
Abstract
Purpose
The variable responses to immunotherapy observed in gastric cancer (GC) patients can be attributed to the intricate nature of the tumor microenvironment. Glutathione (GSH) metabolism significantly influences the initiation and progression of gastric cancer. Consequently, targeting GSH metabolism holds promise for improving the effectiveness of Immune checkpoints inhibitors (ICIs).
Methods
We investigated 16 genes related to GSH metabolism, sourced from the MSigDB database, using pan-cancer datasets from TCGA. The most representative prognosis-related gene was identified for further analysis. ScRNA-sequencing analysis was used to explore the tumor heterogeneity of GC, and the results were confirmed by Multiplex immunohistochemistry (mIHC).
Results
Through DEGs, LASSO, univariate and multivariate Cox regression analyses, and survival analysis, we identified GGT5 as the hub gene in GSH metabolism with the potential to promote GC. Combining CIBERSORT, ssGSEA, and scRNA analysis, we constructed the immune architecture of GC. The subpopulations of T cells were isolated, revealing a strong association between GGT5 and memory CD8+ T cells. Furthermore, specimens from 10 GC patients receiving immunotherapy were collected. mIHC was used to assess the expression levels of GGT5 and memory CD8+ T cell markers. Our results established a positive correlation between GGT5 expression, the enrichment of memory CD8+ T cells, and a suboptimal response to immunotherapy.
Conclusions
Our study identifies GGT5, a hub gene in GSH metabolism, as a potential therapeutic target for inhibiting the response to immunotherapy in GC patients. These findings offer new insights into strategies for optimizing immunotherapy of GC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is one of the most prevalent digestive malignancies, ranking fifth in terms of tumor morbidity and fourth in terms of mortality [1]. Its incidence is particularly high in East Asia [2]. Although reports indicate a decline in its incidence rates over the past few years, advanced GC still has an adverse prognosis, with a five-year survival rate of 10% to 30% [3, 4].
In recent years, immunotherapy has shown promise for improving the clinical outcome of patients with GC. However, due to the strong tumor heterogeneity of GC, tumor immune escape events occur frequently, and the efficacy of immunotherapy remains limited and uncertain. According to the results of CheckMate-649 [5], patients with GC who received Nivolumab in combination with chemotherapy had better clinical benefits than those treated with chemotherapy alone, regardless of the PD-L1 Combined Positive Score (CPS). In the combination treatment group, the two-year overall survival (OS) rate was nearly 40%, progression-free survival (PFS) was significantly increased, and the objective response rate (ORR) was almost 70%. However, the results of KEYNOTE-062 suggested that immunotherapy combined with chemotherapy did not provide any advantage over chemotherapy alone [6]. Therefore, the differences in GC patients' response to immunotherapy need to be addressed. It is essential to investigate the likely mechanisms and determine predictive biomarkers for immunotherapy effectiveness.
The metabolism of tumor cells plays a significant role in regulating the immune response in tumors. Previous studies have already shown that amino acid metabolism is widely involved in immune responses and tumor proliferation in the tumor microenvironment (TME) [7]. Treatment with immune checkpoint inhibitors (ICIs) can enhance the tumor infiltration and anti-tumor function of T cells by reprogramming amino acid metabolism and rebalancing nutrient utilization within the TME [8, 9]. Immunotherapy-induced IFN-γ can inhibit glutathione (GSH) synthesis and cause GSH depletion, inducing tumor ferroptosis [10]. In order to undertake the large amount of energy required for tumor proliferation, invasion, and metastasis, tumor cells significantly enhance the uptake and catabolism of glutamine, competing with T cells [11, 12]. Tumor cells can regulate reactive oxygen species (ROS) metabolism through GSH and NADPH produced by glutamine metabolism to maintain redox homeostasis [13]. Blocking GSH metabolism can not only inhibit tumor proliferation, but also restore anti-tumor immunity. Inhibition of GSH metabolism leads to a decrease in myeloid-derived suppressor cells (MDSCs) and facilitates their conversion into M1 macrophages, which promote the process of cross-presentation for tumor antigens and activate CD8+ T cells, thereby performing anti-tumor functions [14]. Therefore, targeting GSH metabolism has the potential to improve immunotherapy response and survival outcomes in GC patients as a novel strategy. However, the mechanism of GSH metabolism in regulating TME in GC patients requires further exploration.
Considering the issues mentioned above, we investigated genes related to GSH metabolism in the Molecular Signatures Database (MSigDB) and used them to construct a prognostic gene signature based on data from The Cancer Genome Atlas (TCGA) public database. To understand the potential mechanism of immune evasion in GC, we conducted CIBERSORT analysis and tumor immune dysfunction and exclusion (TIDE) analysis. Finally, we conducted a systematic interrogation of different immune cell types related to hub genes through single-cell RNA (scRNA) sequencing analysis, aiming to elucidate the critical role of immune cells related to GSH metabolism in regulating tumor progression in GC. These findings should provide potential strategies to enhance the response to immunotherapy in GC.
Materials and methods
Data source
The bulk RNA-seq data (HTSeq-count format) and clinical information for TCGA, which includes 11,069 samples from 33 types of cancer, were downloaded from the Xena (https://xenabrowser.net/). The external validation cohort of GC was acquired from GSE15459 [15], included 200 primary gastric tumors based on GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array). The single-cell dataset (GSE167297 [16]) was obtained from the GEO database, which includes 15,729 cells from 10 human superficial and deep layers of diffuse-type gastric cancer using 10X Genomics. Gene sets related to GSH metabolism were retrieved from the MSigDB database [17] (https://www.gsea-msigdb.org/gsea/msigdb). Three pathways, namely Kyoto Encyclopedia of Genes and Genomes (KEGG), WikiPathways (WP), and Gene Ontology Biological Process (GOBP), were found to be highly correlated. To further explore the gene signatures of GSH metabolism, we performed an intersection of the three gene sets. All statistical analysis of data was performed using R software (version 4.1.3).
Differential expression analysis and functional enrichment analysis
We compared the differential expression level of gene signatures involving GSH metabolism across cancer types based on the count format data from TCGA database, using the R package "limma." Marker genes were then identified by setting the threshold at a false discovery rate (FDR) < 0.05. To explore highly correlated pathways, we performed GO and KEGG functional enrichment analyses using the R package "clusterProfiler" and filtered the GO and KEGG terms with a cutoff of p value < 0.05 and FDR < 0.05. Finally, we created a circle plot using the R package "GOplot."
Construction of a prognostic model
The "glmnet" R package was used to conduct Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression analysis to select genes significantly associated with GC prognosis. Univariate and multivariate Cox regression analyses were also performed to screen the most independent prognostic factors for GC patients. Ultimately, GGT5 was identified as the hub gene for GSH metabolism in GC, as it was differentially expressed in both tumor and normal tissues and emerged as an independent prognostic factor. The optimal cutoff value for GGT5 was determined using the "surv_cutpoint" function in the "survminer" R package. Patients with GC were then divided into high and low group based on this cutoff value, and Kaplan-Meier (KM) survival curves and the log-rank test were constructed using the "survival" and "survminer" R packages. The HR value was calculated using the “survival” R package. RNA-seq data of 200 gastric cancer patients and their survival data affiliated from GSE15459 were analyzed for external validation. Patients were divided into high and low groups according to the optimal cutoff value of GGT5; then, the KM survival curve was plotted.
Clinical information on gastric cancer was collected from TCGA, including tumor stage, histological grade, T stage, N stage, and M stage. Based on these clinical characteristics, patients were clustered into different risk groups. The expression level of GGT5 was measured in each group and plotted in a violin plot using the "ggplot2" package.
DCA (Decision Curve Analysis) is a method for assessing the net benefit of a forecasting model. We conducted the DCA curve to predict the clinic benefit of GGT5 signature compared with another four published models using the “ggDCA” R package.
Estimation of immune cell infiltration
CIBERSORT analysis was used to identify differential infiltration of immune cells between groups with high/low GGT5 expression. Spearman correlation analysis was then employed to explore immune cell infiltration. The "ggpubr" package was utilized to plot immune cell abundance using a violin plot. Immune cells with statistically significant differences were selected for visualization in scatter plots using the "ggplot2" package. MCP-counter allows quantification of the absolute abundance of 8 immune cell populations and 2 stromal cell populations in a mixed tissue from transcriptome data. EPIC (Estimating the Proportion of Immune and Cancer cells) can accurately detect major cell types in tumors directly based on gene expression levels in tumors. xCell combines the ssGSEA analysis with deconvolution methods to understand the heterogeneity of cells that make up the tumor microenvironment in tumor samples. ESTIMATE infers the ratio of stroma and immune cells in a tumor sample from gene expression characteristics. In our study, we conducted MCP-counter, EPIC, xCell, ESTIMATE analysis to validate the result of CIBERSORT based on the “IOBR” R package.
Additionally, we obtained markers of 28 different immune cell types from the TISIDB [18] database (http://cis.hku.hk/TISIDB), an online portal for studying tumor and immune system interactions. Single-sample gene set enrichment analysis (ssGSEA) was used to investigate the relationship between hub genes and infiltration of the 28 immune cell types using the "GSVA" package.
Tumor immune dysfunction and exclusion (TIDE) and the relationship with immune-related genes
To investigate immune evasion mechanisms in GC, we examined the dysfunction and exclusion of T cells. We employed TIDE analysis to predict the curative effect of immunotherapy. Patients with higher TIDE scores are more likely to experience immune escape and have a poorer response to ICIs. For a deeper understanding of the close relationship between GGT5 and immune infiltration, we visualized the correlation between GGT5 and immune-related genes, such as immune activation genes, immunosuppressive genes, chemokines, and chemokine receptors, using a bubble plot generated by the "corrplot" package.
ScRNA data processing and dimensionality reduction
The GC scRNA-seq data used in this study were obtained from GSE167297, which was published in the GEO database. The dataset consisted of 10 samples, and all raw data were processed using the "Seurat" package. Quality control was performed according to the following criteria: (1) the cell count of each sample was at least 500, and (2) the proportion of mitochondrial genes was less than 5%. Next, the "NormalizeData" function was used to normalize the s4 data, and the "FindVariableFeatures" function was applied to recognize the top 2000 highly variable genes (HVGs). The selected HVGs were further identified, centered, and scaled using the "ScaleData" function.
Cell clustering, annotation, and marker genes identification
According to principal component analysis (PCA), scores were assigned to the Seurat data. Each PC term included half of its features, which combined information related to the features. The "RunPCA" function was used for dimension reduction of highly variable genes (HVGs). Spectral clustering was then employed to identify different cell clusters. The "FindNeighbors" and "FindClusters" functions were subsequently utilized for further clustering, with the resolution parameter set to 0.4. Nonlinear dimensionality reduction was visualized by T-Distributed Random Neighbor Embedding (t-SNE). To cluster different cell types, the "SingleR" package was used to reference a database. The "FindAllMakers" function was then employed to screen for characterized genes with a logfc.threshold of 0.5. The expression level of GGT5 in each different cell type was measured and portrayed as a violin plot. The composition ratio and cell number of each cell type were compared and displayed in a bar chart using the "ggplot2" package. T cells were extracted for further analysis. The NormalizeData, RunPCA, FindNeighbors, and FindClusters functions were reloaded to cluster different T cell subtypes. The tSNE clustering analysis was reperformed to display the expression distribution of T cell subsets. Marker genes for each subgroup of T cells were screened in published articles and the CellMarker database, and the identified clusters were reannotated. The expression value of GGT5 in all T cell subsets and the correlation scores between GGT5 and different CD8 T cells were calculated.
Pseudotime trajectory analysis
To classify the exhaustion of T cells during the process of tumor-educating, the “Monocle 2” package was utilized for trajectory of each single cell. The raw UMI count matrices of 10 gastric cancer patients were imported using Seurat and converted as a cds file with Monocle. Following the standard quality control process, we filtered some low-quality cells, setting the filter parameters as follows: mean expression ≥ 0.1, num_cells_expressed ≥ 10, the q-value of different genes was less than 0.05. Then, we portrayed the trajectory of T cells, CD8+ T cells, and CD4+ T cells into tSNE plots.
Multiplex immunohistochemistry (mIHC) analysis
To investigate the connection between GGT5 expression and memory CD8+ T cells, and the response to immunotherapy, a total of 10 specimens of GC patients undergoing immunotherapy at Beijing Chaoyang Hospital were obtained for this study. Each patient's response to immunotherapy was evaluated based on RECIST criteria, which categorized the patients into either the partial response (PR) group or the non-PR group. mIHC analysis was performed with the Opal™ 4-color Multiplex reagents kit (PerkinElmer, USA). Antibodies for GGT5 (ab283267), CD45RO (ab23), CCR7 (ab253187) were purchased from Abcam Inc. Antibody for CD8 (66868-1-Ig) was purchased from Proteintech. According to the manufacturer’s instructions, nuclear staining was conducted with DAPI (PerkinElmer, USA), GGT5 (ab283267, 1:2000) was stained with opal 620, CD8 (66868-1-Ig, 1:50,000) was stained with opal 570, CD45RO (ab23, 1:5000) was stained with opal 520, CCR7 (ab253187, 1:300) was stained with opal 690. Memory CD8+ T cells were defined as CD8, CD45RO, and CCR7-positive cells. The multiplex fluorophore-stained slides were scanned by Beijing Bodu Hengyi Technology company. Each channel was individually captured and analyzed using NDP View 2 software.
Result
Construction of GSH metabolic signature
The workflow chart of this study is presented in Fig. 1. Initially, the MSigDB database was searched to systematically identify signature genes of GSH metabolism. By intersecting the 3 GSH metabolism gene sets from KEGG, WK, and GOBP databases, a total of 16 genes were selected. Next, these genes were analyzed in 33 types of pan-cancer datasets from TCGA. After excluding cancer types without matched normal samples, 17 cancer types were retained for further analysis, and differential expression gene (DEG) analysis of GSH metabolic genes was performed between the 17 cancer types and normal tissues (Fig. S1a). Of these, 6 GSH metabolism genes were found out to be differentially expressed in GC (p < 0.05) (Fig. 2a). These genes included GGT1, GGT5, GPX1, GPX4, GSS, and GSTA1 (Table 1). Furthermore, LASSO regression analysis was conducted to screen potential prognostic-related genes (Fig. 2b, c), and all six of these genes were found to be prognostic factors and were considered as GSH metabolic signature.

Analysis workflow of this study

The expression mode diagram of glutathione metabolism-related genes of pan-cancer from TCGA and enrichment of GO and KEGG a The bubble plot displays the differentially expression diagram of genes associated with glutathione metabolism between tumor and normal tissues across pan-cancer types. Only the data of FDR < 0.05 were retained and it shows that there are 6 genes differentially expressed in GC. The dot size indicates the FDR and the color represents the fold-change, b, c LASSO regression analysis to screen the prognostic-related genes, d the circle plot for KEGG analysis, terms with p < 0.05 were present. The top 10 terms of BP (e) and MF (f) for GO analysis, of which the p value is less than 0.05, were plotted
GO and KEGG enrichment analysis of key genes
For a deeper understanding of the correlation of the signature and GC, the ‘clusterProfiler’ package was utilized to perform GO and KEGG analysis. The results are presented in Fig. 2d–f. For BP, MF, and KEGG, six of the selected key genes were strongly correlated with GSH metabolism, which validates the screening process. Additionally, KEGG analysis revealed enrichment in arachidonic acid metabolism, taurine and hypotaurine metabolism, and ferroptosis. GO BP terms showed that the top two terms were cellular modified amino metabolic process and sulfur compound metabolic process. In GO MF terms, the top two terms were peroxidase activity and oxidoreductase activity, acting on peroxide as acceptor.
Identification of the key gene of GSH metabolism in GC
To explore the latent mechanism of the key genes in GC, the differential expression levels between GC and paired adjacent normal tissues were measured using ‘limma’ package. The results were presented as violin plots using the ‘ggplot’ package (Fig. 3a). The expression values of GGT5, GPX1, and GSS were found to be significantly higher in GC (p < 0.05), whereas the expression values of GGT1, GPX4, and GSTA1 showed no statistical significance between GC and normal tissues. Furthermore, univariate Cox regression analysis was conducted to determine the prognostic effect (Table 2). This study included age, gender, histologic grade, tumor stage, T stage, N stage, M stage, six key genes, and risk score calculated by LASSO analysis, radiation therapy, reflux history, antireflux treatment, Barrett's esophagus, family history, new tumor event, lymph node count, lymph nodes positive, and signet ring cell carcinoma to identify the risk factors of GC. According to the results, GGT5 was identified as the hub gene for GSH metabolism in GC. Not only was it differentially expressed in tumor tissue, but it also emerged as an independent prognostic gene.

Expression pattern and survival analysis of the key genes for GSH metabolism a The differential expression level of 6 glutathione metabolic signature in GC and adjacent normal tissues shows that expression values of GGT5, GPX1, and GSS are extremely higher in gastric cancer (p < 0.05), while the expression values of GGT1, GPX4, and GSTA1 have no significant meaning between GC and adjacent normal tissues, b the GC samples were classified into high and low groups of GGT5 expression according to the maxstat method. The KM curves including overall survival analysis (p = 0.00029), progression-free interval analysis (0.0041), disease-free interval analysis (p = 0.0033), and disease-specific survival analysis (p = 0.023) were plotted, and these suggest that GGT5 is a risk gene associated with a poor outcome of gastric cancer
Clinical characteristics and outcome related to GGT5 in gastric cancer
To estimate the prognostic value of GGT5, KM curves were portrayed to display the survival rate of GC patients. The results revealed that patients in group of high GGT5 expression had poorer overall survival (OS) than those with low expression (p = 0.00029, HR (95% CI) 0.55 (0.39–0.78)). Similarly, the progression-free interval (PFI) (p = 0.0041, HR (95% CI) 0.60 (0.40–0.91)), disease-free interval (DFI) (p = 0.0033, HR (95% CI) 0.20(0.10–0.41)), and disease-specific survival (DSS) (p = 0.023, HR (95% CI) 0.49 (0.30–0.80)) consistently showed a survival trend (Fig. 3b), indicating that higher expression of GGT5 was closely related to adverse clinical outcomes in GC patients. To validate the result in the TCGA cohort, we collected 200 GC patients from GSE15459; it shows that high GGT5 expression group had a significantly worse OS than the low GGT5 expression group (Fig. S3a, p = 0.00035, HR (95% CI) 0.49(0.32–0.76)).
Besides, DCA curves were used to compare our GGT5 model with another four published models [19,20,21,22]. We found that GGT5 has better clinical application value than other GC signatures (Fig. S3b). Furthermore, the correlation between GGT5 and clinical–pathologic characteristics, including histologic grade, tumor stage, T stage, N stage, and M stage, was measured. According to T stage and histologic grade, GGT5 was expressed at higher levels in T3 + T4 stages than in T1 + T2 stages (p = 0.006) and in G3 grade than in G1 + G2 grades (p = 0.00014), indicating that the value of GGT5 increased with higher clinical stage (Fig. 5a). However, the results between GGT5 and tumor stage, N stage, and M stage showed no statistical significance (Fig. S2b). In conclusion, these findings show that patients with higher expression of GGT5 have poorer clinical outcomes.
Correlation Between GGT5 and Immune Infiltration
The proportion of tumor-infiltrating immune subsets was employed according to the CIBERSORT algorithm, by deconvoluting the expression matrix of 22 immune cells through linear support vector regression [23, 24]. In our study, the GC samples were classified into high/low groups based on the optimal cutoff value of GGT5 and subsequently analyzed for tumor heterogeneity between the two groups. As depicted in Fig. 4a, naïve B cells (p = 0.041), regulatory T cells (Tregs) (p = 0.046), monocytes (p < 0.001), and resting Mast cells (p < 0.001) were observed to have higher expression levels in the high GGT5 group, whereas the low expression group exhibited high expression levels of resting CD4 memory T cells (p = 0.019), activated CD4 memory T cells (p = 0.021), follicular helper T cells (p = 0.001), and M0 macrophages (p = 0.002). EPIC, MCP-counter, xCell, and ESTIMATE were conducted to validate the results above. All of these methods to explore the immune infiltration mode of tumor microenvironment show consistent trend; the CD8+ T cells were observed to have significantly higher expression levels in the GGT5 high expression group (Fig. S3c-d).

The relationship between GGT5 and immune cells of GC a CIBERSORT analysis reveals the relationship of GGT5 with 22 kinds of immune infiltrating cells, it indicates that there are 8 kinds of immune cells differentially expressed in high (red) and low (green) expression group of GGT5 (p < 0.05), b the correlation analysis displays a positive connection between GGT5 with T cells regulatory (Treg), T cells CD8, B cells naïve, monocytes, and mast cells resting, while it has a negative connection with T cells CD4 memory resting
For further validation, we calculated the correlation value of GGT5 and various immune cells, as illustrated in Fig. 4b. Our analysis revealed that GGT5 exhibited a positive correlation with regulatory T cells (Tregs) (R = 0.17, p = 0.00062), CD8 T cells (R = 0.11, p = 0.024), naïve B cells (R = 0.17, p = 0.00052), monocytes (R = 0.25, p = 3.7e−07), and resting Mast cells (R = 0.26, p = 1.7e−07). Conversely, GGT5 showed a negative association with resting CD4 memory T cells (R = -0.2, p = 5e−05). However, the correlation between GGT5 and immune cells like activated CD4 memory T cells, follicular helper T cells, and M0 macrophages was not statistically significant (Fig. S2a). These findings suggest that GGT5 may influence different types of immune cells, particularly T cells, to modulate the TME of GC.
Relationship between GGT5 and immune-related genes and immune therapy
The TIDE scores were utilized to predict the efficiency of immunotherapy on GGT5. A higher TIDE score indicates a greater tendency of immune escape and a poorer response to immune checkpoint inhibitors (ICIs). As shown in Fig. 5b, patients with higher expression of GGT5 had a higher TIDE score (p < 2.2e−16), suggesting that GGT5 overexpression may lead to an adverse response to immunotherapy. To further investigate the role GGT5 in the TME of GC, correlation analysis between GGT5 and immune genes was estimated (Fig. 5c). The results show a positive correlation with immune active genes, immune suppressive genes, chemokines, and chemokine receptors, particularly immune active genes and chemokine receptors. These findings suggest the regulatory role for GGT5 in the TME of GC is possibly related to these immune-related genes.

The relationship of GGT5 with clinicopathological characters and immune features a the relationship between GGT5 with clinicopathological characters of gastric cancer, including histologic grades (p = 2.1e−06) and T stage (p = 0.018), indicates that high expression of GGT5 is highly related to the development of gastric cancer, b the prediction efficiency of response to ICIs based on TIDE scores shows that higher expression of GGT5 is related to a higher TIDE score, also a poorer response to ICIs therapy, c the bubble plots represent the relationship of GGT5 with immune-related genes, including immune-active genes and chemokine receptors. It displays that GGT5 is positively associated with these immune-related genes. The color and size of the dots represent the correlation coefficient
Identification of GGT5-related immune cell infiltration in gastric cancer
For this study, we analyzed 10 gastric cancer samples from GSE167297. After adjusting for batch differences, we integrated the samples and identified the top 2000 highly variable genes (HVGs) based on their mean and dispersion. We then performed principal component analysis (PCA) downsizing and retained the top 20 dimensions of cells. Next, we used t-SNE clustering to visualize the clustering of 15,729 cells from gastric cancer tissues, which were assigned to 14 clusters (Fig. 6a). Using the "SingleR" package and marker genes, we identified 8 cell types, including T cells, B cells, dendritic cells (DC), endothelial cells, epithelial cells, monocytes, NK cells, and smooth muscle cells (Fig. 6b). Notably, T cells had multiple subgroups and a high number of cells, indicating their potential importance in gastric cancer (Fig. 6c).

Overview of single-cell analysis from gastric cancer a a demonstration of t-SNE of gastric cancer samples, 14 cell clusters, and different cell types identified by marker genes, b the expression pattern of each cell type, c the cell ratio in each cluster indicates that T cell has the most multiple subsets, d heatmap shows the top 10 marker genes of clusters, representing the characteristic features of each cell type, e the expression level of GGT5 in each cell type shows that GGT5 has a high expression value in T cells
To validate our findings, we screened the top 10 HVGs of each cluster (Fig. 6d), which were confirmed as marker genes for each cell type. We then portrayed the expression level of GGT5 in each infiltrating cell type using a violin plot (Fig. 6e), which showed that GGT5 expressed particularly higher in T cells. Therefore, we selected T cells for further investigation and sub-division in this study.
We performed PCA downsizing again for selected T cells, retaining only the top 10 dimensions. This led to the identification of 11 clusters (Fig. 7a), which were reannotated using marker genes highly expressed in each cluster and lowly expressed in others, based on the CellMarker database and relevant articles. Next, we measured the activity of GSH metabolism using the KEGG, GO, and WikiPathways databases (Fig. 7g) and found that it was active in T cells of gastric cancer. To increase our understanding of GGT5’s role in T cells, we measured the expression value of GGT5 in each subpopulation available (Fig. 7b) and found that it was most highly expressed in C0-CD8-IL7R, C5-CD8-CCR7, and C8-CD8-TK1 clusters, suggesting a close association between GGT5 and CD8 T cells. To evaluate the differentiation states of different T cells, the continuous trajectory of T cell exhaustion was portrayed by Monocle 2 and visualized as a tSNE plot (Fig. 7c). Additionally, we isolated the CD8 and CD4 T cell clusters to elucidate the association between GGT5 and CD8+ T cells. The results indicate that cluster 0 and 5 were predominantly observed in the early and middle stages of the differentiation trajectory for both the entire T cell exhaustion system and CD8+ T cells (Fig. 7d). The expression level of GGT5 in CD8+ T cells was significantly higher than in CD4+ T cells (Fig. 7e), with concentration primarily in the early and middle phases of the T cell trajectory (Fig. 7f). This suggests that elevated GGT5 expression might contribute to T cell exhaustion.

Overview of single cells from T cells a the t-SNE plot shows 11 clusters and different subsets of T cells, b the expression value of GGT5 in each subgroup of T cells indicates that GGT5 is highly expressed in C0-CD8-IL7R, C5-CD8-CCR7, and C8-CD8-TK1. c, d different trajectories of T cells, CD8+ T cells, and CD4+ T cells, with the shade and color for the pseudotime and clusters calculated by monocle, e the expression level of GGT5 in CD8+ T cells and CD4+ T cells, f pseudotime analysis of GGT5 ± T cells, g the degree of activity of 3 GSH metabolism gene sets from the MSigDB database based on the AUC value shows that glutathione metabolism is actively expressed in T cells
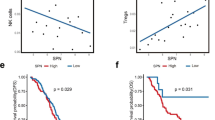
To further interrogate the association between GGT5 and CD8+ T cells, we obtained markers for 28 immune cell types and conducted ssGSEA analysis to explore the interplay among these immune cells, GGT5, and gastric cancer tumor stage using the TISIDB database (Fig. 8a). Our analysis revealed that higher GGT5 expression and advanced tumor stage were linked to increased enrichment of memory CD8+ T cells. Furthermore, correlation analysis revealed a robust positive relationship between GGT5 expression and central memory CD8+ T (TCM) cells, along with effector memory CD8+ T (TEM) cells (Fig. 8b).

a ssGSEA analysis of 28 kinds of immune cells in high/low expression of GGT5 and different stage of GC patients, suggesting that memory CD8+ T cells, especially TCM and TEM, are more prevalent in GGT5 high tumors and advanced stages of GC, b the correlation of GGT5 with TCM and TEM, showing a positive association with TCM and TEM, c mIHC analysis shows expression level of CD45RO, CD8, CCR7, GGT5 in GC patients receiving immunotherapy. Group 1 represents 3 patients which are evaluated as PR according to RECIST. Group 2 represents patients which are evaluated as non-PR. mIHC expression analysis for CD45RO, CD8, CCR7, and GGT5 reveals that the tumors of GC patients non-responsive to immunotherapy have enhanced expression of GGT5 and memory CD8+ T cell markers. Magnification, × 400
Multiplex immunohistochemistry (mIHC) analysis of GC tissue specimens
Multiplex immunohistochemistry (mIHC) analysis was performed among 10 GC tissue specimens to further explore the association between GGT5 expression with memory CD8+ T cells and immunotherapy. The response of all the 10 patients to immunotherapy was documented (Table S2). The most representative mIHC staining images of GGT5, CD8, CD45RO, and CCR7 are shown in Fig. 8. Multiplexed fluorophore-stained slides of 4 other patients were not presented due to the markedly low expression of each maker and diminished fluorescence intensity. Patients in Group 1 (patient1, 9, 10) were categorized as PR according to the RECIST, and patients in Group 2 (patient 2, 3,7) were designated as non-PR. Obviously, GGT5 expression displayed a positive correlation with memory CD8+ T cells. Furthermore, patients with typically poorer responses to immunotherapy (Group 2) exhibited higher levels of GGT5 expression and a greater concentration of memory CD8+ T cells. Hence, our mIHC results not only confirmed the significant correlation between GGT5 and memory CD8+ T cell enrichment but also revealed that patients with elevated GGT5 expression had a diminished response to immunotherapy.
Discussion
Reactive oxygen species (ROS) exert severe damage on DNA in the cell [25, 26]. As an adaptive response, tumor cells possess high levels of molecules that scavenge ROS. GSH metabolism plays an antioxidant role that helps to eliminate free radicals and detoxify agents, and is commonly elevated during oxidative stress [27]. Disturbances in GSH metabolic homeostasis play a crucial role in the occurrence and progression of diseases, including cancers [28]. Furthermore, GSH metabolism can promote metastasis and is positively associated with therapeutic resistance in various cancer types [27, 29, 30]. Therefore, GSH metabolism has become a crucial target for cancer research and treatment.
In this study, genes related to GSH metabolism were screened to identify the active metabolic gene. GGT5 was finally identified as the hub gene of GSH metabolism and predicted to regulate the TME of GC. The mechanism of how GGT5 affects GC was explored at both the bulk RNA and scRNA levels. We found that: (1) GGT5 is an independent prognostic gene marker for GC and can promote the tumor microenvironment; (2) GGT5 has a highly positive connection with immune-related genes, and high expression of GGT5 can alleviate the effect of ICIs on cancers; (3) GGT5 plays its role mainly by promoting memory CD8+ T cells.
The GGT family acts in a crucial role in extracellular metabolism of GSH, which can maintain the balance of GSH metabolism via cleaving GSH peptides. It has been suggested that GGT family is elevated in multiple cancers and can lead to poor clinic outcome, such as stomach, ovarian, pancreatic, breast cancer [31,32,33,34]. Moreover, an accumulation of evidence suggests that GGT family plays an important role in tumorigenesis and progression of stomach cancer [35]. Not only is GGT5 associated with poor outcome in GC, but also with the ability to regulate the TME [36, 37]. In this study, GGT5 expression was measured based on TCGA and found to have higher expression in GC compared to normal tissues. Moreover, higher GGT5 expression in gastric cancer is correlated with poor clinical outcomes based on OS, DFI, PFI, and DSS analyses. Furthermore, the clinical correlation analysis revealed that GGT5 expression is elevated in higher grades of gastric cancer, such as histologic grade 3 and T stages III-IV. These findings suggest an indispensable role of GGT5 in tumor invasion. However, the specific mechanisms of how GGT5 affects GC is still unclear.
It has been largely noted that in response to pathogens or dangerous environments, immune cells release leukotrienes (LTs) to defend against damage [38]. GGT5, a γ-glutamyl leukotrienes, can metabolize LTC4 to LTD4 [39, 40], making it a key player in the active role of asthma pathogenesis [41] and indicating that it serves a significant role in the immune response. TIDE analysis predicted that high GGT5 expression is associated with a poorer response to ICIs in gastric cancer, prompting further investigation into how GGT5 affects the TME of GC. In this study, the relationship between GGT5 and immune genes of GC was investigated, revealing a positive relation between GGT5 and immune-active genes and chemokine receptors, particularly CXCL12, CXCR4, and TGFB1. CXCL12 is a small protein that binds with CXCL4 on tumor cells, triggering the anti-apoptosis pathway via Bcl-2 upregulation signal pathways and the EMT pathway via the Rho-ROCK pathway [42]. Elevated expression of CXCR4 and/or CXCL12 expression has been examined in larger tumors and lymphatic invasion samples in gastric cancer [43], as well as decreased response to radio- and chemo-therapy in various cancer types [44]. TGFB1 facilitates the progression and evasion of cancers, suppressing antitumor immune responses. TGFB1 has been shown to play a role in the immune escape of gastric cancer, with the mechanism likely involving the suppression of CD8+ T cells and increases in regulatory T (Treg) cells [45, 46]. Therefore, GGT5 may exert a pro-tumor effect by regulating immune genes such as CXCL12 and CXCL4 in the tumor microenvironment. This finding prompts further investigation into the involvement of immune cells. In tumor metabolism, GGT5 exhibits catalytic activity in both hydrolysis and transpeptidation reactions. As one of the few extracellular enzymes identified with the capability to cleave gamma-glutamyl bonds, GGT5 assumes a significant role in the initial stage of glutathione metabolism, involving the cleavage of gamma-glutamyl bonds [47]. Consequently, attenuating the expression of GGT5 would impede GSH metabolism and enhance anti-tumor capabilities. The application of GGsTOP, a remarkably selective and potent irreversible inhibitor of GGT activity, can markedly lower the expression of GGT5, hinder the proliferation and chemotherapy resistance of lung adenocarcinoma cells [48]. These findings underscore the importance of GGT5 as a novel target for anti-tumor therapy. Exploration of the GGT5 amino acid sequence reveals distinctive residues that interact with the glutathione active site, providing a theoretical groundwork for the future development of drugs targeting GGT5 [49].
In our study, CIBERSORT analysis revealed the infiltration of 8 kinds of immune cells between different expression group of GGT5 in gastric cancer. To better understand the underlying mechanism of GGT5 and TME, further investigation of the relationship between GGT5 and these immune-related cells is necessary. The correlation analysis indicated that GGT5 has a positive correlation with CD8 T cell, Tregs, B cells naïve, monocytes, and resting Mast cells, while it has a negative correlation with resting CD4 memory T cells. Similar trend was found in EPIC, MCP-counter, xCell, and ESTIMATE analysis. It has been reported that CD8+ T cells play a crucial role in tumor immune resistance with PD-L1, and increases in CD8+ T cells are associated with poor PFS and OS of gastric cancer [50]. Tregs, promoted by the TNFR2 signaling pathway, are identified as immune suppressors, and their accumulation in the TME of GC is considered a risk factor in prognosis [51]. Also, the infiltration of CD4+ T cells is higher in patients with T3 stage of GC compared to that in T1 and T2 stage, implying a critical effect of CD4+ T cells in the prognosis of gastric cancer [52]. We next conducted scRNA analysis to identify the heterogeneity of immune cell types within the TME of gastric cancer, which were initially clustered into eight cell types. By combining the results of immune infiltration and scRNA analysis, we explored the specialty of T cells, which prompted further investigation. A previous study demonstrated that GSH metabolism is dispensable for early T cell activation but can promote T cell growth [53]. In our study, we evaluated the involvement of GSH metabolism in T cells and subsequently identified characteristic T cell subsets. Our results indicated that GGT5 is mainly involved in three CD8+ T cell subsets named C0-CD8-IL7R, C5-CD8-CCR7, and C8-CD8-TK1. Furthermore, we isolated CD8+ T cells and CD4+ T cells; it was found that GGT5 was extremely higher in CD8+ T cells than that in CD4+ T cells. These results exhibit a strong correlation with CD8+ T cells. According to previous studies, the loss of IL-7 and IL-7R can lead to severe immune deficiency, and IL-7R acts a vital role in the generation and long-term maintenance of memory CD8+ T cells [54]. Deficiency in CCR7 can impair the function of memory CD8+ T cells to sustain IL-7 and IL-7R, which has a negative effect on the survival and homeostasis of memory CD8+ T cells [55]. TK1, which is associated with CD8+ T cells [56], is a marker of abnormal tumor cell proliferation. Its concentration increases with lesion growth, disease progression, and distant metastasis [57]. In summary, our findings indicate a connection between GGT5 and memory CD8+ T cells. Next, we investigated GGT5 and 28 immune cell types based on ssGSEA analysis. Our findings demonstrated that memory CD8+ T cells, including TCM and TEM, were more abundant in the group with higher GGT5 expression and in the higher tumor stage group. This suggests a role for GGT5 in maintaining CD8+ T cells, especially TCM and TEM cells.
Three kinds of memory T cells have now been reported in peripheral blood: central memory T cells (TCM) located in secondary lymphoid tissues and reactivated during secondary infection; effector memory T cells (TEM) that circulate in various tissues and are cytotoxic; and tissue-resident memory T cells (TRM) that persist in tissues without circulating [58, 59]. It has been shown that cancer patients treated with ICIs show an increasing frequency of memory T cells, both TCM and TEM cells, and that infiltration of memory CD8+ T cells is associated with poor prognosis [60, 61]. Our findings in this study are consistent with these previous results. We conducted mIHC analysis to investigate the expression mode of GGT5 in GC tissues and its correlation with the response to immunotherapy and found that patients with a poor response to immunotherapy have higher GGT5 expression and more memory CD8+ T cell accumulation. In addition, our analysis using monocle revealed that memory T cell clusters, specifically C0-CD8-IL7R and C5-CD8-CCR7, predominantly accumulate in the early and middle stages of T cell differentiation. Unlike effector T cells, memory T cells do not fully transition but persist, contributing to long-term immunological memory. Consequently, memory T cells are positioned within the intermediate stage of CD8+ T cell differentiation, according to the linear model, owing to their shared characters of transcriptional, phenotypic, and epigenetics with both effector and naïve T cells [62]. Notably, the impact of GGT5 on T cells has not been previously reported, and our findings indicate that the expression of GGT5 mostly accumulates during the early and middle period of the T cell trajectory, mirroring the distribution of memory CD8+ T cells. This finding implies a potential role for GGT5 in sustaining the longevity of memory CD8+ T cells throughout the progress of gastric cancer.
Previous studies have already shown that a reduction in GSH levels and a rise in ROS levels can impair the formation and maintenance stage of memory CD8 T cells. In contrast, exogenous GSH can shield TCM and TEM cells from apoptosis [63, 64]. Nevertheless, the connection between GGT5, an active gene in GSH metabolism, and memory CD8+ T cells has rarely been explored and reported. Our study is the first to propose that GGT5 wields a remarkable effect on the initiation and progression of gastric cancer, while concurrently regulating the immune milieu by preserving live memory CD8+ T cells. Nevertheless, our study still has several shortcomings, which are detailed below: (1) Future clinical investigations involving multiple centers and extensive cohorts are imperative to validate this predictive signature before deeming it clinically beneficial; (2) additional experiments, including both overexpression and knockdown studies, as well as immune/tumor co-culture experiments, are essential to clarify the functional impact of GGT5 on the development and progression of gastric cancer; (3) future progress of foundational experiments concerning molecular structure research and the exploration of GGT5's druggable potential is crucial for its potential application in clinical therapy.
Conclusion
This study shows that GGT5 serves as an active gene of GSH metabolism and promotes progression, drug resistance, and immune microenvironmental remodeling in gastric cancer. Although some of these processes have been reported before, the specific mechanism of how this occurs still remain unknown. Moreover, the interplay between GGT5 and memory CD8+ T cells and their influence on gastric cancer has rarely been studied. A deeper comprehension of the role of GGT5 in these pathways may offer valuable insights for the treatment of gastric cancer.
Data Availability
All experimental data required to prove the conclusions of the study have been included in this paper and/or the supplemental materials. Other data relating to our study could be accessible from the corresponding author. Publicly supporting data from TCGA could be downloaded from https://xenabrowser.net/. GEO data are available from GSE167297 and GSE15459.
Abbreviations
- GC:
-
Gastric cancer
- GSH:
-
Glutathione
- ICIs:
-
Immune checkpoint inhibitors
- ROS:
-
Reactive oxygen species
- TCM:
-
Central memory CD8+ T cells
- TEM:
-
Effector memory CD8+ T cells
- TME:
-
Tumor microenvironment
References
Sung H et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249
Smyth EC et al (2020) Gastric cancer. Lancet 396(10251):635–648
Machlowska J et al (2020) Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int J Mol Sci 21(11):4012
Shitara K et al (2017) Phase II study of adjuvant chemotherapy of S-1 plus oxaliplatin for patients with stage III gastric cancer after D2 gastrectomy. Gastric Cancer 20(1):175–181
Janjigian YY et al (2021) First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398(10294):27–40
Shitara K et al (2020) Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: the KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol 6(10):1571–1580
Vettore L, Westbrook RL, Tennant DA (2020) New aspects of amino acid metabolism in cancer. Br J Cancer 122(2):150–156
Lim S et al (2017) Interplay between immune checkpoint proteins and cellular metabolism. Cancer Res 77(6):1245–1249
Wang W, Zou W (2020) Amino acids and their transporters in T cell immunity and cancer therapy. Mol Cell 80(3):384–395
Wang W et al (2019) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569(7755):270–274
Zhang J, Pavlova NN, Thompson CB (2017) Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J 36(10):1302–1315
Reinfeld BI et al (2021) Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593(7858):282–288
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947
Leone RD et al (2019) Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366(6468):1013–1021
Ooi CH et al (2009) Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet 5(10):1000
Jeong HY et al (2021) Spatially distinct reprogramming of the tumor microenvironment based on tumor invasion in diffuse-type gastric cancers. Clin Cancer Res 27(23):6529–6542
Subramanian A et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545–15550
Ru B et al (2019) TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics 35(20):4200–4202
Yan LH et al (2015) Overexpression of CDX2 in gastric cancer cells promotes the development of multidrug resistance. Am J Cancer Res 5(1):321–332
Jo JH et al (2019) Novel gastric cancer stem cell-related marker LINGO2 is associated with cancer cell phenotype and patient outcome. Int J Mol Sci 20(3):555
Shen JJ et al (2020) Serum proteome profiling reveals SOX3 as a candidate prognostic marker for gastric cancer. J Cell Mol Med 24(12):6750–6761
Barker N et al (2010) Lgr5 stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6(1):25–36
Wu Y et al (2022) B3GNT3 as a prognostic biomarker and correlation with immune cell infiltration in lung adenocarcinoma. Ann Transl Med 10(6):295
Xu Z et al (2022) Improving ovarian cancer treatment decision using a novel risk predictive tool. Aging (Albany NY) 14(8):3464–3483
Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 91(23):10771–10778
Olinski R et al (1992) DNA base modifications in chromatin of human cancerous tissues. FEBS Lett 309(2):193–198
Lushchak VI (2012) Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012:736837
Forman HJ, Zhang H, Rinna A (2009) Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 30(1–2):1–12
Huang ZZ et al (2001) Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J 15(1):19–21
Carretero J et al (1999) Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin Exp Metastasis 17(7):567–574
Xiao Y et al (2019) Serum gamma-glutamyltransferase and the overall survival of metastatic pancreatic cancer. BMC Cancer 19(1):1020
Grimm C et al (2013) Association of gamma-glutamyltransferase with severity of disease at diagnosis and prognosis of ovarian cancer. Br J Cancer 109(3):610–614
Staudigl C et al (2015) Prognostic relevance of pretherapeutic gamma-glutamyltransferase in patients with primary metastatic breast cancer. PLoS ONE 10(4):e0125317
Xu XS et al (2015) Model based on alkaline phosphatase and gamma-glutamyltransferase for gallbladder cancer prognosis. Asian Pac J Cancer Prev 16(15):6255–6259
Wang Q et al (2017) Tumor and serum gamma-glutamyl transpeptidase, new prognostic and molecular interpretation of an old biomarker in gastric cancer. Oncotarget 8(22):36171–36184
Wang Y et al (2022) Identification of GGT5 as a novel prognostic biomarker for gastric cancer and its correlation with immune cell infiltration. Front Genet 13:810292
Liu C et al (2022) Classification of stomach adenocarcinoma based on fatty acid metabolism-related genes frofiling. Front Mol Biosci 9:962435
Lukic A et al (2016) Pulmonary epithelial cancer cells and their exosomes metabolize myeloid cell-derived leukotriene C4 to leukotriene D4. J Lipid Res 57(9):1659–1669
Carter BZ et al (1997) Metabolism of leukotriene C4 in gamma-glutamyl transpeptidase-deficient mice. J Biol Chem 272(19):12305–12310
Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88
Peters-Golden M, Henderson WR Jr (2007) Leukotrienes. N Engl J Med 357(18):1841–1854
Daniel SK, Seo YD, Pillarisetty VG (2020) The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Semin Cancer Biol 65:176–188
Yasumoto K et al (2006) Role of the CXCL12/CXCR4 axis in peritoneal carcinomatosis of gastric cancer. Cancer Res 66(4):2181–2187
Jung K et al (2017) Targeting CXCR4-dependent immunosuppressive Ly6C(low) monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci USA 114(39):10455–10460
Guan Z et al (2015) TGF-beta induces HLA-G expression through inhibiting miR-152 in gastric cancer cells. J Biomed Sci 22:107
Tuncel T et al (2013) Immunoregulatory function of HLA-G in gastric cancer. Asian Pac J Cancer Prev 14(12):7681–7684
Wickham S et al (2011) Gamma-glutamyl compounds: Substrate specificity of gamma-glutamyl transpeptidase enzymes. Anal Biochem 414(2):208–214
Wei JR, Dong J, Li L (2020) Cancer-associated fibroblasts-derived gamma-glutamyltransferase 5 promotes tumor growth and drug resistance in lung adenocarcinoma. Aging-Us 12(13):13220–13233
Heisterkamp N et al (2008) The human gamma-glutamyltransferase gene family. Hum Genet 123(4):321–332
Thompson ED et al (2017) Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut 66(5):794–801
Qu Y et al (2022) The effects of TNF-alpha/TNFR2 in regulatory T cells on the microenvironment and progression of gastric cancer. Int J Cancer 150(8):1373–1391
Jiang S et al (2021) GARP Correlates With Tumor-Infiltrating T-Cells and Predicts the Outcome of Gastric Cancer. Front Immunol 12:660397
Mak TW et al (2017) Glutathione primes T cell metabolism for inflammation. Immunity 46(4):675–689
Osborne LC et al (2007) Impaired CD8 T cell memory and CD4 T cell primary responses in IL-7R alpha mutant mice. J Exp Med 204(3):619–631
Jung YW et al (2016) CCR7 expression alters memory CD8 T-cell homeostasis by regulating occupancy in IL-7- and IL-15-dependent niches. Proc Natl Acad Sci USA 113(29):8278–8283
Ren J et al (2020) A novel prognostic model of early-stage lung adenocarcinoma integrating methylation and immune biomarkers. Front Genet 11:634634
Wang Y et al (2022) Clinical value on combined detection of serum CA724, DKK1, and TK1 in diagnosis of gastric cancer. J Oncol 2022:6941748
Nose Y et al (2023) The tissue-resident marker CD103 on peripheral blood T cells predicts responses to anti-PD-1 therapy in gastric cancer. Cancer Immunol Immunother 72(1):169–181
Ando M et al (2020) Memory T cell, exhaustion, and tumor immunity. Immunol Med 43(1):1–9
Takeuchi Y et al (2018) Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int Immunol 30(1):13–22
Ribas A et al (2016) PD-1 Blockade expands intratumoral memory T cells. Cancer Immunol Res 4(3):194–203
Henning AN, Roychoudhuri R, Restifo NP (2018) Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 18(5):340–356
Ma R et al (2018) A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat Cell Biol 20(1):21–27
Gupta S et al (2007) Differential sensitivity of naive and subsets of memory CD4+ and CD8+ T cells to hydrogen peroxide-induced apoptosis. Genes Immun 8(7):560–569
Funding
This work was financially supported by grants from the Beijing Municipal Science and Technology Commission (NO. Z221100007422109), Interdisciplinary Clinical Research Innovation Team Project (CYDXK202206), the National Natural Science Foundation of China (No. 82003057), and Beijing Hospitals Authority Youth Programme (QML20210307).
Author information
Authors and Affiliations
Contributions
Wenjing Zhao and Ziwei Liang contributed equally to this study. Wenjing Zhao, Ziwei Liang, and Jiannan Yao contributed to the conception and design of the study. Wenjing Zhao, Yongshi Yao, Ling Duan and Guangyu An conducted the statistical analysis. Ziwei Liang, Jiannan Yao, and Yang Ge performed the experiments. All authors wrote the paper; Jiannan Yao and Guangyu An revised the paper. All authors contributed to the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no competing interest.
Ethical Approval
All patient tissues obtained in this study were paraffin-embedded fixed tissues after surgery, and no other intervention or experiment would be carried out on the patients. Ethical approval was obtained from the Ethics Committee of Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China. The ethics application number is 2023–1-29–2.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, W., Liang, Z., Yao, Y. et al. GGT5: a potential immunotherapy response inhibitor in gastric cancer by modulating GSH metabolism and sustaining memory CD8+ T cell infiltration. Cancer Immunol Immunother 73, 131 (2024). https://doi.org/10.1007/s00262-024-03716-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03716-3