
Abstract
In multiple myeloma (MM), B cell maturation antigen (BCMA)-directed CAR T cells have emerged as a novel therapy with potential for long-term disease control. Anti-BCMA CAR T cells with a CD8-based transmembrane (TM) and CD137 (41BB) as intracellular costimulatory domain are in routine clinical use. As the CAR construct architecture can differentially impact performance and efficacy, the optimal construction of a BCMA-targeting CAR remains to be elucidated. Here, we hypothesized that varying the constituents of the CAR structure known to impact performance could shed light on how to improve established anti-BCMA CAR constructs. CD8TM.41BBIC-based anti-BCMA CAR vectors with either a long linker or a short linker between the light and heavy scFv chain, CD28TM.41BBIC-based and CD28TM.CD28IC-based anti-BCMA CAR vector systems were used in primary human T cells. MM cell lines were used as target cells. The short linker anti-BCMA CAR demonstrated higher cytokine production, whereas in vitro cytotoxicity, T cell differentiation upon activation and proliferation were superior for the CD28TM.CD28IC-based CAR. While CD28TM.CD28IC-based CAR T cells killed MM cells faster, the persistence of 41BBIC-based constructs was superior in vivo. While CD28 and 41BB costimulation come with different in vitro and in vivo advantages, this did not translate into a superior outcome for either tested model. In conclusion, this study showcases the need to study the influence of different CAR architectures based on an identical scFv individually. It indicates that current scFv-based anti-BCMA CAR with clinical utility may already be at their functional optimum regarding the known structural variations of the scFv linker.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) represents an incurable plasma cell neoplasm originating in the bone marrow. Almost all patients ultimately relapse with especially poor survival noted in patients with a high-risk cytogenetic profile or treatment-resistant disease [1]. Improved understanding of this disease has resulted in continuous evolution of treatment options leading to a new therapeutic repertoire [1]. Among them, chimeric antigen receptor (CAR) T cell therapy has emerged as a novel treatment modality with potential for long-term disease control. B cell maturation antigen (BCMA) is the most investigated target antigen for MM, mainly expressed by plasma cells and some mature B cells [2]. The U.S. Food and Drug Administration (FDA) approved the anti-BCMA CAR T cell products [3] idecabtagene vicleucel (ide-cel) [4] and ciltacabtagene autoleucel (cilta-cel) [5] for relapsed/refractory MM in 2021 and 2022, respectively. The first approved product ide-cel consists of a BCMA targeting single chain variable fragment (scFv), a CD8 alpha hinge/spacer and transmembrane domain (TM), a CD137 (41BB) intracellular costimulatory domain (IC or ICD), and a CD3zeta cytoplasmic domain (CD3ζ) [3, 4]. In terms of efficacy, only 33% of the patients treated with ide-cel exhibited a complete remission and 26% of all patients achieved MRD negativity [4]. ScFv-based anti-BCMA CAR T cell therapy remains a promising treatment approach, although coming with limitations including therapeutic resistance and toxicity [2, 4,5,6]. Besides the disease and patient’s characteristics, the CAR design influences its functionality [7,8,9,10]. The scFv, length and composition of the hinge/spacer, TM domain and ICD can impact CAR T cell functionality independently of each other [7, 11, 12].
It has been reported that a short amino acid linker (short linker) between the light (VL) and the heavy (VH) chain of the scFv part of 41BBIC-based anti-CD22 CAR T cells can lead to intermolecular CAR interactions that promote autonomous 41BB signaling, priming T cells for enhanced anti-tumoral effects [13]. In contrast, short linker versions of anti-CD33 and anti-CD19 CAR T cells did not exert such an effect [13]. This suggests that shortening the linker length of the scFv can induce CAR intermolecular pairing, but that this may vary between constructs and target antigens. It is thus promising to assess a short linker with a scFv-based BCMA-targeting CAR.
Another key aspect of CAR T cell functionality is the utilized costimulatory domain: CD28 is frequently employed both in preclinical models and in clinical products and leads to a more profound and rapid therapeutic impact, while 41BB (CD137) exhibits prolonged T cell persistence that is conducive to durable remissions [14]. However, this outcome is not uniformly observed across studies [15, 16], again indicating inter-CAR variability. Although CAR architecture targeting the same antigen can have a divergent impact on performance and efficacy, the optimal composition of such an scFv-based anti-BCMA CAR has not been investigated.
We aimed to perform a BCMA-target specific comparison of such CAR variations to evaluate the impact on anti-BCMA CAR T cell activity and to determine an optimized scFv-based anti-BCMA CAR design.
Materials and methods
Cell lines
MM cell lines RPMI 8226, U-266 (U266), JJN-3 (JJN3) and JK-6 (JK6) have been obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ). Acute monocytic leukemia cell (AML) line THP-1 have been previously described [17]. All cell lines were cultured in a humidified incubator (37 °C, 5% CO2) and in RPMI 1640 medium containing 10–20% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. All cell lines were lentivirally transduced with a pCDH-EF1a-eFly-eGFP plasmid [18]. After transduction, enhanced green fluorescent protein (eGFP)-positive cells were single cell sorted using a BD FACSAria III cell sorter, and expression of firefly luciferase (fLuc) was verified using a Bio-Glo luciferase assay system. Cells were frozen in 90% FBS with 10% DMSO and stored at − 80 °C or in liquid nitrogen for long-term storage. Cell lines were regularly checked for mycoplasma contamination. Authentication of cell lines by short tandem repeat (STR) profiling analysis was conducted in house.
Generation of CAR constructs
All CAR constructs were second-generation CAR vector systems as previously described [17, 19,20,21] and either generated using conventional cloning techniques or codon-optimized and cloned into pMP71 retroviral vectors using commercial cloning services (Twist Bioscience, USA). c-Myc tags were used to detect CAR expression. Anti-BCMA CAR constructs were based on the patented sequence of the approved construct idecabtagene vicleucel [22]. CD8TM.41BBIC-based CAR vectors with either a short (referred as short linker CD8TM.41BBIC) or a long (referred as long linker CD8TM.41BBIC) amino acid linker in the extracellular antigen recognition domain between the VL and VH chain of the scFv section, CD28TM.41BBIC-based and CD28TM.CD28IC-based CAR vector systems were generated and used in primary human T cells. Besides the short linker construct, the other anti-BCMA CARs have the same long linker based on ide-cel. The long linker CD8TM.41BBIC therefore served as universal control. Anti-CD19 CAR T cells were designed based on anti-CD19-CAR-FMC63-28Z CAR T cells and used as negative controls [23], as previously described [17]. Single-cell clones were generated and screened for the highest level of virus production by determining T cell transduction efficiency, as previously described [20].
CAR T cell generation and expansion
For virus production, retroviral pMP71 vectors carrying the CAR sequence (kindly provided by C. Baum, Hannover) were stably introduced into packaging cell lines 293Vec-Galv and 293Vec-RD114, as previously described [24]. T cells were isolated from peripheral blood mononuclear cells (PBMCs) of healthy donors by density gradient centrifugation, enriched by anti-CD3 microbeads (Miltenyi Biotec, Germany) and activated by Dynabeads human T-Activator CD3/CD28 (Life Technologies, Germany) before transduction. Human T cell transduction has been previously described [17, 19,20,21]. Untransduced T cells (UT) were isolated and activated like CAR T cells without transduction on day 2. T cells were expanded 1–2 weeks in human T cell medium (hTCM) containing RPMI 1640 with 2.5% human serum, 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, 1% sodium pyruvate and 1% non-essential amino acid solution supplemented with recombinant human IL-2 (PeproTech, Germany/Novartis, Switzerland) used in a final concentration of 180 U/ml and IL-15 (PeproTech, Germany/Miltenyi Biotech, Germany) used in a final concentration of 2 ng/ml. Expansion steps were performed in the same manner for both untransduced and CAR T cells.
T cell stimulation and killing assay using tumor cells
For co-culture experiments, 1.5 − 2.5 × 104 tumor cells were plated in flat bottom 96-well plates. T cell numbers transduced with the indicated CAR constructs or untransduced T cells were added at different effector cell to target cell ratios (E:T ratios). Co-cultures were performed in hTCM without cytokines. Cytokines were washed out before the assay. Killing was assessed by luciferase-based killing assays using Bio-Glo™ Luciferase Assay System according to manufacturer’s protocol (Promega Corporation, USA).
T cell stimulation assay using plate-bound recombinant protein
Flat bottom 96-well plates were coated overnight at 4 °C with Fc-tagged recombinant human BCMA (SinoBiological, China) with 2.0 µg/ml diluted in 50 µl PBS per well (0.1 µg/well). The next day, the protein-containing PBS was removed, plates were blocked with 70 µl 2% bovine serum albumin (BSA) dissolved in PBS for 30 min and washed after removal of the blocking solution with 100 µl PBS. After removal of the PBS serving as washing solution, 105 T cells were resuspended in hTCM without cytokines and added into the protein-coated wells. Following an incubation time, culture supernatants were obtained for storage at − 20 °C, and cells were used for flow cytometry-based analysis.
Proliferation assay
For long-term co-culture experiments, 2.5 × 104 U266 tumor cells were plated in flat bottom 96-well plates. T cell numbers transduced with the indicated CAR constructs or untransduced T cells (UT) were added at an E:T ratio of 2:1. Co-culture was performed in hTCM without cytokines for > 200 h. No further addiction or re-challenge of T cells with tumor cells was performed. At the end of cultivation time, proliferation and immunophenotype were assessed by flow cytometry and cytokine production was detected in culture supernatants by ELISA after storage at -20 °C.
Cytokine protein level quantification
IFN-γ, interleukin (IL)-2 and granzyme B (Grzm B) release in co-culture supernatants was quantified by enzyme-linked immunosorbent assay (ELISA) according to manufacturer´s protocol (IFN-γ and IL-2: BD Biosciences, USA; Grzm B: R&D Systems, USA).
Flow cytometry
For staining, cells were transferred into U-bottom 96-well plates and washed with ice-cold PBS and centrifuged (400 g, 5 min, 4 °C). Blocking of Fc receptors with TruStain FcX (BioLegend, USA) was performed (15 min). Dead cells were excluded after staining with a fixable viability dye (eFluor™ 780; eBioscience, USA). Surface proteins were stained (20 min, 4 °C). The following fluorophore-conjugated antibodies reactive to human antigens were used: anti-CCR7 (G043H7), anti-PD-1 (EH12.2H7), anti-CD8 (SK1), anti-TIM-3 (F38-2E2), anti-CD45RA (HI100), anti-CD3 (HIT3a or OKT3), anti-CD62L (DREG-56), anti-CD69 (FN50), anti-CD45 (HI30) and anti-CD4 (A161A1) (all from BioLegend). Anti-c-Myc (SH1-26E7.1.3) from Miltenyi Biotec was used for CAR detection. Target antigen expression was detected by BCMA-PE (19F2), CD19-PerCP-Cy5.5 (HIB19), and CD33-PE (P67.6). Quantification of absolute cell counts was carried out by using Count Bright™ Absolute Counting Beads (Thermo Fisher Scientific, USA). Cells were analyzed on a BD FACSCanto™ or BD LSRFortessa™ II flow cytometer, and data were analyzed with FlowJo software (version 10.7.2).
Animal experiments
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were originally bought from Charles River Laboratories or Janvier or bred at the local facilities. Animals were housed in specific pathogen-free facilities and in groups of 2–5 animals per cage. Mice were held in facilities with a 12 h dark/12 h light cycle including a 30 min twilight phase at noise levels below 50 dB. Air velocity was held below 0.2 m/s. Air humidity in the facilities was between 45 and 60%, and the average temperature was held between 20 and 22 °C. We used 5–10 week-old female mice (n = 30) as recipients of matching appropriate tumor cell lines as described for each experiment. For bioluminescence imaging (BLI), mice were anesthetized using an isoflurane–oxygen mixture (1.5–2.5%) following intraperitoneal (i.p.) injection of BLI substrate (Xenolight D-Luciferin Potassium Salt, PerkinElmer, USA) into each mouse, according to the manufacturer’s protocol. An in vivo imaging system platform Lumina X5 (IVIS, PerkinElmer, USA) was used to measure BLI signal. The xenograft model using 2 × 106 U266 tumor cells was established by i.v. injection. 2 × 106 active CAR T cells were transferred at indicated times and numbers. Mice that had to be removed from animal experiments due to non-tumor-related toxicities (for example, did not have measurable BLI signal at the exclusion timepoint) were censored. Censored mice are indicated in the respective BLI images. All experiments were carried out randomized. For survival analyses, defined criteria (decrease in body weight or in general health condition) were taken as surrogates for survival and recorded in Kaplan–Meier plots. Percentage of tumor growth inhibition (TGI) was calculated with the formula [1 − (change of tumor volume in treatment group/change of tumor volume in control group)] × 100. Detailed experimental procedure can be found in the results section as a scheme before each experiment.
Preparation of single-cell suspensions
Harvested organs and material of mice were processed to single-cell suspensions as previously described [20, 21]. Blood and bone marrow were diluted with PBS followed by erythrocyte lysis. Spleens were passed through 30 µm cell strainers, followed by erythrocyte lysis. Flow cytometry analysis is described above.
Statistical analysis
At least three biological replicates for each experiment were analyzed unless otherwise indicated in the figure legends. Duplicates or triplicates for each group were performed. Two-tailed Student’s t-test was used for comparisons between two groups, while two-way ANOVA with Bonferroni posttest (multiple time points) or one-way ANOVA with Tukey’s posttest (single time points) was used for comparisons across multiple groups. A log-rank (Mantel-Cox) test was used to compare survival curves. Sample size was determined by t-test (two-tails) using the software G*Power 3.1 with given alpha, power, and effect size. All statistical tests were performed with GraphPad Prism 9 (GraphPad Software Inc., USA). p-values < 0.05 were considered statistically significant and represented as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Graphs were designed with GraphPad Prism 9 (GraphPad Software Inc., USA) and Adobe Illustrator v26.0.2 (Adobe, USA). Where not otherwise indicated, results are presented as mean ± standard error of the mean (SEM).
Results
Comparative analysis of four anti-BCMA CAR constructs using different target cell lines
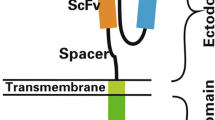
In contrast to CD19-positive diseases, only 41BBIC-based anti-BCMA CAR products are approved (Suppl. Figure 1A) [3,4,5]. Here, we compared scFv-based BCMA-CAR constructs with either CD28 or CD8 as TM and 41BB or CD28 as ICD (Fig. 1A). We designed a novel anti-BCMA CAR with a short instead of a long linker between the VL and VH chain of the scFv (Fig. 1A). The long linker design is based on ide-cel; therefore, the long linker CD8TM.41BBIC anti-BCMA CAR was used as universal control. All constructs were successfully transduced (mean transduction efficiency > 60%) into primary human T cells (Fig. 1B + C). Primary myeloma cells vary in BCMA surface expression [25]. Therefore, several MM cell lines were used as target cell lines with a range of different BCMA, CD19 and CD33 expression profiles (Fig. 1D). While RPMI 8226 and U266 having the highest BCMA expression, for JJN3 and JK6 expression was low (Fig. 1D and Suppl. Figure 1B). All cell lines were CD19 negative so that an anti-CD19 CAR could be used as a negative control CAR for testing specificity (Fig. 1A-C). AML cell line THP-1 was used as an antigen-negative non-B cell or plasma cell-based control cell line (Fig. 1D and Suppl. Figure 1B).

Evaluation of different anti-BCMA CAR constructs. A Schematic of CD28TM.CD28IC-based (red), CD28TM.41BBIC-based (yellow), and CD8TM.41BBIC-based anti-BCMA CAR constructs with either a long (blue) or short (green) linker between the light (VL) and the heavy (VH) chain of the scFv part. Anti-CD19 CAR T cells were used as negative control CAR T cells (gray). B + C Transduction efficiency in primary human T cells. Data for B one representative donor or C for n = 12 (anti-BCMA CAR) and n = 10 (anti-CD19 CAR) shown as means ± SEM. D Expression of human BCMA, CD19 and CD33 was detected on different multiple myeloma cells (RPMI 8226, U266, JJN3, JK6) and on the AML cell line THP-1. Expression was compared to isotype controls
CD28 as costimulatory domain mediated a stronger activation then 41BB
All anti-BCMA constructs showed specific activation by recombinant BCMA after 24 h of stimulation, measured by expression of the T cell activation markers CD69 and CD107a (LAMP-1) as well as T cell exhaustion marker PD-1, which is also a marker of early T cell activation (Fig. 2A–F). Expression of CD69 with 93 ± 3% (Fig. 2A) and CD107a with 87 ± 4% (Fig. 2B) was the highest for CD28TM.CD28IC-based CAR T cells (Fig. 2A–B). PD-1 expression was relatively low compared to CD69 and CD107a (Fig. 2C). CD28TM.CD28IC-based CAR T cells also revealed a higher proportion of multifunctional (CD69 + CD107a + PD-1 +) cells with 46 ± 7% upon activation compared to 41BBIC-based anti-BCMA CAR constructs with between 8 and 12% (p < 0.01; Fig. 2G). Increased expression of T cell activation markers led to higher IFN-γ production of CD28TM.CD28IC-based CAR T cells (Fig. 2H) when stimulated with 2.0 µg/ml (p < 0.05) of recombinant BCMA. There were no differences between 41BBIC-CAR variants or scFv linker lengths (Fig. 2A-H).

Specific CAR T cell activation by BCMA. A + D CD69, B + E CD107a (LAMP-1), C + F PD-1 expression as % of CD3+ T cells A-C and as MFI D-F, G CD69 ± CD107a (LAMP-1) ± PD-1 expression and H IFN-γ ELISA after stimulation of anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green), anti-CD19 CAR T cells (gray) and untransduced T cells (black) with 2.0 µg/ml recombinant BCMA (n = 3 donors). Each experiment of subfigures A-H was performed in triplicates. Values in all graphs represent means ± SEM (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Only selected p-values are shown. Statistical comparison was performed by a 2-way ANOVA with Bonferroni multiple comparison correction
CD28TM.CD28IC-based CAR T cells showed higher in vitro cytotoxic capacity
We next compared the in vitro cytotoxic capacity. In general, all constructs were very potent against the used MM cells with an E:T ratio of 0.25:1 (Fig. 3A-D), particularly after 48 h and 72 h of co-culture reducing the window to perform comparative analysis (Suppl. Figure 2 + 3). CD28TM.CD28IC-based CAR T cells showed a higher cytotoxic capacity compared to the other constructs (Fig. 3A–D) particularly with RPMI 8226 (CD28TM.CD28IC vs CD28TM.41BBIC vs long linker CD8TM.41BBIC vs short linker CD8TM.41BBIC: 44 ± 2% vs 14 ± 5% vs 35 ± 4% vs 30 ± 3% for 24 h). BCMA low-expressing JJN3 and JK6 cell lines were killed similarly by the constructs and particularly JJN3 showed unspecific killing by anti-CD19 CAR and untransduced T cells due to higher intrinsic T cell sensitivity unrelated of targeting (Fig. 3 and Suppl. Figure 2). Similar effects were seen with E:T ratios of 2:1, 1:1, and 0.5:1 (Suppl. Figure 3). No clear differences were seen for the two linker lengths. As expected, AML cell line THP-1 could not be sufficiently killed (Suppl. Figure 4A).

Highest cytotoxic capacity with CD28TM.CD28IC-based anti-BCMA CAR T cells. A-D Anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green) were co-cultured with A RPMI 8226, B U266, C JJN3 or D JK6 tumor cell lines for 24 h at an indicated E:T ratio of 0.25:1. Anti-CD19 CAR T cells (gray) were used as negative controls, respectively. Cell lysis was quantified by luciferase-based killing assay. Subfigures show data of one representative donor out of three independent experiments. Each experiment was performed in triplicates. Values in all graphs represent means ± SEM (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Only p-values for comparison of different anti-BCMA CAR constructs were shown except U266. Cytotoxic capacity of anti-BCMA CAR in comparison to anti-CD19 CAR T cells for RPMI 8226, JJN3 and JK6 were significant (p < 0.05). Statistical comparison was performed by a 2-way ANOVA with Bonferroni multiple comparison correction
Shortening the scFv linker of 41BBIC-based anti-BCMA CAR increased cytokine production
Shortening of the scFv linker led to an increased in vitro IFN-γ production (Fig. 4). After 72 h of co-culture in an E:T ratio of 2:1, short linker CAR T cells displayed a higher IFN-γ production compared to long linker versions in presence of RPMI 8226 (Fig. 4A), U266 (Fig. 4B), JJN3 (Fig. 4C) and JK6 (Fig. 4D). Donor variability led to less distinct effects when pooled data were analyzed (Suppl. Figure 5). Also, highest granzyme B and IL-2 production was seen with short linker CD8TM.41BBIC CAR T cells when co-cultured with RPMI 8226 for 72 h (Suppl. Figure 6A + B). Co-culture with THP-1 revealed a similar cytokine production as T cell only conditions (Suppl. Figure 4B-E).

Highest IFN-γ production with short linker CD8TM.41BBIC-based anti-BCMA CAR. A-D IFN-γ ELISA with 24 h, 48 h, and 72 h co-culture supernatants of anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green), anti-CD19 CAR T cells (gray) and untransduced T cells (black) with A RPMI 8226, B U266, C JJN3, or D JK6 in an E:T ratio of 2:1 for one representative donor out of three different donors. Each experiment was performed in triplicates. Values in all graphs represent means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Only p-values for comparison of different anti-BCMA CAR constructs were shown. Statistical comparison was performed by a 2-way ANOVA with Bonferroni multiple comparison correction
Long-term co-culture revealed higher proliferative capacity of CD28IC-based CAR T cells
Previous reports have demonstrated that 41BBIC-based CAR T cells typically exhibit a longer persistence compared to CD28IC-based CAR T cells [14]. We assessed the impact of a long-term co-culture (> 200 h) with MM cells. Remarkably, the CD28TM.CD28IC-based anti-BCMA construct showed better proliferative capacity (p < 0.05) of all CD3 + T cells (33 ± 16) and CAR T cells (71 ± 34) compared to 41BBIC-based T cells (< 10) and CAR T cells (< 12) (Fig. 5A). No differences were seen of long and short linker versions (Fig. 5A). In contrast, the different CAR formats showed similar expression levels of PD-1 (Fig. 5B) and cytokine release (Fig. 5C). Analysis of T cell subtypes revealed no relevant changes during long-term co-culture (Fig. 5D).

CD28TM.CD28IC-based anti-BCMA CAR T cells performed superior in long-term co-cultures. A Proliferative capacity, B PD-1 expression, C IFN-γ release and D T cell subtypes of anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green), anti-CD19 CAR T cells (gray) and untransduced T cells (black) co-cultured for more than 200 h with U266 measured by FACS. A Proliferation is shown as T cells per bead, CAR T cells per bead or untransduced T cells per bead ratio and was normalized to conditions without tumor measured by FACS. Data is shown for three independent donors (n = 3). Each experiment was performed in triplicates. Values in all graphs represent means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Statistical comparison was performed by a 2-way ANOVA with Bonferroni multiple comparison correction
CD8TM.41BBIC CAR T cells had a higher proportion of central memory-like T cells
We further assessed the phenotypic composition of the T cell product. For analysis of T cell subtypes, the CD8 to CD4 T cell ratio was similar for CD28IC-based and 41BBIC-based anti-BCMA CAR T cells after co-culture with MM cells (Fig. 6A). After co-culture with tumor cells, a trend towards a higher proportion of central memory-like T cells for CD8TM.41BBIC-based CAR T cells was seen compared to the CD28TM-based CAR T cells (Fig. 6B). Single marker analysis of activation and exhaustion marker expression was similar for all conditions (Fig. 6C), while the proportion of multifunctional T cells (CD69+ 41BB+ PD-1+) and exhausted T cells (PD-1+ TIM-3+) was highest for CD28TM.CD28IC CAR T cells, albeit without reaching statistical significance (Fig. 6D + E).

Phenotypic changes of CAR T cells after activation. Phenotype of anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green), anti-CD19 CAR T cells (gray) and untransduced T cells (black) are shown after 48 h of co-culture in the presence of U266, JJN3 and JK6 or no tumor cells (T cell only conditions) of three donors (n = 3). A CD8+ and CD4+ T cell subtypes are shown. B T cell subpopulations for T cell differentiation status are shown. Naïve-like T (TN) cells were defined as CD45RA+CCR7+ , central memory-like T (TCM) cells as CD45RA-CCR7+ , effector memory-like T (TEM) cells as CD45RA-CCR7- and effector-like T (TEff) cells as CD45RA+CCR7-. Changes in T cell subtypes and T cell subpopulations for anti-BCMA CAR T cells were not significant (ns). C Expression of 41BB (CD137), CD69, PD-1 and TIM-3 is shown. Only p-values for comparison of different anti-BCMA CAR constructs were shown. Comparison was not significant (ns). D Expression of CD69 ± 41BB ± PD-1 is shown. Changes for anti-BCMA CAR T cells were not significant (ns). E Expression of PD-1 ± TIM-3 is shown. Changes for anti-BCMA CAR T cells were not significant (ns). Each experiment was performed in triplicates. Values in all graphs represent means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Selected p-values are shown in the figures or described in the Results section. Statistical comparison was performed by a 2-way ANOVA with Bonferroni multiple comparison correction
Prolonged in vivo persistence and tumor control of CD28TM.41BBIC-based CAR T cells
To assess in vivo persistence and proliferation of anti-BCMA CAR T cell constructs, U266-bearing immunodeficient NSG mice were treated with 2 × 106 active anti-BCMA CAR T cells or PBS (Fig. 7A). In the first days after treatment, CD28TM.CD28IC-based CAR T cells cleared the tumor slightly quicker than the 41BBIC-based CAR constructs (Fig. 7B-D). However, tumor clearance was comparable for all tested CAR constructs with a complete clearance of the tumor after 13 days compared to the PBS group (p < 0.0001) (Fig. 7B-D) and tumor growth inhibition (TGI) of nearly 100% (Fig. 7E).

In vivo efficiency of anti-BCMA CAR T cells. A Experimental layout for B-E. Anti-BCMA CAR T cells (CD28TM.CD28IC: red; CD28TM.41BBIC: yellow; long linker CD8TM.41BBIC: blue; short linker CD8TM.41BBIC: green) or PBS (black) were intravenously (i.v.) injected in U266-bearing NSG mice. B BLI images, C + D quantification of tumor burden and E percentage of tumor growth inhibition (% of TGI) in U266 tumor-bearing mice after treatment with different CAR T cells (n = 5 mice per group). F Experimental layout for (G–O). G BLI images, H quantification of tumor burden, I survival curves and J + K body weight analysis of U266 bearing mice re-challenged 46 days after initial T cell injection with again tumor cells (n = 5 mice per group) and treatment naïve tumor only control mice (n = 5 mice per group). PBS-treated mice of A-B underwent no re-challenge. L BCMA + tumor cells per bead ratio for blood, bone marrow and spleen at time point of termination of the experiment on day 97 (+ 51), M T cells per bead ratio for blood in the first week and second week after CAR T cell injection. N T cells per bead ratio and O CAR T cells per bead ratio for cells detected in blood, bone marrow and spleen at time point of termination of the experiment. Values in all graphs represent means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Selected p-values are shown in the figures. Statistical comparison except for survival curves was performed by a 2-way ANOVA with Bonferroni multiple comparison correction. For Kaplan–Meier curves, statistical significance was calculated with log-rank test
To better evaluate T cell persistence, we re-challenged the anti-BCMA CAR T cell-treated survivor mice again with U266 cells on day 46 (+ 0) after initial T cell injection (Fig. 7F). Mice initially treated with PBS (black color) were not re-challenged (Fig. 7B, right column). As control mice for the re-challenge experiment (gray color), U266 tumor cells were injected into new NSG mice that had neither been treated with T cells nor PBS (Fig. 7G, right column). Remarkably, the CD28TM.41BBIC CAR T cells showed the best tumor control (Fig. 7G + H). Despite short linker CD8TM.41BBIC CAR T cells, anti-BCMA CAR T cell-treated groups showed better tumor control than the tumor only control group (gray; p < 0.05) in the re-challenge setting. No advantage in survival was seen as most of the mice were still alive at time point of termination (Fig. 7I). Weight analysis revealed (Fig. 7J + K) a significant reduction for CD28TM.41BBIC-based CAR T cells. At termination, BCMA+ tumor cells were only detectable in the bone marrow of the original PBS-treated mice (day 97) and the tumor only control mice for the re-challenge experiment (day 51 after re-challenge) (Fig. 7L). Bleeding of mice in the first and second week after T cell injection did not indicate differences for T cell detection (Fig. 7M). Upon termination of the experiment, detection of both T and CAR T cells in blood, bone marrow and spleen was only possible for the CD28TM.41BBIC and long linker CD8TM.41BBIC anti-BCMA CAR T cells (Fig. 7N + O), which was particularly evident for CAR T cells in the spleen (p < 0.05). In summary, shortening of the scFv linker did not lead to an advantage in an in vivo setting.
Discussion
While commercial scFv-based anti-BCMA CAR T cell products have achieved excellent clinical results in pivotal studies, therapeutic failure and relapse are still observed [6]. CAR T cell therapy has evolved in recent years due to continuous improvements in CAR vector design [7, 8]. However, the optimal construction of an anti-BCMA CAR has yet to be defined. To evaluate the impact of CAR structure variations, we set out to perform a BCMA-target specific comparison on CAR T cell activity in vitro and in vivo so as to determine an optimized scFv-based anti-BCMA CAR design. We evaluated different scFv linker lengths and costimulatory domains.
In preclinical studies, anti-BCMA CAR T cell product ide-cel (bb2121) showed low antigen-independent signaling and potent in vitro killing of multiple myeloma tumor cells across a range of BCMA expression levels [26]. In comparison with studies testing CD28-based anti-BCMA CAR T cells, 41BB-based CAR T cells displayed improved persistence and a lower rate of toxicities including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [27]. Even though this and other clinical studies revealed promising results leading to approval of two commercial 41BB-based anti-BCMA CAR products, durable persistence of scFv-based anti-BCMA CAR T cells is generally poor and recurrence is common (for ide-cel median progression-free survival was 8.5 months and the median overall survival was 12.5 months in the real-world setting) [28]. Therefore, scFv-based anti-BCMA CAR constructs still require optimization.
Optimization of CAR design to enhance synthetic receptor-driven T cell functionality has been an important focus of the field [7,8,9,10]. Even though commercial CAR T cell products and preclinical CAR T designs share common features, all of them slightly differ from each other. Each part including the scFv, the hinge/spacer and TM domain and the ICD of the CAR can influence the functionality [11, 12]. While first-generation CAR T cells had only the CD3ζ chain, introduction of ICD particularly of CD28 and 41BB (CD137) markedly improved CAR T cell functionality [7]. CD28IC-containing CAR constructs were associated with enhanced levels of Th2-like cytokines like IL-4 and IL-10, and their kinetics of activation are more rapid than CAR vectors using 41BB as costimulatory domains [29,30,31]. Rapid activation of CD28IC-containing CAR T cells may make these cells more likely to initiate early-onset CRS in treated patients. Comparison of CAR constructs with identical scFvs in tumor treatment models revealed that 41BBIC-containing CAR T cells developed into more central memory-like T cells [14] with enhanced respiratory capacity, increased fatty acid oxidation, enhanced mitochondrial activity and better persistence compared to CD28IC-containing CAR T cells [14]. For now, two commercial 41BBIC-based anti-BCMA CAR products are approved. However, as persistence of these CAR T cells is generally poor [28], further improvement is required. We tested different combinations of CD28 and CD8 for TM domains as well as CD28 and 41BB for ICD for scFv-based anti-BCMA CAR constructs based on the design of the commercially available ide-cel product. In our study, CD28-based CAR T cells outperformed the 41BBIC-based CAR T cells regarding in vitro cytotoxicity, proliferative capacity, and overall activation status. Overall cytotoxicity after 48 and 72 h of co-culture was very high for all constructs. The long-term cytotoxic experiments cannot reasonably differentiate between the role of proliferation; therefore, proliferation might be a confounding factor regarding cytotoxicity. However, we observed better persistence of 41BBIC-based CAR T cells in vivo, as expected. These discrepancies may be model-related and therefore might be evaluated also with other cell lines in vivo or simply related to the utilized scFv as all constructs are based on the ide-cel scFv. By changing the scFv, the effect could be different due to a different ability for tonic signaling as well as avidity and affinity towards the target antigen.
Besides therapy-associated toxicities, another reason for leaking CAR T cell efficiency is tonic signaling by antigen-independent clustering of a CAR on the T cell [9, 10]. Multiple domains of the CAR have been associated with provoking or at least contributing to tonic signaling [10]. It can mediate spontaneous release of effector cytokines and the expression of surface markers associated with T cell exhaustion [10]. This led to poor in vivo activity and lack of in vivo function particularly when CD28 is used as ICD [10] due to distinct receptor biochemistry (not linker-driven clustering) of these CD28IC-based CARs [32, 33]. Replacement of a CD28 ICD with a 41BB ICD prevented the development of CAR T cell dysfunction suggesting a protective impact of tonic 41BBIC-based CAR signaling. Singh and colleagues could demonstrate that antigen-independent tonic signaling improved the function of 41BBIC-based anti-CD22 CAR T cells and can be enhanced by shortening the linker length between the VL and VH chain of the scFv part [13]. This shortening of the linker does not universally lead to clustering in every CAR product [13]. As such, it was recommended that each new CAR and target antigen should likely be evaluated individually for clustering and tonic signaling [13]. Thus, we evaluated the effect of the linker shortening in CD8TM.41BBIC-containing scFv-based anti-BCMA CAR T cells. We could not see a superior effector function of short linker CAR T cells except for a trend towards an increased in vitro cytokine production. This trend was not superior in a long-term co-culture setting measuring IFN-γ. The decay of the protein over time will underestimate the absolute amount produced. While decay will not be influenced by the CAR design, allowing direct comparison between constructs, it does not correct for cell numbers, nor for potential differences between cell populations in IFN-γ production. The findings of superior cytokine production in short-term co-cultures are in line with short linker anti-CD19 CAR T cells showing no tonic signaling with equivalent in vitro and in vivo cytotoxicity against CD19 + tumor cells but higher cytokine production compared to the long linker versions [13]. The superior short linker anti-CD22 CAR had a VH–VL composition [13], while the here tested anti-BCMA CAR had a VL–VH composition which may also influence the ability to perform intermolecular VL and VH pairing [34]. Each variable domain of the scFv contains three complementarity-determining regions (CDRs) forming loops and making up the unique antigen-binding site [35]. Additionally, four framework regions (FRs) in the scFv comprising beta-strands and additional loop regions are crucial for scFv stability [35]. The VH–VL orientation is often favored because it leaves more distance between the scFv linker and the third CDR of the heavy chain, which usually is crucial for antigen-binding. However, VL–VH orientation has been associated with increased expression and antigen-binding in some cases [36]. In other cases, similar surface expression and antigen-binding have been reported for either orientation [37]. Therefore, an ideal orientation of the VL and VH domains in scFvs still must be evaluated. The linker length itself heavily influences the oligomerization behavior of scFvs.
The scFv of most CARs is murine-derived and therefore can lead to host immunogenic responses and engineered T cell disappearance. Development of less immunogenic CAR constructs like CARs composed of camelid heavy-chain variable domains (VHH) has the potential to overcome this obstacle. Replacement of the targeting domain of a CAR with a single-chain antibody such as VHHs can also prevent tonic signaling as domain swapping and oligomerization is intrinsically not possible [38, 39]. Because VHH-based CARs are intrinsically not capable to oligomerize, shortening their linker length probably will not exert a major effect.
Another reason for the divergent results could be the target antigen itself as BCMA is a cell surface receptor and therefore has a different structure and function as CD19 and CD22, respectively. Since BCMA is primarily expressed on normal and malignant plasma cells and some mature B cells, it represents a suitable target for MM [40]. In this work, low BCMA expressing cell lines were also cleared by anti-BCMA CAR T cells. Primary myeloma cells vary in BCMA surface expression [25, 41]. Preclinical data of ide-cel (bb2121) revealed robust in vitro killing of MM cells independent of the BCMA expression levels or the presence of soluble BCMA [26, 27] also described by others [41]. Additionally, in the CARTITUDE-1 trial testing cilta-cel (NCT03548207), response rates were also independent of baseline BCMA expression [5]. Also, tumor cell killing is dictated by T cell and tumor cell intrinsic characteristics. In this specific setting, it seems that JJN3 and JK6 have a higher intrinsic T cell sensitivity unrelated to antigen targeting compared with RPMI 8226 or U266. We reasoned that these differences are not linked to BCMA nor BCMA targeting. A further promising target for MM is GPRC5D [42, 43]. Profound comparison of different CAR constructs designs and scFv linker lengths should also be performed with this clinically relevant target [42, 43]. It could be shown that structural alterations (biallelic loss of BCMA or GPRC5D) and antigen loss resulting from non-truncating missense point mutations in BCMA extracellular domain also mediate resistance to targeted immunotherapies [44].
Here, we used a c-Myc tag in the CAR construct to detect transduction efficiency. Antigen-based CAR expression is difficult to standardize and can be very CAR-dependent. Adding a c-Myc tag to the CAR sequence does not seem to negatively influence CAR performance in different CAR constructs [17, 19, 21]. Any minute alteration to a CAR sequence may potentially have functional consequences. The strategy was used by many in the meantime [45,46,47]. However, a systematic comparison on the CAR performance has not been performed to date.
In this present study, detailed analysis of in vitro and in vivo functionality of the anti-BCMA CAR constructs was performed; however, no further analysis concerning biophysical properties of CAR binding was conducted (i.e., affinity, avidity, and antigen density). Affinity and avidity can also substantially influence efficacy [48, 49]. It could be shown that moderate-affinity antigen-binding domain CAR have less off-target toxicity but were more effective [48]. The positive effects of short linker-mediated tonic signaling were reduced when using an affinity enhancement approach [13]. This could further help to understand the role of the CAR design and its interaction with the target antigen for CAR T cell effector function.
Our findings on anti-BCMA CAR T cells are not only relevant for MM but also for B cell NHL, as BCMA is also expressed in a relevant percentage of lymphoma samples [26]. It was shown that anti-BCMA CAR T cells do not react with normal PBMCs [26]. However, as many CD19, CD20 and/or CD22 targeting CAR T cell products are currently under investigation and four different anti-CD19 CAR T cell products are already approved, the role of BCMA as target antigen for the treatment of B cell lymphoma is for now less clear compared to CD19. Nonetheless, CD19/BCMA dual-targeting CAR approaches have been already tested for relapsed/refractory B cell NHL [50].
Anti-BCMA CAR are not functionally very sensitive to alterations in structure when BCMA-targeting scFv is conserved. While CD28 and 41BB (CD137) costimulation come with different in vitro and in vivo characteristics, we did not observe consistent domain-specific differences in terms of outcomes in the examined murine model. Shortening of the amino acid linker between the VL and VH chain of the scFv part of 41BBIC-based CAR is promising for CAR T cell therapy. However, the short scFv linker strategy could not relevantly improve the clinically approved scFv-based anti-BCMA CAR construct in this study. This may indicate that the scFv linker design of this CAR cannot be further improved by altering the length of the amino acid linker as proposed here and would require more elaborate modules such as cytokine signaling (so-called armored or fourth-generation CARs). This study showcases the need to study the influence of different CAR architectures based on an identical scFv individually. Optimizing anti-BCMA CAR constructs may improve outcomes; however, there are many variables that limit the success.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary files. Additional materials and data are available upon reasonable request.
Abbreviations
- BCMA:
-
B cell maturation antigen
- BLI:
-
Bioluminescence imaging
- BSA:
-
Bovine serum albumin
- CAR:
-
Chimeric antigen receptor
- CD3ζ:
-
CD3zeta
- CDR:
-
Complementarity-determining region
- CRS:
-
Cytokine release syndrome
- DMEM:
-
Dulbecco's Modified Eagle's Medium
- DSMZ:
-
German Collection of Microorganisms and Cell Cultures GmbH
- ECD:
-
Extracellular domain
- eGFP:
-
Enhanced green fluorescent protein
- E:T ratio:
-
Effector cell to target cell ratio
- FDA:
-
U.S. Food and Drug Administration
- FBS:
-
Fetal bovine serum
- FITC:
-
Fluorescein isothiocyanate
- fLuc:
-
Firefly luciferase
- FR:
-
Framework region
- Grzm B:
-
Granzyme B
- hTCM:
-
Human T cell medium
- ICANS:
-
Immune effector cell-associated neurotoxicity syndrome
- IC:
-
Intracellular costimulatory domain
- ICD:
-
Intracellular costimulatory domain
- IL:
-
Interleukin
- Ig:
-
Immunoglobulin
- i.v.:
-
Intravenous
- i.p.:
-
Intraperitoneal
- MM:
-
Multiple myeloma
- NHL:
-
Non-Hodgkin lymphoma
- NSG:
-
NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz
- PBMC:
-
Peripheral blood mononuclear cell
- PBS:
-
Phosphate-buffered saline
- ROB:
-
Regierung von Oberbayern
- RPMI:
-
Roswell Park Memorial Institute
- scFv:
-
Single chain variable fragment
- SEM:
-
Standard error of the mean
- STR:
-
Short tandem repeat
- TCM :
-
Central memory-like T cell
- TEff :
-
Effector-like T cell
- TEM :
-
Effector memory-like T cell
- TGI:
-
Tumor growth inhibition
- TH :
-
T helper cell
- TLS:
-
Tumor lysis syndrome
- TM:
-
Transmembrane domain
- TME:
-
Tumor microenvironment
- TN :
-
Naïve-like T cell
- TSCM :
-
Stem cell memory-like T cell
- UT:
-
Untransduced T cell
- V H :
-
Heavy chain of the scFv part
- VHH:
-
Camelid heavy-chain variable domains
- V L :
-
Light chain of the scFv part
References
Chim CS, Kumar SK, Orlowski RZ et al (2018) Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 32:252–262. https://doi.org/10.1038/leu.2017.329
Teoh PJ, Chng WJ (2021) CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J 11:84. https://doi.org/10.1038/s41408-021-00469-5
Cappell KM, Kochenderfer JN (2023) Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol 20:359–371. https://doi.org/10.1038/s41571-023-00754-1
Munshi NC, Anderson LD Jr, Shah N et al (2021) Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 384:705–716. https://doi.org/10.1056/NEJMoa2024850
Berdeja JG, Madduri D, Usmani SZ et al (2021) Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 398:314–324. https://doi.org/10.1016/S0140-6736(21)00933-8
Rejeski K, Jain MD, Smith EL (2023) Mechanisms of resistance and treatment of relapse after CAR T-cell therapy for large b-cell lymphoma and multiple myeloma. Transplant Cell Ther. https://doi.org/10.1016/j.jtct.2023.04.007
Stock S, Schmitt M, Sellner L (2019) Optimizing manufacturing protocols of chimeric antigen receptor T cells for improved anticancer immunotherapy. Int J Mol Sci. https://doi.org/10.3390/ijms20246223
Stock S, Kluver AK, Fertig L, Menkhoff VD, Subklewe M, Endres S, Kobold S (2023) Mechanisms and strategies for safe chimeric antigen receptor T-cell activity control. Int J Cancer. https://doi.org/10.1002/ijc.34635
Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, Kobold S (2020) Determinants of response and resistance to CAR T cell therapy. Semin Cancer Biol 65:80–90. https://doi.org/10.1016/j.semcancer.2019.11.004
Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S (2019) Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. https://doi.org/10.3390/cells8050472
Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR (2015) The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3:125–135. https://doi.org/10.1158/2326-6066.CIR-14-0127
Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, Brenner MK, Leen AM, Vera JF (2016) Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 5:e1253656. https://doi.org/10.1080/2162402X.2016.1253656
Singh N, Frey NV, Engels B et al (2021) Antigen-independent activation enhances the efficacy of 4–1BB-costimulated CD22 CAR T cells. Nat Med 27:842–850. https://doi.org/10.1038/s41591-021-01326-5
Kawalekar OU, O’Connor RS, Fraietta JA et al (2016) Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44:380–390. https://doi.org/10.1016/j.immuni.2016.01.021
Cheng Z, Wei R, Ma Q et al (2018) In vivo expansion and antitumor activity of coinfused CD28- and 4–1BB-engineered CAR-T cells in patients with B cell leukemia. Mol Ther 26:976–985. https://doi.org/10.1016/j.ymthe.2018.01.022
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS (2016) Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126:3130–3144. https://doi.org/10.1172/JCI83092
Gottschlich A, Thomas M, Grunmeier R et al (2023) Single-cell transcriptomic atlas-guided development of CAR-T cells for the treatment of acute myeloid leukemia. Nat Biotechnol. https://doi.org/10.1038/s41587-023-01684-0
Sandhofer N, Metzeler KH, Rothenberg M et al (2015) Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 29:828–838. https://doi.org/10.1038/leu.2014.305
Lesch S, Blumenberg V, Stoiber S et al (2021) T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat Biomed Eng 5:1246–1260. https://doi.org/10.1038/s41551-021-00737-6
Cadilha BL, Benmebarek MR, Dorman K et al (2021) Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci Adv. https://doi.org/10.1126/sciadv.abi5781
Stock S, Benmebarek M, Kluever A et al (2022) Chimeric antigen receptor T cells engineered to recognize the P329G-mutated Fc part of effector-silenced tumor antigen-targeting human IgG1 antibodies enable modular targeting of solid tumors. J Immunother Cancer. https://doi.org/10.1136/jitc-2022-005054
Morgan R, Friedman K (2020) BCMA Chimeric antigen receptors. US patent US 10,646,558 B
Kochenderfer JN (2015) Chimeric antigen receptors targeting CD19. Patent Cooperation Treaty WO 2015(187528):A1
Ghani K, Wang X, de Campos-Lima PO, Olszewska M, Kamen A, Riviere I, Caruso M (2009) Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther 20:966–974. https://doi.org/10.1089/hum.2009.001
Seckinger A, Delgado JA, Moser S et al (2017) Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody em801 for multiple myeloma treatment. Cancer Cell 31:396–410. https://doi.org/10.1016/j.ccell.2017.02.002
Friedman KM, Garrett TE, Evans JW, Horton HM, Latimer HJ, Seidel SL, Horvath CJ, Morgan RA (2018) Effective targeting of multiple b-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther 29:585–601. https://doi.org/10.1089/hum.2018.001
Raje N, Berdeja J, Lin Y et al (2019) Anti-BCMA CAR T-Cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med 380:1726–1737. https://doi.org/10.1056/NEJMoa1817226
Hansen DK, Sidana S, Peres LC et al (2023) Idecabtagene vicleucel for relapsed/refractory multiple myeloma: Real-world experience from the myeloma car t consortium. J Clin Oncol 41:2087–2097. https://doi.org/10.1200/JCO.22.01365
Milone MC, Fish JD, Carpenito C et al (2009) Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17:1453–1464. https://doi.org/10.1038/mt.2009.83
Finney HM, Akbar AN, Lawson AD (2004) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 172:104–113. https://doi.org/10.4049/jimmunol.172.1.104
van der Stegen SJ, Hamieh M, Sadelain M (2015) The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14:499–509. https://doi.org/10.1038/nrd4597
Frigault MJ, Lee J, Basil MC et al (2015) Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res 3:356–367. https://doi.org/10.1158/2326-6066.CIR-14-0186
Long AH, Haso WM, Shern JF et al (2015) 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21:581–590. https://doi.org/10.1038/nm.3838
Schirrmann T, Menzel C, Hust M, Prilop J, Jostock T, Dubel S (2010) Oligomeric forms of single chain immunoglobulin (scIgG). MAbs 2:73–76. https://doi.org/10.4161/mabs.2.1.10784
Chowdhury PS, Vasmatzis G (2003) Engineering scFvs for improved stability. Methods Mol Biol 207:237–254. https://doi.org/10.1385/1-59259-334-8:237
Zhang K, Geddie ML, Kohli N et al (2015) Comprehensive optimization of a single-chain variable domain antibody fragment as a targeting ligand for a cytotoxic nanoparticle. MAbs 7:42–52. https://doi.org/10.4161/19420862.2014.985933
Fujiwara KStacarts, Masutani M, Tachibana M, Okada N, (2020) Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun 527:350–357. https://doi.org/10.1016/j.bbrc.2020.03.071
Ajina A, Maher J (2018) Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol Cancer Ther 17:1795–1815. https://doi.org/10.1158/1535-7163.MCT-17-1097
Rajabzadeh A, Rahbarizadeh F, Ahmadvand D, Kabir Salmani M, Hamidieh AA (2021) A VHH-Based Anti-MUC1 chimeric antigen receptor for specific retargeting of human primary T Cells to MUC1-positive cancer cells. Cell J 22:502–513. https://doi.org/10.22074/cellj.2021.6917
Tai YT, Anderson KC (2015) Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 7:1187–1199. https://doi.org/10.2217/imt.15.77
Zhao WH, Liu J, Wang BY et al (2018) A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol 11:141. https://doi.org/10.1186/s13045-018-0681-6
Mailankody S, Devlin SM, Landa J et al (2022) GPRC5D-targeted CAR T cells for myeloma. N Engl J Med 387:1196–1206. https://doi.org/10.1056/NEJMoa2209900
Smith EL, Harrington K, Staehr M et al (2019) GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aau7746
Lee H, Neri P, Ahn S et al (2022) Role of TNFRSF17 and GPRC5D structural and point mutations in resistance to targeted immunotherapies in multiple myeloma (MM). Blood 140:252–253. https://doi.org/10.1182/blood-2022-167602
Nunoya JI, Masuda M, Ye C, Su L (2019) Chimeric antigen receptor T cell bearing herpes virus entry mediator co-stimulatory signal domain exhibits high functional potency. Mol Ther Oncolytics 14:27–37. https://doi.org/10.1016/j.omto.2019.03.002
Feng Y, Liu X, Li X, Zhou Y, Song Z, Zhang J, Shi B, Wang J (2021) Novel BCMA-OR-CD38 tandem-dual chimeric antigen receptor T cells robustly control multiple myeloma. Oncoimmunology 10:1959102. https://doi.org/10.1080/2162402X.2021.1959102
Yekehfallah V, Pahlavanneshan S, Sayadmanesh A, Momtahan Z, Ma B, Basiri M (2022) Generation and functional characterization of PLAP CAR-T cells against cervical cancer cells. Biomolecules. https://doi.org/10.3390/biom12091296
Mao R, Kong W, He Y (2022) The affinity of antigen-binding domain on the antitumor efficacy of CAR T cells: Moderate is better. Front Immunol 13:1032403. https://doi.org/10.3389/fimmu.2022.1032403
Greenman R, Pizem Y, Haus-Cohen M et al (2021) Shaping functional avidity of CAR T cells: affinity, avidity, and antigen density that regulate response. Mol Cancer Ther 20:872–884. https://doi.org/10.1158/1535-7163.MCT-19-1109
Chen X, Ping Y, Li L et al (2022) P1218: first-in-human study of CD19/BCMA dual targeting fast CAR T GC012F for patients with relapsed/refractory B cell non-hodgkin’s lymphoma. Hemasphere. 6:1104–1105. https://doi.org/10.1097/01.HS9.0000847736.14020.3e
Acknowledgements
SS was supported by the Else Kröner-Fresenius Clinician Scientist Program Cancer Immunotherapy, the Munich Clinician Scientist Program (MCSP), the DKTK School of Oncology, the Förderprogramm für Forschung und Lehre (FöFoLe) and the Momente Mentoring Program of the Medical Faculty of the LMU Munich. LF, LM, VDM and RG were supported by the Förderprogramm für Forschung und Lehre (FöFoLe) of the LMU Munich. DMCDS was supported by the DKTK School of Oncology. We acknowledge the iFlow Core Facility of the LMU University Hospital of the LMU Munich for assistance with the generation of flow cytometry data. We acknowledge the Core Facility Flow Cytometry of the LMU University Hospital of the LMU Munich, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)–499549016. Figures were created with BioRender.com.
Funding
Open Access funding enabled and organized by Projekt DEAL. SS declares research funding from the Else Kröner-Fresenius-Stiftung, the DKTK School of Oncology, the Novartis InCa Förderpreis 2022 for young researchers, the Förderprogramm für Forschung und Lehre (FöFoLe) of the Medical Faculty of the LMU Munich (grant number 1168) and the SFB-TRR 338 (start-up funding). AG was supported by Förderprogramm für Forschung und Lehre (FöFoLe) of the LMU Munich (grant number 1138), the Bavarian Center for Cancer Research (BZKF), the Deutsche Forschungsgemeinschaft (DFG; grant number GO 3823/1–1). KR and DMCDS received a fellowship from DKTK School of Oncology and KR was funded by the Else Kröner Forschungskolleg (EKFK) within the Munich Clinician Scientist Program (MCSP). DMCDS received funding from the Förderprogramm für Forschung und Lehre (FöFoLe) of the LMU Munich (grant number 1089). This work was supported by a Deutsche Forschungsgemeinschaft (DFG) research grant provided within the Sonderforschungsbereich SFB-TRR 388/1 2021 – 452881907 (to MS and ST), and the DFG research grant 451580403 (to MS). The work was also supported by the Bavarian Elite Graduate Training Network (to MS), the Wilhelm Sander-Stiftung (project no. 2018.087.1, to MS), the Else Kröner-Fresenius-Stiftung (to MS), the Bavarian Center for Cancer Research (BZKF; to MS) and by the Bruno and Helene Jöster Foundation (to MS). Funding was received from the DFG research grant 391587558 (to ST and MB), the Thomas Kirch Stiftung (to ST and MB) and Myeloma Study Group within the Bavarian Cancer Research Center (BZKF; to ST). This study was furthermore supported by the Marie Sklodowska-Curie Program Training Network for Optimizing Adoptive T Cell Therapy of Cancer funded by the H2020 Program of the European Union (Grant 955575, to SK); by the Hector Foundation (to SK); by the International Doctoral Program i-Target: Immunotargeting of Cancer funded by the Elite Network of Bavaria (to SK and SE); by Melanoma Research Alliance Grants 409510 (to SK); by the Else Kröner-Fresenius-Stiftung (2021_EKFK_01, to SK); by the German Cancer Aid (AvantCAR.de, to SK); by the Ernst-Jung-Stiftung (to SK); by the LMU Munich’s Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (to SE and SK); by the Bundesministerium für Bildung und Forschung (to SE and SK); by the Go-Bio Initiative (to SK), by the m4 award of the Bavarian Ministry for Economical Affairs (to SK); by the Wilhelm Sander-Stiftung (2022.051.1, to SK); by the European Research Council Grant 756017, 101100460 and 101124203 (to S.K.); Deutsche Forschungsgemeinschaft (DFG; KO5055-2–1 and 510821390, to S.K.); by the SFB-TRR 338/1 2021–452881907 (to SK); by the Fritz Bender Foundation (to SK); by the Bavarian Research Foundation (BAYCELLator, to SK); by the Deutsche José Carreras Leukämie-Stiftung (to SK); by the Bruno and Helene Jöster Foundation (to SK); by the BZKF (TANGO, to SK) and by Monika Kutzner Foundation (to SK). The remaining authors declare that no funds, grants, or other support were received during the preparation of this manuscript relevant for this project.
Author information
Authors and Affiliations
Contributions
SS and LF designed the experiments, performed the experiments, and analyzed the data; SS designed the figures, wrote the primary manuscript and revised the manuscript; AG, JD, FM, LM, VDM, and RG assisted with the experiments; KR, DMCDS, and ST supported the project. SS and SK edited the manuscript; KR, DMCDS, ST, MB, SE, MS, and SK reviewed the manuscript; SK supervised the project; and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
SS, LF, AG, JD, FM, LM, VDM, RG, DMCDS and MB have no relevant financial or non-financial interests to disclose relevant for this work. KR received research funding Kite/Gilead, honoraria from BMS/Celgene and Novartis and travel support from Kite/Gilead and Pierre-Fabre. KR serves as a consultant for BMS/Celgene. ST received honoraria from AMGEN, BMS/Celgene, GSK, Janssen, Pfizer, Sanofi, and Takeda. MS received industry research support from Amgen, Bristol-Myers Squibb/Celgene, Gilead, Janssen, Miltenyi Biotec, Morphosys, Novartis, Roche, Seattle Genetics, and Takeda, and serves as a consultant/advisor to AvenCell, CDR-Life, Ichnos Sciences, Incyte Biosciences, Janssen, Molecular Partners, and Takeda. She serves on the speakers’ bureau at Amgen, AstraZeneca, BMS/Celgene, Gilead, GSK, Janssen, Novartis, Pfizer, Roche, and Takeda. SK has received honoraria from TCR2 Inc., Novartis, Galapagos, Cymab, Miltenyi, BMS, and GSK. SK and SE are inventors of several patents in the field of immuno-oncology. SK and SE received license fees from TCR2 Inc. and Carina Biotech. SK and SE received research support from TCR2 Inc., Tabby Therapeutics, Plectonic GmbH, Catalym GmbH, and Arcus Bioscience for work unrelated to the manuscript.
Ethics approval
Approval was obtained from the ethics committee of LMU Munich. The procedures used in this study adhere to the tenets of the Declaration of Helsinki. All animal experimental studies were approved and performed in accordance with guidelines and regulations implemented by the Regierung von Oberbayern (ROB).
Consent to publish
The manuscript does not contain any individual person’s data in any form. No patient consent is necessary for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stock, S., Fertig, L., Gottschlich, A. et al. Comparative performance of scFv-based anti-BCMA CAR formats for improved T cell therapy in multiple myeloma. Cancer Immunol Immunother 73, 100 (2024). https://doi.org/10.1007/s00262-024-03688-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03688-4