
Abstract
Background
Immunotherapy has changed the paradigm of treating non-small cell lung cancer (NSCLC). But, selecting patients who will achieve long-term benefits from treatment remains unsatisfactory. Here, we investigated the possible use of the soluble form of CD8 antigen (sCD8) in predicting durable disease control after PD-1/PD-L1 blockade. CD8 is a marker of the cytotoxic T lymphocytes. Its soluble form (sCD8) is secreted under activation of the immune system but also has immunosuppressive properties. The data about serum sCD8 in patients dosed with anti-PD-1/PD-L1 drugs are lacking.
Methods and results
We included 42 NSCLC patients and collected samples at baseline and for the first 3 months of atezolizumab immunotherapy. The serum sCD8 concentrations were measured with the ELISA kit and correlated with treatment outcomes. Patients with durable (≥ 12 months) disease control presented lower serum sCD8 than those without long-term benefits. The sCD8 levels measured at the end of cycle 2 (sCD8.2) were the earliest time point that successfully differentiated patients (3.76 vs. 9.68 ng/mL, respectively, p < 0.001). Individuals with low sCD8.2 (≤ 4.09 ng/mL) presented longer progression-free survival (HR = 0.061, p < 0.001) and overall survival (HR = 0.104, p < 0.05) compared to individuals with high sCD8.2 (median values unreached vs. 4.4 months and 14.4 months for PFS and OS, respectively).
Conclusions
Serum sCD8 could be an early biomarker of durable disease control after anti-PD-L1 treatment. Higher sCD8 in patients with worse outcomes could suggest the inhibitory effect of sCD8 on cytotoxic T-cells activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy has changed the paradigm of treating non-small cell lung cancer (NSCLC). Introducing monoclonal antibodies targeting the programmed cell death 1 receptor/programmed cell death ligand-1 (PD-1/PD-L1) axis has significantly improved the treatment outcomes in both men and women [1]. However, the percentage of patients who do not respond to treatment is still high, with almost half of the patients presenting progression as their best response [2]. A recent meta-analysis showed that the median progression-free survival (PFS) was only 3.4 months, while the overall survival (OS)—10 months in real-world settings, with PD-1/PD-L1 inhibitors as the second-line therapy in advanced and metastatic NSCLC [2]. Thus, in the era of personalized medicine, the selection of patients who will achieve long-term benefits from anti-PD-1/PD-L1 therapy remains poor and unsatisfactory.
Over the last several years, multiple biomarkers have been tested to identify patients who would benefit from PD-1/PD-L1 inhibitors [3]. The only approved biomarker that has been included so far in the immunotherapy protocols, i.e., the expression of PD-L1 in lung cancer tissue, requires invasive biopsy. Moreover, the efficacy of this biomarker remains debatable, which implies that more specific and selective biomarkers are needed. Among others, the blood-based biomarkers are under intensive investigation as blood is easy to collect and enables multiple non-invasive sampling [4]. Here, we explored the usefulness of one another circulating molecule that has never been tested in relation to the PD-1/PD-L1 blockade, i.e., the soluble form of a cluster of differentiation 8 (CD8) protein (sCD8).
The CD8 antigen is a well-recognized marker of the cytotoxic T lymphocytes (CTLs, cytotoxic CD8+ T-cells) [5], which are considered a backbone of cancer immunotherapy [6]. The CD8 surface glycoprotein exists either as a heterodimer (built from one alpha and one beta chain) or a homodimer (composed of two alpha chains) and plays a crucial role in the neoplastic cells’ killing [5, 7]. Namely, a heterodimer CD8αβ acts as a co-receptor for the T-cell receptor (TCR): along with the TCR, it binds to the peptide-loaded major histocompatibility complex class I (MHC-I) at the surface of the antigen-presenting cells, which enhances the TCR signaling [5, 6] (see also Fig. 4a in the ‘Discussion’ section). The process initiates in the lymphoid tissue, where activated naïve CD8+ T-cells rapidly proliferate and differentiate into the effector CTLs, subsequently migrating into the target sites (e.g., the tumor microenvironment) [8, 9]. Conversely to CD8αβ, a homodimer CD8αα might negatively regulate the T-cell activation via being a TCR co-repressor [7]. Moreover, it modulates the activation of the natural killer cells through the inhibitory receptor KIR3DL1 [5].
Except for the membrane-bound CD8, also the soluble form of this protein has been identified, which originates from the alternative splicing [10] or membrane shedding [11]. Both the monomeric [10,11,12] and the dimeric forms [12,13,14] of sCD8 have been detected in human sera [12, 14] and in vitro experiments [10, 11, 13, 14]. The sCD8 is secreted upon the activation of CD8+ T lymphocytes [13, 15]; thus, its elevated levels might be a surrogate marker of immunological activation. In line with this hypothesis, some authors reported abnormal (too high) concentrations of sCD8 in conditions linked with the enhanced activity of the immune system, e.g., systemic lupus erythematosus [16, 17], rheumatoid arthritis [18], Graves’ disease [19], or acute renal allograft rejections [20]. On the other hand, the potential immunosuppressive role of sCD8 has been highlighted by others [21,22,23,24], suggesting its immunomodulatory character.
Even though immunotherapy aims to support the activation of the immune system [25], there are no published reports evaluating the role of sCD8 protein (i.e., a molecule clearly linked with the immune system) in the successful treatment with anti-PD-1/PD-L1 agents, neither in lung cancer nor in other malignancies. Thus, the focus of our study was to assess the usefulness of circulating sCD8 as a biomarker of long-term benefits from therapy with immune checkpoint inhibitors. We enrolled NSCLC patients treated with the anti-PD-L1 drug, atezolizumab (ATEZO), and prospectively collected several blood samples. The results of our study might serve as a basis for future research that would help to understand the exact role of serum sCD8 in the successful therapy with PD-1/PD-L1 inhibitors and—ultimately—improve treatment outcomes in cancer patients.
Materials and methods
Patient selection and study design
This was a prospective, observational, single-center study designed to evaluate the usefulness of selected serum soluble proteins as predictors of successful immunotherapy in patients with advanced NSCLC. Between February 2019 and June 2020, serial blood samples were collected at baseline and during the first 3 months of immunotherapy from 42 adult patients eligible for therapy with ATEZO within the Polish national NSCLC drug program. The detailed inclusion criteria for the program were described previously [26]. Subjects’ characteristic was presented in the ‘Results’ section. Patients received treatment at the Eugenia and Janusz Zeyland Wielkopolska Center of Pulmonology and Thoracic Surgery in Poznan, Poland. Participating in the project had no impact on medical decisions. The study was conducted in accordance with the Declaration of Helsinki and the approval of the Bioethics Committee at Poznan University of Medical Sciences (decisions 80/19 and 251/19). All subjects signed informed consent.
The anti-PD-L1 treatment (1200 mg of intravenous ATEZO every three weeks) was continued until disease progression, death, unacceptable (≥ grade 3) toxicity, or significant deterioration of the quality of life. Response to ATEZO was evaluated every 3 months according to the Response Evaluation Criteria in Solid Tumors (RECIST, v1.1) [27]. Clinical responses were categorized as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). Patients who continued therapy were followed up for at least 24 months from the start of treatment. The primary endpoint was the long-term disease control rate, i.e., the percentage of patients with at least SD after 12 months from the start of treatment (patients with SD, PR, or CR who were still on ATEZO treatment at the evaluation time point, i.e., one year from the start of immunotherapy). The secondary endpoints were: progression-free survival (PFS), defined as the time from the first ATEZO dose to confirmed progression or death, and overall survival (OS), defined as the time from the start of immunotherapy to death.
Sample collection
Blood samples (n = 173) were collected at baseline (first day of ATEZO administration, pre-dose) and at the end of cycles 1–4. After clotting, serum was separated by centrifugation at 1700 × g for 15 min, aliquoted, and kept at − 80 °C until used.
Determination of serum sCD8 levels
The concentrations of sCD8 were determined by sandwich ELISA using a kit from MyBioSource (San Diego, CA, USA; Cat. No. MBS016364). All experiments were performed according to the manufacturer’s instructions. The optical density was read at 450 nm with the BioTek 800TS plate reader (BioTek Instruments, USA), and the log–log regression was used to calculate the sCD8 levels. The calibration range was 3.12–100 ng/mL. Samples were prepared in duplicate, and their mean was used for statistical analyses.
Statistical analysis
The statistical analysis was performed with MedCalc 20.106 software (MedCalc Software Ltd, Ostend, Belgium). For all analyses, a p value < 0.05 was considered significant. The normal distribution of variables was assessed with the Shapiro–Wilk test; the normally distributed data were then compared with the t-Student test and expressed as mean ± standard deviation (sd), while the non-normally distributed data with Mann–Whitney U test and expressed as median (interquartile range). Categorical data were compared with Fisher’s exact test and expressed as numbers (%). The changes in sCD8 level over time compared to baseline were assessed with the Wilcoxon test.
The receiver operator characteristic curve (ROC) analysis with the Youden index was used to determine the optimal cutoff for the sCD8 protein. The durable disease control, i.e., at least SD 12 months from the start of ATEZO treatment, was chosen as an outcome for ROC analysis. The Kaplan–Meier method was then implemented to investigate the association between low/high sCD8 levels and progression or survival, and the differences between the groups were assessed with a log-rank test. Subjects who did not experience the event of interest (death or progression for analyses with OS and PFS, respectively) were censored in Kaplan–Meier analyses. The exception was patients who died within the next 3 months after the last dose of ATEZO—all these patients were considered progressed regardless of the reason for ending immunotherapy. The univariable and multivariable Cox proportional hazard models were used to confirm the prognostic value of sCD8 protein on OS and PFS. All variables from the univariable Cox analyses with a p < 0.1 were included in the multivariable model.
Results
Baseline patients’ characteristics
Table 1 presents the characteristics of the study population. The baseline data were retrospectively extracted from the hospital records and a short questionnaire completed by participants on the day of enrollment. All individuals were of Caucasian origin and had undergone one prior systemic treatment. The vast majority of patients were diagnosed with adenocarcinoma (64.3%) and stage IV of the disease (95.2%). Following the requirements of the NSCLC drug program in Poland, patients with adenocarcinoma did not have mutations in EGFR (epidermal growth factor receptor) or ALK (anaplastic lymphoma kinase) genes. The PD-L1 status was unknown for most participants as PD-L1 expression in cancer tissue does not play a role in the qualification for ATEZO treatment (supplementary file in ref. [26]).
Clinical benefits and survival
Figure 1 demonstrates the results of the treatment response recorded during the study period. Short-term clinical benefits (evaluated 3 months from the start of treatment) were observed in 50.0% of patients, while durable disease control (≥ 12 months)—in 21.4%. Almost 40% of patients who showed benefits (at least SD) at their first response evaluation progressed within the next 3 months. The objective response rate was 14.3%, but PR lasted only 3 months in one patient, while in the other two, it was delayed ≥ 12 months. Five patients (11.9%) continued ATEZO immunotherapy for more than two years.

Flowchart of response to ATEZO in 42 NSCLC patients included in the study. Patients who stopped immunotherapy are highlighted in red. ATEZO atezolizumab, NSCLC non-small cell lung cancer, PD progressive disease, PR partial response, SD stable disease
Two patients (4.8%) stopped immunotherapy due to severe (grade ≥ 3) toxicity (both received only one dose of ATEZO), and the other two (4.8%) due to progression and toxicity. Twenty-two patients (52.4%) discontinued treatment due to confirmed progression, three (7.1%)—because of the unacceptable quality of life, and eight (19.0%)—due to death, including seven individuals who died before their first response evaluation. The median PFS in the whole study population was 4.5 (3.0–12.1) months, and the median OS was 16.8 (4.6–28.7) months. At the time of data analysis, five patients (11.9%) were still on the ATEZO treatment, and twelve patients (28.6%) were alive. One subject (2.4%) was lost to follow-up after discontinuation of ATEZO immunotherapy.
Determination of sCD8 levels
In total, 173 serum samples were analyzed for sCD8 concentrations. The sCD8 levels in two samples (1.2%) exceeded the upper limit of quantitation (100 ng/mL); thus, a new (previously unfrozen) aliquot of those sera was diluted per the manufacturer’s protocol and re-analyzed. Thirteen samples (7.5%) were below the lower limit of quantitation (3.12 ng/mL), with the range of the observed concentrations 2.16–3.08 ng/mL and a median value of 2.59 ng/mL (95% confidence interval, 95% CI: 2.44–2.85 ng/mL). The sCD8 concentration in all BQL (below the quantification limit) samples was at least twice the kit’s sensitivity (1 ng/mL); therefore, we decided to include all BQL samples in statistical tests. As discussed in literature [28], BQL data carry relevant information, so ignoring those samples, especially in the biomarker study, could bias the obtained results.
We decided to exclude four samples from statistical analysis; the reason was a long delay in the next dose of ATEZO in one patient, which could have affected the sCD8 concentrations. The second dose of ATEZO was administered to this patient ten instead of three weeks after the first dose; thus, only the baseline serum sample from this patient (sCD8.0) was used in statistics.
Serum sCD8 level in the study population
The median baseline sCD8 level (sCD8.0) was 7.44 (4.45–14.58) ng/mL in all patients. After the start of immunotherapy, the median sCD8 concentrations were: 9.51 (4.62–14.67) ng/mL at the end of cycle one (sCD8.1); 6.66 (4.64–14.25) ng/mL at the end of cycle two (sCD8.2); 6.00 (4.14–16.70) ng/mL at the end of cycle three (sCD8.3), and 7.41 (5.21–14.16) ng/mL at the end of cycle four (sCD8.4), i.e., about 3 months from the start of treatment.
The sCD8 concentrations at baseline and during immunotherapy were independent of age (< 65 vs. ≥ 65), BMI (< 25 vs. ≥ 25), gender, NSCLC subtype (adenocarcinoma vs. others), and smoking status (current smokers vs. others) (Supplementary Materials, Fig. S1–S5).
Serum sCD8 levels and long-term disease control
Figure 2 presents the changes in the sCD8 levels within the first 3 months of ATEZO treatment in relation to durable (≥ 12 months) disease control. Baseline sCD8 values were not different between the groups. After the start of immunotherapy, there were no significant changes in the sCD8 concentrations compared to baseline in either group (p > 0.05 from the Wilcoxon test). However, patients who achieved long-term disease control presented significantly lower sCD8 concentrations at the end of cycles 2 (sCD8.2), 3 (sCD8.3), and 4 (sCD8.4) than patients who lacked the long-term benefits from treatment (p < 0.05 from Mann–Whitney U test). The median sCD8 values were: 5.81 versus 8.68 ng/mL at baseline (p > 0.05); 5.97 versus 9.89 ng/mL (p = 0.06) for sCD8.1; 3.76 versus 9.68 ng/mL (p < 0.001) for sCD8.2; 3.36 versus 8.70 ng/mL (p < 0.05) for sCD8.3; 4.48 versus 8.85 ng/mL (p < 0.05) for sCD8.4. There were no differences in sCD8 levels between patients who achieved and lacked the objective response during immunotherapy (data not shown).

Serum sCD8 during the first 3 months of immunotherapy with atezolizumab (1200 mg Q3W) in relation to long-term (≥ 12 months) disease control. Data are presented as medians (interquartile ranges). Patients with long-term benefits from immunotherapy are marked with triangles/dashed lines, while those who lacked long-term benefits from treatment are marked with circles/solid lines. Time points with a significant difference between the groups are highlighted with asterisks. Specific time points referred to: sCD8.0—baseline, sCD8.1 to sCD8.4—concentrations of sCD8 at the end of cycles 1–4; disease control was defined as at least stable disease
Cutoff selection
As sCD8 concentrations at the end of the second cycle were the earliest time point that successfully identified patients with durable disease control, the sCD8.2 levels were selected for survival analysis. The ROC analysis was performed to find the best sCD8.2 value to predict the long-term benefits from treatment (supplementary Fig. S6). We found sCD8.2 level ≤ 4.09 ng/mL (AUC = 0.928, 95% CI: 0.785–0.988, p < 0.001) as a threshold with the highest sensitivity (75.0%, 95% CI: 34.9–96.8%) and specificity (96.2%, 95% CI: 80.4–99.9%), and further classified patients into low- and high-sCD8.2 level subgroups (n = 7 and n = 27, respectively). The Kaplan–Meier and Cox proportional hazard regression analyses were subsequently performed to test the usefulness of the ROC-derived cutoff for PFS and OS.
Survival analysis
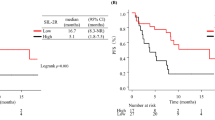
Kaplan–Meier analyses confirmed better outcomes in subjects with low sCD8.2 concentrations (Fig. 3). Patients with high sCD8.2 (> 4.09 ng/mL) achieved a median PFS of 4.4 months (95% CI: 3.0–6.0 months) and a median OS of 14.4 months (95% CI: 7.3–20.9 months), while patients with low sCD8.2 (≤ 4.09 ng/mL) reached neither the median PFS nor OS (with a 95% CI follow-up for this subgroup 16.3–34.5 months).

Kaplan–Meier curves for a progression-free survival (PFS) and b overall survival (OS) in non-small cell lung cancer patients dosed with atezolizumab stratified by high (> 4.09 ng/mL) and low (≤ 4.09 ng/mL) serum sCD8 levels at the end of cycle 2 (sCD8.2). Log-rank test showed significant differences in PFS (p < 0.001) and OS (p = 0.007) between patients with low- and high-sCD8.2
The univariable Cox proportional hazard analyses (Table 2) confirmed the low sCD8.2 level as a prognostic factor of longer PFS and OS. For PFS, the multivariable Cox regression was also performed to account for the other covariates’ influence. Patients with low sCD8 concentrations at the end of cycle 2 presented 93.9% lower hazard of progression (adjusted HR = 0.061, 95% CI: 0.012–0.304, p < 0.001), and 89.6% lower hazard of death (HR = 0.104, 95% CI: 0.014–0.777, p < 0.05) compared to patients with high sCD8.2.
Discussion
This is the first study to demonstrate the circulating soluble sCD8 protein as a predictor of successful anti-PD-1/PD-L1 therapy in cancer. NSCLC patients with durable (≥ 12 months) disease control presented lower on-treatment levels of serum sCD8 compared to those who lacked the long-term benefits from treatment. We identified the sCD8 concentrations measured at the end of cycle 2 to be predictive in terms of treatment efficacy: individuals with low sCD8.2 (≤ 4.09 ng/mL) presented longer PFS (adjusted HR = 0.061, p < 0.001) and OS (HR = 0.104, p < 0.05) compared to individuals with high sCD8.2. Thus, sCD8 seems to be a promising early biomarker of successful NSCLC therapy with ATEZO. Further studies are warranted to confirm our observation in a larger cohort of patients, in other types of cancer, and other PD-1/PD-L1 inhibitors.
Data regarding sCD8 protein and lung cancer are scarce. The serum sCD8 levels remain unchanged in NSCLC compared to healthy controls [29,30,31] and do not depend on the disease stage (III or IV) [29] or NSCLC subtype [29, 30]. Our study confirmed this observation: we found no differences in sCD8 levels between patients with adenocarcinoma and other NSCLC subtypes (supplementary Fig. S4). Moreover, the protein levels were similar in females and males (supplementary Fig. S3), normal weight versus overweight/obese individuals (supplementary Fig. S2), and younger and older patients (supplementary Fig. S1), which also corroborates with the available literature [14, 32, 33]. Active smoking did not impact the sCD8 levels (supplementary Fig. S5), which supports the observations from the general population [34] and contradicts those presented for pregnant women [35].
We demonstrated that NSCLC patients who achieved long-term disease control presented lower serum sCD8 at the end of cycles 2–4. Although no published papers assessed the circulating sCD8 levels in cancer patients on PD-1/PD-L1 inhibitors, lower baseline sCD8 was previously shown in responders to immunotherapy with interferon-alpha (IFN-α) [36] and higher on-treatment sCD8—in patients with progressive disease dosed with IFN-γ [37]. In contrast, other authors highlighted better outcomes with a higher baseline sCD8 [38] or a higher increase in serum sCD8 during treatment [39], but those patients were dosed with a different drug (interleukin-2, IL-2). The ambiguous results could suggest that the direction of correlation between sCD8 levels and immunotherapy success is not that obvious and might depend on additional factors, such as a type of malignancy and the used immunomodulator. Accordingly, higher sCD8 levels were favorable in melanoma and renal cell carcinoma patients but only during treatment with IL-2 [38, 39] and not with IFN-γ [37]. Moreover, different behavior of sCD8 was noted during immunotherapy with various drugs: there was a trend toward the sCD8 levels increasing after dosing with recombinant IL-2 [31, 39,40,41] or successful treatment with IFN-γ [37], but a similar trend was not observed in our cohort (Fig. 2). Interestingly, in patients on IFN-α, the sCD8 concentrations increased or decreased after successful immunotherapy depending on the initial sCD8 values [36].
Poor anti-PD-L1 treatment outcomes in patients with high serum sCD8 might be due to the inhibitory effect of sCD8 on immunological activation. Indeed, despite considering the soluble form of CD8 as a marker of immunological activation (e.g., [13, 15, 17, 20]), sCD8 also inhibited (in a dose-dependent manner [42]) the CTLs in vitro [22, 42] and attenuated the function of CD8+ T-cells in vivo [21]. A potential immunosuppressive role of sCD8 was previously suggested in some autoimmunological diseases, e.g., rheumatoid arthritis [18] or Hashimoto’s disease [24, 43]. In rheumatoid arthritis, despite the elevated sCD8 levels (reflecting, most likely, the immunological activation), the increase in sCD8 preceded the clinical improvement (suggesting the suppression of the immunological response) [18]. Similarly, patients with severe Hashimoto’s disease presented lower serum sCD8 compared to the mild form of the disease [24], and those with thyrotoxicosis (i.e., a condition in which thyrocytes are getting damaged mostly by cytotoxic T-cells) showed lower sCD8 compared to healthy controls [43].
The CD8 antigen serves as a co-receptor for TCR (Fig. 4a): it enhances the binding of MHC-I to TCR and increases TCR signaling [5], so the efficient (undisturbed) interaction between membrane-bound CD8 and MHC-I seems to be necessary to properly activate the naïve CD8+ T-cells. On the other hand, the sCD8 protein binds to MHC-I [42] and HLA-I (i.e., a ‘human version’ of MHC) [23], which may interfere with the binding of the CTL-derived membrane-bound CD8 to MHC-I/HLA-I (Fig. 4b). This interaction, in turn, could result in an ineffective antigen presentation and failed immunological activation [23], which may be the case in our patients who lacked the long-term benefits from treatment. As immunotherapy works through the activation of the immune system (and, compared to chemotherapy, does not kill cancer cells directly), any disturbances in the function of immune system might lead to worse treatment outcomes in patients dosed with PD-1/PD-L1 inhibitors.

Schematic presentation of the interaction between TCR and MHC-I during antigen presentation a in case of a proper (i.e., undisturbed) activation of the immune system b proposed mechanism in case of the sCD8 excess. a MHC-I (built from a three-domain α chain and a β2m chain) presents a peptide antigen to a heterodimeric TCR; the CD8 heterodimer serves as a co-receptor for TCR: it enhances T-cell antigen recognition by binding to MHC-I [6]. b The sCD8 protein binds to MHC-I, which might block the interaction between membrane-bound CD8 and MHC-I; as various sCD8 forms have been reported by different authors in human blood (for further details, please go to the main text), at the moment it is unclear which specific sCD8 form interacts with MHC-I. β2m β2-microglobulin, CD8 cluster of differentiation 8, MHC-I major histocompatibility complex class I, sCD8 soluble CD8 protein, TCR T-cell receptor
The interesting question remains which sCD8 form is mainly secreted in NSCLC patients undergoing immunotherapy with PD-1/PD-L1 inhibitors. The in vitro studies showed that the molecular weight of sCD8 secreted by the human T-cell lines was 27 kDa [11], 30 kDa (a form generated via the alternative splicing) [10], 52 kDa [14], or 54 kDa [13], indicating both the monomeric and dimeric forms. Moreover, Pui and colleagues [14] demonstrated the elevated levels of 52 kDa homodimer of sCD8 in lymphoid malignancies, while Schlesinger and colleagues [12] revealed that three different sCD8 forms were detected in human sera, i.e., 28–30 kDa, 57–62 kDa, and 66–70 kDa, the last one more widely distributed in the HIV-positive individuals. This observation suggests that a different proportion of various sCD8 molecules might be secreted under specific clinical conditions, e.g., in patients with the chronically stimulated immune system. The ELISA kit used by our research team had both the capture and detection antibody directed against CD8α, suggesting that it could—hypothetically—detect sCD8α monomer as well as sCD8 multimers, depending on the binding site.
Strengths and limitations
We acknowledge that our study had some limitations. Firstly, due to its pilot character, the sample size was small, and we could not include a validation cohort. Moreover, we did not define an adequately powered sample size a priori as the study was a pioneer in demonstrating the usefulness of sCD8 as a biomarker of successful anti-PD-L1 therapy. Also, the number of samples collected from specific patients was sometimes incomplete, as a few patients died or stopped receiving ATEZO before their first response evaluation. Lastly, the biological mechanism underlying better outcomes in patients with low sCD8 levels has not been investigated, and further experiments are necessary to reveal the role of sCD8 in successful cancer immunotherapy. If poor immunotherapy outcomes resulted from the inhibitory effect of sCD8 during T-cells activation, the agents selectively directed against sCD8 could potentially reverse that effect. This question, at the moment, remains open.
Despite all these limitations, our pilot study demonstrated, for the first time, that circulating sCD8 levels could successfully indicate patients with durable disease control after PD-L1 blockade and that the topic is worth exploring. The strength of the study was its longitudinal character, which allowed us to identify the earliest time point that differentiated patients into those with and without long-term benefits based on the sCD8 levels. Also, the vast majority of samples were collected before the COVID-19 pandemic outbreak, while the sample collection process was definitely ended before the number of COVID-19 cases in Poland became significant; thus, our results were unaffected by SARS-CoV-2 infection. As sCD8 increases in viral diseases (elevated levels were observed, e.g., in mononucleosis [44] and dengue hemorrhagic fever [45]), this condition was mandatory to obtain the unbiased results.
Conclusions
The serum on-treatment levels of sCD8 could be considered an early biomarker of durable (≥ 12 months) disease control in patients treated with anti-PD-L1 drugs. Low sCD8 after two cycles of ATEZO predicted longer PFS and OS in NSCLC, suggesting a possible inhibitory effect of high sCD8 concentrations on the activation of the immune system. Further studies are warranted to confirm our observations in a larger study population, as well as in patients receiving other anti-PD-1/PD-L1 agents and suffering from other types of cancer. However, the results of our study are promising and provide a step toward a better understanding of phenomena associated with successful anti-PD-1/PD-L1 immunotherapy.
Data availability
All relevant data are included in this article and presented as figures, tables, or supplementary materials. The data are available from the corresponding author upon reasonable request.
Abbreviations
- ALK :
-
Anaplastic lymphoma kinase gene
- ATEZO:
-
Atezolizumab
- BMI:
-
Body mass index
- BQL:
-
Below the quantification limit
- CD8:
-
Cluster of differentiation 8
- CI:
-
Confidence interval
- CR:
-
Complete response
- CTLs:
-
Cytotoxic T lymphocytes
- EGFR :
-
Epidermal growth factor receptor gene
- HIV:
-
Human immunodeficiency virus
- HLA-I:
-
Human leukocyte antigen class I
- HR:
-
Hazard ratio
- IFN:
-
Interferon
- IL-2:
-
Interleukin-2
- MHC-I:
-
Major histocompatibility complex class I
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PD:
-
Progressive disease
- PD-1:
-
Programmed cell death 1 receptor
- PD-L1:
-
Programmed cell death ligand-1
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- ROC:
-
Receiver operator characteristic curve
- sCD8:
-
Soluble form of CD8 antigen
- SD:
-
Stable disease
- sd:
-
Standard deviation
- TCR:
-
T-cell receptor
References
Madala S, Rasul R, Singla K et al (2022) Gender differences and their effects on survival outcomes in lung cancer patients treated With PD-1/PD-L1 checkpoint inhibitors: a systematic review and meta-analysis. Clin Oncol. https://doi.org/10.1016/j.clon.2022.03.010
Mencoboni M, Ceppi M, Bruzzone M et al (2021) Effectiveness and safety of immune checkpoint inhibitors for patients with advanced non small-cell lung cancer in real-world: review and meta-analysis. Cancers 13:1388. https://doi.org/10.3390/cancers13061388
Niu M, Yi M, Li N et al (2021) Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. Exp Hematol Oncol 10:18. https://doi.org/10.1186/s40164-021-00211-8
Oitabén A, Fonseca P, Villanueva MJ et al (2022) Emerging blood-based biomarkers for predicting immunotherapy response in NSCLC. Cancers 14:2626. https://doi.org/10.3390/cancers14112626
Geng J, Raghavan M (2019) CD8αα homodimers function as a coreceptor for KIR3DL1. Proc Natl Acad Sci U S A 116:17951–17956. https://doi.org/10.1073/pnas.1905943116
Raskov H, Orhan A, Christensen JP, Gögenur I (2021) Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer 124:359–367. https://doi.org/10.1038/s41416-020-01048-4
Cheroutre H, Lambolez F (2008) Doubting the TCR coreceptor function of CD8alphaalpha. Immunity 28:149–159. https://doi.org/10.1016/j.immuni.2008.01.005
Kaech SM, Wherry EJ, Ahmed R (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2:251–262. https://doi.org/10.1038/nri778
Farhood B, Najafi M, Mortezaee K (2019) CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol 234:8509–8521. https://doi.org/10.1002/jcp.27782
Giblin P, Ledbetter JA, Kavathas P (1989) A secreted form of the human lymphocyte cell surface molecule CD8 arises from alternative splicing. PNAS 86:998–1002. https://doi.org/10.1073/pnas.86.3.998
Fujimoto J, Stewart SJ, Levy R (1984) Immunochemical analysis of the released Leu-2 (T8) molecule. J Exp Med 160:116–124. https://doi.org/10.1084/jem.160.1.116
Schlesinger M, Chu FN, Badamchian M et al (1994) A distinctive form of soluble CD8 is secreted by stimulated CD8+ cells in HIV-1-infected and high-risk individuals. Clin Immunol Immunopathol 73:252–260. https://doi.org/10.1006/clin.1994.1195
Tomkinson BE, Brown MC, Ip SH et al (1989) Soluble CD8 during T cell activation. J Immunol 142:2230–2236
Pui CH, Ip SH, Dodge RK et al (1988) Serum levels of CD8 antigen in childhood lymphoid malignancies: a possible indicator of increased suppressor cell activity in poor-risk patients. Blood 72:1015–1021
Kim HS, Degiannis D, Raskova J, Raska K (1991) Cyclosporine A and prednisolone inhibit lectin- and alloantigen-induced release of sCD8: correlation with proliferative responses. Clin Immunol Immunopathol 60:27–39. https://doi.org/10.1016/0090-1229(91)90109-n
Sawada S, Hashimoto H, Iijima S et al (1993) Immunologic significance of increased soluble CD8/CD4 molecules in patients with active systemic lupus erythematosus. J Clin Lab Anal 7:141–146
Spronk PE, ter Borg EJ, Huitema MG et al (1994) Changes in levels of soluble T-cell activation markers, sIL-2R, sCD4 and sCD8, in relation to disease exacerbations in patients with systemic lupus erythematosus: a prospective study. Ann Rheum Dis 53:235–239. https://doi.org/10.1136/ard.53.4.235
Symons JA, Wood NC, di Giovine FS, Duff GW (1990) Soluble CD8 in patients with rheumatic diseases. Clin Exp Immunol 80:354–359. https://doi.org/10.1111/j.1365-2249.1990.tb03292.x
Balázs C, Bokk A, Farid NR (1994) Serum soluble CD8 concentration is an indicator of disease activity in patients with Graves’ disease. Thyroid 4:27–30. https://doi.org/10.1089/thy.1994.4.27
Grunewald RW, Fiedler GM, Stock B et al (2000) Soluble CD-4 and CD-8 as markers of immunological activation in renal transplant recipients. Nephrol Dial Transplant 15:71–77. https://doi.org/10.1093/ndt/15.1.71
Peng Y, Falck-Pedersen E, Elkon KB (2000) Soluble CD8 attenuates cytotoxic T cell responses against replication-defective adenovirus affording transprotection of transgenes in vivo. J Immunol 165:1470–1478. https://doi.org/10.4049/jimmunol.165.3.1470
Sewell AK, Gerth UC, Price DA et al (1999) Antagonism of cytotoxic T-lymphocyte activation by soluble CD8. Nat Med 5:399–404. https://doi.org/10.1038/7398
Morgan CL, Price CP, Cohen SBA et al (1999) Soluble CD8 stabilizes the HLA class I molecule by promoting β2M exchange: Analysis in real-time. Hum Immunol 60:442–449. https://doi.org/10.1016/S0198-8859(99)00014-2
Yamamoto N, Watanabe M, Matsuzuka F et al (2004) Lower concentration of serum soluble CD8 in severe Hashimoto’s disease. Clin Exp Immunol 137:601–605. https://doi.org/10.1111/j.1365-2249.2004.02576.x
Salmaninejad A, Valilou SF, Shabgah AG et al (2019) PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy. J Cell Physiol 234:16824–16837. https://doi.org/10.1002/jcp.28358
Siemiątkowska A, Bryl M, Kosicka-Noworzyń K et al (2021) Serum sCD25 protein as a predictor of lack of long-term benefits from immunotherapy in non-small cell lung cancer: a pilot study. Cancers 13:3702. https://doi.org/10.3390/cancers13153702
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Lee JW, Devanarayan V, Barrett YC et al (2006) Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res 23:312–328. https://doi.org/10.1007/s11095-005-9045-3
Orditura M, De Vita F, Roscigno A et al (1998) Soluble interleukin-2 receptor and soluble CD8 antigen levels in serum from patients with solid tumors. Int J Mol Med 2:75–79. https://doi.org/10.3892/ijmm.2.1.75
Vibe-Petersen J, Tvede N, Diamant M et al (1991) Soluble interleukin-2 receptor and soluble CD8 antigen levels in serum from patients with non-resectable lung cancer. Cancer Immunol Immunother 33:121–127. https://doi.org/10.1007/BF01742540
Lissoni P, Rovelli F, Tisi E et al (1992) Effects of interleukin-2 immunotherapy on soluble lymphocyte markers in cancer patients. Int J Biol Markers 7:260–262
Gause A, Verpoort K, Roschansky V et al (1991) The clinical significance of serum CD8 antigen levels in adult patients with Hodgkin’s disease. Ann Oncol 2:579–583. https://doi.org/10.1093/oxfordjournals.annonc.a058024
Macs M, Vandoolaeghe E, Ranjan R et al (1996) Increased serum soluble CD8 or suppressor/cytotoxic antigen concentrations in depression: suppressive effects of glucocorticoids. Biol Psychiat 40:1273–1281. https://doi.org/10.1016/0006-3223(95)00627-3
Maes M, Bosmans E, Ranjan R et al (1996) Lower plasma CC16, a natural anti-inflammatory protein, and increased plasma interleukin-1 receptor antagonist in schizophrenia: effects of antipsychotic drugs. Schizophr Res 21:39–50. https://doi.org/10.1016/0920-9964(96)00029-1
Burns DN, Nourjah P, Wright DJ et al (1999) Changes in immune activation markers during pregnancy and postpartum. J Reprod Immunol 42:147–165. https://doi.org/10.1016/S0165-0378(98)00085-0
Ho AD, Grossman M, Trümper L et al (1990) Clinical implications of increased plasma levels of CD8 in patients with hairy cell leukemia. Blood 75:1119–1124. https://doi.org/10.1182/blood.V75.5.1119.1119
Höbarth K, Hallas A, Steiner G et al (1996) Circulating immune markers in advanced renal cell carcinoma during immunotherapy with interferon gamma. Urol Res 24:101–106. https://doi.org/10.1007/BF00431087
Abbate I, Correale M, Musci MD et al (1993) Modification of soluble immunological parameters during treatment with interleukin-2. Int J Biol Markers 8:227–232. https://doi.org/10.1177/172460089300800405
Martens A, Janssen RA, Sleijfer D et al (1993) Early sCD8 plasma levels during subcutaneous rIl-2 therapy in patients with renal cell carcinoma correlate with response. Br J Cancer 67:1118–1121. https://doi.org/10.1038/bjc.1993.205
Lim SH, Newland AC, Kelsey S et al (1992) Continuous intravenous infusion of high-dose recombinant interleukin-2 for acute myeloid leukaemia—a phase II study. Cancer Immunol Immunother 34:337–342. https://doi.org/10.1007/BF01741555
Chi K-H, Myers JN, Chow KC et al (2001) Phase II trial of systemic recombinant interleukin-2 in the treatment of refractory nasopharyngeal carcinoma. Oncology 60:110–115. https://doi.org/10.1159/000055306
Kern P, Hussey RE, Spoerl R et al (1999) Expression, purification, and functional analysis of murine ectodomain fragments of CD8alphaalpha and CD8alphabeta dimers. J Biol Chem 274:27237–27243. https://doi.org/10.1074/jbc.274.38.27237
Watanabe M, Amino N, Hochito K et al (1997) Opposite changes in serum soluble CD8 in patients at the active stages of graves’ and hashimoto’s diseases. Thyroid 7:743–747. https://doi.org/10.1089/thy.1997.7.743
Yoneyama A, Nakahara K, Higashihara M, Kurokawa K (1995) Increased levels of soluble CD8 and CD4 in patients with infectious mononucleosis. Br J Haematol 89:47–54. https://doi.org/10.1111/j.1365-2141.1995.tb08912.x
Kurane I, Innis BL, Nimmannitya S et al (1991) Activation of T lymphocytes in dengue virus infections. High levels of soluble interleukin 2 receptor, soluble CD4, soluble CD8, interleukin 2, and interferon-gamma in sera of children with dengue. J Clin Invest 88:1473–1480. https://doi.org/10.1172/JCI115457
Acknowledgements
We thank all patients and hospital staff involved in the study.
Funding
This work was supported by the National Science Center in Poland (Narodowe Centrum Nauki) [Grant No. 2019/03/X/NZ6/00406].
Author information
Authors and Affiliations
Contributions
AS was involved in conceptualization, funding acquisition, investigation, data interpretation, project administration, writing—the original draft; MB helped in patient recruitment, treatment response evaluation, supervision of the clinical part of the study; KKN contributed to investigation, data interpretation, and manuscript revision; JT was involved in investigation; IGG helped in patient recruitment and treatment response evaluation; FKG contributed to supervision of the analytical aspects of the study and manuscript revision. MB and KKN contributed equally to this work. All authors read and accepted the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
AS received a grant from the National Science Center in Poland (2019/03/X/NZ6/00406) during the conduct of the study; MB reports personal fees from Hoffmann La Roche, Astra Zeneca, Boehringer-Ingelheim, MSD, BMS, and Pfizer outside the submitted work; JT reports personal fees from GlaxoSmithKline outside the submitted work; KKN, IGG, and FKG declare no conflict of interest. The funder had no role in the design of the study or data interpretation.
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki and the approval of the Bioethics Committee at Poznan University of Medical Sciences (decisions 80/19 and 251/19). All subjects signed informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Siemiątkowska, A., Bryl, M., Kosicka-Noworzyń, K. et al. Low on-treatment levels of serum soluble CD8 (sCD8) predict better outcomes in advanced non-small cell lung cancer patients treated with atezolizumab. Cancer Immunol Immunother 72, 1853–1863 (2023). https://doi.org/10.1007/s00262-023-03377-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-023-03377-8