at 298 and 102 K. A Cryojet system from Oxford instruments delivered a nitrogen stream in the cooling experiment at 102 K.
For both experiments (298 and 102 K), the frame width of the ω scans was 1º and counting time per frame was 21 s. A sphere of intensity data were collected to 60º 2θ with 100 % completeness. The intensity data were corrected for Lorentz polarization and an empirical absorption correction was applied using CrysalisRED software (Agilent Technologies). The crystal structure was solved and refined using the SHELX program (Sheldrick 2008) within the WinGX environment (Farrugia 1999). The details of data collection are given in Table 1.
Results and discussion
Chemical composition
EMPA data of seven analyses from the single crystal are listed in Table 2. The synthesised Na–H3O jarosite has an average composition of [Na0.57(3) (H3O)0.36 (H2O)0.07]A Fe2.93(3) (SO4)2 (OH)5.70 (H2O)0.30 if deprotonation of H3O at the A site and protonation of all four coordinating OH groups around a Fe vacancy are included in the formula calculation (Nielsen et al. 2008). Compositional variations between the single analyses are 5 % for Na2O, 1 % for Fe2O3 and 1 % for SO3. The totals including calculated water contents of the single analysis are 100.4–101.3 wt% (Table 2). It indicates that the employed EMPA method using low voltage and current, a large beam spot size, and short counting times could successfully minimize potential beam damage due to Na and H2O contents of the crystal.
Crystal structure refinement
Details of data collection and structure refinement are summarized in Table 1. The room-temperature structure was solved in space group \(R\overline {3}m \) using direct methods. The space group choice \(R\overline {3}m \) (R int = 0.034) is in agreement with previous single-crystal studies of Na jarosite and H3O jarosite (Grohol et al. 2003; Nestola et al. 2013; Majzlan et al. 2004; Plášil et al. 2014; Spratt et al. 2014).
All non-hydrogen atoms could be located and their displacement factors were refined anisotropically. The hydrogen of the hydroxyl group was identified from the difference Fourier map and introduced in a subsequent refinement step. Hydrogen from H3O and H2O at the A site could not be identified; a result that was expected because motion and disorder makes it difficult to locate hydrogen within the cavity surrounding the A site (e.g. Spratt et al. 2014; Nielsen et al. 2008, 2011). The estimated residual electron density maxima for hydrogen should be very low (<0.25 e/Å3) considering a H3O/H2O content of <50 % at the A site in this study and a fractional occupancy of 0.5 at the proposed m site for hydronium hydrogen (Wyckoff site 18 h, full site occupancy is 0.5; e.g. Plášil et al. 2014).
The occupancies of the A and B site were studied in two different ways. Firstly, EMPA data were used to set the occupancies for the A site (Na = 0.57 and O4 = 0.43) and for the B site (Fe = 0.98). The refinement results using these fixed site occupancies are shown in Tables 3 and 4. Secondly, occupancies were refined for the A site (Na + O4 = 1) and B site (Fe + vacancies = 1), and yielded occupancies of Na = 0.53(3) and Fe = 0.97(1) with a marginal change of the refinement statistics (R 1 = 0.0179, wR 2 = 0.0370, Goof = 1.109).
An identical refinement strategy was carried out with the XRD data from the low-temperature experiment at 102 K. No phase transition was observed, and the structure was refined in \(R\overline {3}m \). The final refinement resulted in R 1 = 0.0184, wR 2 = 0.0367 and GooF = 1.124 and data are shown in Tables 3 and 4. An additional refinement of the low-temperature analysis using A and B site occupancies as free variables converged to Na = 0.51(3) Fe = 0.97(1) with a rather similar R 1 = 0.0184, wR 2 = 0.0361 and GooF = 1.121. The Fe occupancies refined by the room- and low-temperature XRD data conform closely with the EMPA data. The refined Na occupancies by XRD are slightly lower than the EMPA data. It is assumed that non-refinable small contributions from the hydronium/water hydrogen could affect the refinement of the A site occupancy.
The hydronium oxygen atom O4 was located at the \(\overline {3}m \) site (3a Wyckoff site, coordinates 0, 0, 0) which is in agreement with H3O jarosite studies (Plášil et al. 2014; Majzlan et al. 2004). However, a potential displacement of O4 along the c axis was discussed in previously published work (Wills and Harrison 1996; Majzlan et al. 2004; Spratt et al. 2014) because the hydronium ion has usually 3m symmetry, which is inconsistent with \(\overline {3}m \) symmetry of hydronium in H3O jarosite. An attempt was made to refine O4 at a 3m site (coordinates 0, 0, z). The refinement converged to (0, 0, 0) which confirmed that the most probable position of the hydronium oxygen O4 is the \(\overline {3}m \) site. Furthermore, anisotropic displacement factors with elongation along the a–b plane do not support a displacement of the hydronium oxygen along the c axis direction (Table 3, U 11 = U 22 > U 33).
Crystal structure at 298 K: comparison with H3O jarosite
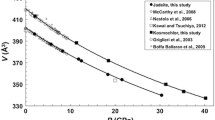
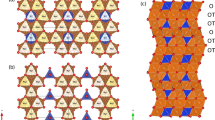
The major units of the jarosite structure are Fe(OH)4O2–SO4 layers stacked in c direction and AO12 polyhedra intercalated between successive Fe(OH)4O2–SO4 layers (Figs. 1, 2). In our study, the A site is occupied with Na+ and oxygen [from H3O+ + H2O] in a 57:43 ratio. The ionic radius of Na+ is smaller than H3O+ ([12]Na+ = 1.39 Å, Shannon 1976; [12]H3O+ = 1.52 Å, Okada et al. 1987). Compared to H3O jarosite (e.g. Majzlan et al. 2004), incorporation of the smaller Na+ cation causes a decrease in the unit cell parameters, in which c decreases to a greater extent (−1.7 %) than a (−0.4 %), confirming the well-known observation that substitution of univalent cations at the A site in jarosite results in strong variation of the c parameter while the a parameter experiences only minor changes (e.g. Menchetti and Sabelli 1976). Our unit cell data fall close to the linear a–c trend refined from powder data of Na–H3O jarosite solid solutions (Fig. 3 in Basciano and Peterson 2008).

Crystal structure of the jarosite group. (a) View along a direction. (b) View along c direction, A sites and one neighbouring Fe(OH)4O2–SO4 layer is shown
The decrease in unit cell parameters is controlled by changes of the A site environment (Fig. 2). The A site is coordinated to 12 oxygens (O2 and O3) forming an icosahedron. O2 corners (×6) are shared by FeO6 and SO4 polyhedra, whereas O3 corners (×6) are shared by two FeO6 polyhedra and hydrogen. All A–O bond lengths decrease compared to H3O jarosite (ΔA–O3 = −0.062 Å, ΔA–O2 = −0.036 Å). In contrast, SO4 and FeO6 polyhedra show only minor changes and their bond lengths remain constant (this study: <S–O> = 1.476 Å, <Fe–O> = 2.008 Å; H3O jarosite: <S–O> = 1.476 Å, <Fe–O> = 2.009 Å, Majzlan et al. 2004). As a result, the AO12 polyhedron for Na–H3O jarosite becomes more regular and its edge lengths become more similar (Fig. 3a, b). Here, the longer polyhedron edges of AO12, that are not shared with FeO6 octahedra, change most with Na incorporation.

Topology of the A site environment. Neighbouring FeO6 octahedra share a face with the AO12 icosahedron. SO4 tetrahedra share corners with FeO6 and AO12. The largest flexibility of the structure lies along the non-face sharing AO12 edges

AO12 polyhedron of Na–H3O jarosite. View along c direction. fs indicates faces that are shared with a FeO6 octahedron and therefore less flexible. The regularity of the AO12 icosahedron increases with Na incorporation and lower temperature. Shortening is more pronounced for edges that are not shared with FeO6 octahedra (a) H3O jarosite at 298 K (Majzlan et al. 2004). (b) Na–H3O jarosite at 298 K. (c) Na–H3O jarosite at 102 K
Crystal structure at 298 K: comparison with general trends in the jarosite group
It is useful to plot structural parameters versus unit cell parameter c. Figures 4, 5, and 6 show plots of bond lengths, bond length distortion and quadratic elongation versus c for our study and recent crystal structure studies of the jarosite group. Arrows in the figures indicate changes with temperature. Bond length distortion was calculated after Baur (1974). Quadratic elongation was used as a measure of polyhedral distortion (Robinson et al. 1971). Here, calculations of the ideal coordination polyhedron were based on icosahedra for AO12, octahedra for FeO6 and tetrahedra for SO4.

Change of bond lengths versus unit cell parameter c for jarosite group minerals. Room-temperature data are shown except assigned otherwise. Variation in composition and temperature show similar effects on bond lengths. Literature data: Ag jarosite Groat et al. (2003); Na jarosite Nestola et al. (2013); H3O jarosite Spratt et al. (2014) (173 K), Plášil et al. (2014), Majzlan et al. (2004); K jarosite Mills et al. (2013) 133–297 K, Xu et al. (2010a) 298–575 K; NH4 jarosite (Basciano and Peterson 2007)

Bond length distortion of jarosite group minerals versus unit cell parameter c. Room-temperature data are shown except assigned otherwise. Similar distortion effects can be observed with decreasing size of the A cations and with lowering of the temperature. Literature data: Ag jarosite Groat et al. (2003); Na jarosite Nestola et al. (2013); H3O jarosite Spratt et al. (2014) (173 K), Plášil et al. (2014), Majzlan et al. (2004); K jarosite Mills et al. (2013) 133–297 K, Xu et al. (2010a) 298–575 K; NH4 jarosite (Basciano and Peterson 2007)

Quadratic elongation versus c axis for jarosite group minerals. Room-temperature data are shown except assigned otherwise. Variation in composition and temperature affects mainly distortion of the AO12 polyhedron. Literature data: Ag jarosite Groat et al. (2003); Na jarosite Nestola et al. (2013); H3O jarosite Spratt et al. (2014) (173 K), Plášil et al. (2014), Majzlan et al. (2004); K jarosite Mills et al. (2013) 133–297 K, Xu et al. (2010a) 298–575 K; NH4 jarosite (Basciano and Peterson 2007)
Figures 4, 5, and 6 reveal that most trends are a linear function of the c parameter reflecting the simple response of the jarosite structure for A site substitutions. The A–O bond lengths decrease with smaller cation size, whereas Fe–O and S–O bonds remain relatively unchanged (Fig. 4). Our data match the linear trend very well. The linear change for A–O bond lengths is anisotropic, with A–O3 decreasing more strongly than A–O2 with smaller cation size, resulting in increasing bond length distortion around the A site (Fig. 5). On the other hand, polyhedron distortion (quadratic elongation) of the AO12 icosahedron decreases with smaller cation size (Fig. 6). The AO12 polyhedron becomes more regular with decreasing cation size as length differences between polyhedron edges get smaller. As shown above for H3O jarosite and Na–H3O jarosite (Fig. 3a, b), increased regularity is mainly a result of shortening of the less constrained polyhedron edges that are not shared with FeO6 octahedra. In contrast, the bond length distortion and polyhedral distortion of FeO6 and SO4 polyhedra remain unchanged throughout the jarosite group and are independent of substitution effects on the A site (Figs. 5, 6). A possibility to modify the relative rigidity of the Fe(OH)4O2–SO4 network would be ordering of vacancies at the B site which can be observed in monoclinic jarosite. Although not naturally occurring, monoclinic Na–H3O and K–H3O jarosite with C2/m symmetry could be synthesised (Scarlett et al. 2010; Grey et al. 2011, 2013). Vacancy ordering occurs at the Fe1 site, which is one of the two non-equivalent Fe sites. A major effect of the ordered disruption of the structure is an increase of the average bond length around the vacancy-bearing site <Fe1–O>, whereas the bond length of <Fe2–O> remains close to rhombohedral values of <Fe–O> (e.g. <Fe1–O> = 2.04 Å, <Fe2–O> = 2.01 Å, sample Najar-D1, Grey et al. 2011; <Fe–O> = 2.008 Å, this study).
A large thermal displacement U eq of 0.052 A2 was observed in this study for the A site (Na/O4). Similar high values were reported by Nestola et al. (2013) for Na jarosite (\(U_{eq}^{Na}\) = 0.053 Å2). Excepting Ag jarosite (\(U_{eq}^{Ag}\) = 0.031 Å2 Groat et al. 2003), there is a general trend of increasing U eq for smaller A cations (Fig. 7), reflecting greater thermal motion of smaller cations within the large A site. Anisotropic displacement factors for the A site are more pronounced in the a–b plane (Table 3, U 11 = U 22 > U 33), which can also be observed in Ag, H3O and K jarosites (Groat et al. 2003; Plášil et al. 2014; Mills et al. 2013). Smaller A cations induce a major shortening in the c direction via the A–O3 bond and a minor shortening along the a–b directions (via A–O2), possibly indicating that insufficient shortening of the A–O2 bond triggers increasingly stronger thermal motion in the a–b plane for smaller cations.

Crystal structure at 102 K
The low-temperature structure of Na–H3O jarosite at 102 K showed a volume decrease of 0.5 % (ΔV = 3.9 Å3) compared to the structure at room temperature. The volume reduction is mainly controlled by a change of the unit cell along the c axis which decreases by 0.45 %, whereas the a axis decreases only by 0.03 %. The strong axial anisotropy upon cooling mainly correlates with bond length changes in the AO12 polyhedron (Table 4). Upon cooling, the average bond length <A–O> decreases −0.010 Å whereas <Fe–O> and <S–O> show only minor changes (<0.001 and 0.003 Å, respectively). The main shortening occurs for A–O3 bonds which are major connectors of the A site in c direction. To compensate for lower temperatures, bond length distortion in the AO12 polyhedron increases slightly from 0.038 at 298 K to 0.039 at 102 K (Fig. 5). Polyhedral distortion of the AO12 icosahedron is decreasing from 1.0085 to 1.0080 (Fig. 6). It indicates that the AO12 polyhedron becomes more regular at lower temperature, in similar fashion as with incorporation of smaller A cations. By comparing Fig. 3b, c it becomes clear that the cell edges of the icosahedron are less different upon cooling. In contrast, FeO6 octahedra and SO4 tetrahedra do not change significantly at lower temperatures.
Comparable temperature effects were observed for K jarosite (297–133 K, Mills et al. 2013; 298–575 K, Xu et al. 2010a) and can also be inferred for H3O jarosite by comparing studies undertaken at room temperature (Majzlan et al. 2004; Plášil et al. 2014) and 173 K (Spratt et al. 2014). Arrows in Figs. 4, 5, and 6 confirm the presence of comparable trends for K, H3O and Na–H3O jarosites, e.g. with lower temperatures: (1) bond length shortening within the AO12 polyhedron, (2) increasing bond length distortion of A–O bonds, (3) decreasing quadratic elongation (polyhedron distortion) for AO12 towards a more regular icosahedron, and (4) negligible changes in FeO6 and SO4 polyhedra. The controlling factor of the A site deformation on the jarosite structure can also be observed with increasing pressure. A high-pressure study of Na jarosite (up to 8.8 GPa, Nestola et al. 2013) showed that the main deformation mechanism is governed by shortening of Na–O bonds with only minor or no shortening of Fe–O and S–O bonds. The large contraction of the NaO12 polyhedron with pressure is mainly responsible for strong shortening along the c axis and minor shortening along the a axis. A large axial cell anisotropy with increasing pressure was also reported for K jarosite (up to 8.1 GPa, Xu et al. 2010b).
Implications for identification of jarosite minerals
The jarosite crystal structure is clearly affected by non-ambient conditions. Structural data are required for in situ XRD identification of jarosite in potentially low-temperature environment, such as on the Mars surface. Understanding how the jarosite structure varies upon cooling is relevant because XRD patterns of jarosite group minerals at low temperature could mimic room-temperature structures with smaller cations on the A site; e.g. an initially assigned Na jarosite could actually be a Na–H3O jarosite analysed at low temperature. Splitting of jarosite peaks in XRD patterns (e.g. indices 033 and 027, Scarlett et al. 2010) could indicate formation of monoclinic jarosite.
The structure data for room temperatures and low temperature are still limited given the wide range of solid solutions in the jarosite group. However, the available data for K, H3O and Na–H3O jarosite comprise important compositions commonly observed in nature. Structural changes within this compositional range are predictable given the linearity of the structural trends (Figs. 4, 5, 6). Therefore, interpolation of structural models for K–H3O–Na jarosite solid solutions at room temperature and low temperature should be a reasonable approach if there is lack of data for specific compositions.
Conclusion
It can be concluded that temperature- and compositional-dependent changes of bond lengths and distortion parameters follow similar patterns for all minerals in the jarosite group (Figs. 4, 5, 6). Structural changes upon cooling resemble effects caused by the incorporation of smaller cations at the A site. Co-linear trends with composition and temperature suggest that the jarosite lattice activates a simple mechanism which is controlled by the flexibility of the large AO12 polyhedra and the rigidity of Fe(OH)4O2–SO4 layers. Similar structural effects can also be observed with increasing pressure (Nestola et al. 2013). The major flexibility of the AO12 polyhedron can be found around edges that are not shared with FeO6 octahedra. The environment around these non-sharing edges of the AO12 polyhedron is mainly responsible for structural changes with composition and temperature.
References
Basciano LC, Peterson RC (2007) The crystal structure of ammoniojarosite, (NH4) Fe3 (SO4)2 (OH)6 and the crystal chemistry of the ammoniojarosite—hydronium jarosite solid solution series. Mineral Mag 71:427–441
Basciano LC, Peterson RC (2008) Crystal chemistry of the natrojarosite–jarosite and natrojarosite–hydronium jarosite solid-solution series: a synthetic study with full Fe site occupancy. Am Mineral 93:853–862
Baur WH (1974) The geometry of polyhedral distortions. Predictive relationships for the phosphate group. Acta Crystallogr A B30:1195–1215
Bayliss P, Kolitsch U, Nickel EH, Pring A (2010) Alunite supergroup: recommended nomenclature. Mineral Mag 74:919–927
Desborough GA, Smith KS, Lowers HA, Swayze GA, Hammarstrom JM, Diehl SF, Leinz RW, Driscoll RL (2010) Mineralogical and chemical characteristics of some natural jarosites. Geochim Cosmochim Acta 74:1041–1056
Dutrizac JE, Jambor, JL (2000) Jarosites and their application in hydrometallurgy. In: Alpers CN, Jambor JL, Nordstrom DK (eds) Sulfate minerals—crystallography, geochemistry, and environmental significance 40: 405–452. Reviews in Mineralogy and Geochemistry, Mineralogical Society of America, Chantilly, Virginia
Farrugia LJ (1999) WinGX: an integrated system of Windows Programs for the solution, refinement and analysis of single-crystal X-ray diffraction data. J Appl Crystallogr 32:837–838
Grey IE, Scarlett NVY, Bordet P, Brand HEA (2011) Jarosite–butlerite intergrowths in non-stoichiometric jarosites: crystal chemistry of monoclinic natrojarosite-hydronium jarosite phases. Mineral Mag 75:2775–2791
Grey IE, Scarlett NVY, Bordet P, Brand HEA (2013) Crystal chemistry and formation mechanism of non-stoichiometric monoclinic K-jarosites. Mineral Mag 77:249–268
Groat LA, Jambor JL, Pemberton BC (2003) The crystal structure of argentojarosite, AgFe3(SO4)2(OH)6. Can Mineral 41:921–928
Grohol D, Nocera DG, Papoutsakis D (2003) Magnetism of pure Fe jarosites. Phys Rev B Condens Matter Mater Phys 67: 064401/1–064401/13
Hudson-Edwards KA (2003) Sources, mineralogy, chemistry and fate of heavy metal-bearing particles in mining-affected river systems. Mineral Mag 67(2):204–217
Jamieson HE, Robinson C, Alpers CN, Nordstrom DK, Poustovetov A, Lowers HA (2005) The composition of coexisting jarosite-group minerals and water from the Richmond Mine, California. Can Mineral 43:1225–1242
Klingelhöfer G, Morris RV, Bernhardt B, Schroder C, Rodionov D, de Souza PAJ, Yen AS, Gellert R, Evlanov EN, Zubkov B, Foh J, Bonnes U, Kankeleit E, Gutlich P, Ming DW, Renz F, Wdowiak TJ, Squyres SW, Arvidson RE (2004) Jarosite and hematite at Meridiani Planum from Opportunity’s Mössbauer spectrometer. Science 306:1740–1745
Majzlan J, Stevens R, Boerio-Goates J, Woodfield BF, Navrotsky A, Burns PC, Crawford MK, Amos TG (2004) Thermodynamic properties, low-temperature heat-capacity anomalies, and single-crystal X-ray refinement of hydronium jarosite, (H3O)Fe3(SO4)2(OH)6. Phys Chem Miner 31:518–531
Menchetti S, Sabelli C (1976) Crystal chemistry of the alunite series; crystal structure refinement of alunite and synthetic jarosite. Neues Jahrbuch fuer Mineralogie Monatshefte 9:406–417
Milliken RE, Swayze GA, Arvidson RE, Bishop JL, Clark RN, Ehlmann BL, Green RO, Grotzinger JP, Morris RV, Murchie SL, Mustard JF, Weitz C (2013) Opaline silica in young deposits on Mars. Geology 36:847–850
Mills SJ, Nestola F, Kahlenberg V, Christy AG, Hejny C, Redhammer G (2013) Looking for jarosite on Mars: the low temperature crystal structure of jarosite. Am Mineral 98:1966–1971
Morris RV, Klingelhöfer G, Schröder C, Rodionov DS, Yen A, Ming DW, de Souza PA Jr., Wdowiak T, Fleischer I, Gellert R et al (2006) Mössbauer mineralogy of rock, soil, and dust at Meridiani Planum, Mars: opportunity’s journey across sulfate-rich outcrop, basaltic sand and dust, and hematite lag deposits. J Geophys Res 111: E12S15. DOI: 10.1029/2006JE002791
Nestola F, Mills SJ, Periotto B, Scandalo L (2013) The alunite supergroup under high pressure: the case of natrojarosite, NaFe3(SO4)2(OH)6. Mineral Mag 77:3007–3017
Nielsen UG, Majzlan J, Grey CP (2008) Determination and quantification of the local environments in stoichiometric and defect jarosite by solid-state 2H NMR spectroscopy. Chem Mater 20:2234–2241
Nielsen UG, Heinmaa I, Samoson A, Majzlan J, Grey CP (2011) Insight into the local magnetic environments and deuteron mobility in jarosite (AFe3(SO4)2(OD, OD2)6, A = K, Na, D3O) and hydronium alunite ((D3O)Al3(SO4)2(OD)6), from variable-temperature 2H MAS NMR spectroscopy. Chem Mater 23:3176–3187
Okada K, Soga H, Ossaka J, Otsuka N (1987) Syntheses of minamiite type compounds, M0.5Al3(SO4)2(OH)6 with M = Sr2+, Pb2+and Ba2+. Neues Jahrbuch für Mineralogie Monatshefte, 64–70
Plášil J, Škoda R, Fejfarova K, Čejka J, Kasatkin AV, Dušek M, Talla D, Lapčák L, Machovič V, Dini M (2014) Hydroniumjarosite, (H3O)+Fe3(SO4)2(OH)6, from Cerros Pintados, Chile: single-crystal X-ray diffraction and vibrational spectroscopic study. Mineral Mag 78:535–547
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–570
Scarlett NVY, Grey IE, Brand HEA (2010) Ordering of iron vacancies in monoclinic jarosites. Am Mineral 95:1590–1593
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr A A32:752–767
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A A64:112–122
Spratt HJ, Avdeev M, Pfrunder MC, McMurtrie J, Rintoul L, Martens WN (2014) Location of hydrogen atoms in hydronium jarosite. Phys Chem Miner 41:505–517
Stoffregen RE, Alpers CN, Jambor JL (2000) Alunite–jarosite crystallography, thermodynamics and geochronology. In: Alpers CN, Jambor JL, Nordstrom DK (eds) Sulfate minerals-crystallography, geochemistry, and environmental significance 40: 453–479. Reviews in Mineralogy and Geochemistry, Mineralogical Society of America, Chantilly, Virginia
Wills AS, Harrison AH (1996) Structure and magnetism of hydronium jarosite, a model Kagomé antiferromagnet. J Chem Soc Faraday Trans 92:2161–2166
Xu H, Zhao Y, Vogel SC, Hickmott DD, Daemen LL, Hartl MA (2010a) Thermal expansion and decomposition of jarosite: a high-temperature neutron diffraction study. Phys Chem Miner 37:73–82
Xu H, Zhao Y, Zhang J, Wang Y, Hickmott DD, Daemen LL, Hartl MA, Wan L (2010b) Anisotropic elasticity of jarosite: a high-P synchrotron XRD study. Am Mineral 95:19–23
Acknowledgments
The authors thank Mark Welch for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Najorka, J., Lewis, J.M.T., Spratt, J. et al. Single-crystal X-ray diffraction study of synthetic sodium–hydronium jarosite. Phys Chem Minerals 43, 377–386 (2016). https://doi.org/10.1007/s00269-016-0802-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0802-0