
Abstract
Adoptive cell transfer (ACT) of tumor-specific T lymphocytes represents a relevant therapeutic strategy to treat metastatic melanoma patients. Ideal T-cells should combine tumor specificity and reactivity with survival in vivo, while avoiding autoimmune side effects. Here we report results from a Phase I/II clinical trial (NCT02424916, performed between 2015 and 2018) in which 6 metastatic HLA-A2 melanoma patients received autologous antigen-specific T-cells produced from PBMC, after peptide stimulation in vitro, followed by sorting with HLA-peptide multimers and amplification. Each patient received a combination of Melan-A and MELOE-1 polyclonal specific T-cells, whose specificity and anti-tumor reactivity were checked prior to injection, with subcutaneous IL-2. Transferred T-cells were also characterized in terms of functional avidity, diversity and phenotype and their blood persistence was evaluated. An increase in specific T-cells was detected in the blood of all patients at day 1 and progressively disappeared from day 7 onwards. No serious adverse events occurred after this ACT. Clinically, five patients progressed and one patient experienced a partial response following therapy. Melan-A and MELOE-1 specific T-cells infused to this patient were diverse, of high avidity, with a high proportion of T lymphocytes co-expressing PD-1 and TIGIT but few other exhaustion markers. In conclusion, we demonstrated the feasibility and safety of ACT with multimer-sorted Melan-A and MELOE-1 specific T cells to metastatic melanoma patients. The clinical efficacy of such therapeutic strategy could be further enhanced by the selection of highly reactive T-cells, based on PD-1 and TIGIT co-expression, and a combination with ICI, such as anti-PD-1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although having provided unprecedented benefit in the treatment of solid tumors, the use of anti-PD-1 antibodies, alone or in combination with other ICIs, does not exceed a therapeutic efficacy of 40% (monotherapy) to 60% (in combination). One possible reason for the therapeutic failure of ICIs in a significant proportion of patients may be the lack of a tumor-specific T-repertoire that can be mobilized by anti-IC antibodies. In this context, adoptive transfer of tumor-specific T cells could be a realistic option, especially in combination with ICI. For these strategies, ideal effector T-cells should combine different properties, such as tumor specificity and reactivity together with survival and tumor infiltration after transfer to autologous patients. In this context, we previously documented clinical responses in half of metastatic melanoma patients after adoptive transfer of T-cell clones specific for the Melan-A/MART-1 antigen [1, 2]. The lack of therapeutic efficacy in the other half of patients may have resulted from the injection of poorly reactive T cell clones in vivo due to the exhaustion status of infused T cells. Indeed, during the production process, the successive culture steps and the high number of cell divisions, required to get sufficient number of therapeutic T cell clones may cause exhaustion of some of these T cell clones. In addition, the infusion of T-cells specific for one single tumor antigen could promote tumor escape by favoring the growth of Melan-A negative tumor cells. These considerations led us to develop in 2015 a new ACT trial that combines tumor-reactivity of infused T-cells, polyclonality, and a shorter production process to improve T-cells survival in vivo. Moreover, we targeted two antigens to minimize tumor escape mechanisms by targeting a single specificity. At that time, anti-PD-1 had not yet become the standard of care for metastatic melanoma, so we did not immediately consider a combination therapy, this study being primarily a feasibility and safety study.
The two targeted antigens, Melan-A and MELOE-1, shared common features regarding their frequent expression in melanoma tumors, the recognition of two immunodominant HLA-A2 epitopes by melanoma-specific CD8 T cells involved in melanoma immuno-surveillance [3, 4], and of vast and diverse specific TCR repertoires in all HLA-A2 melanoma patients [5,6,7]. Indeed, blood frequencies of Melan-A and MELOE-1 specific T cells are, respectively, around 10–4 and 10–5 among CD8 T cells, which makes it possible to sort them from peripheral blood following a single step of in vitro peptide stimulation. These exceptionally high frequencies among T-cell repertoires specific for tumor antigens can be partly explained by a very strong bias in V-alpha usage in TCRs specific for these two epitopes [8, 9]. In addition, these two T cell repertoires also contain high avidity T cells making them relevant for a use in adoptive transfer. In terms of safety, Melan-A expression being restricted to the melanocytic lineage, the injection of T-cells specific reactive against this antigen may cause vitiligo by killing normal melanocytes which is an acceptable adverse effect considering the severity of metastatic melanoma [2]. Furthermore, results from previous ACT trials targeting Melan-A support the absence of other serious side effects [10, 11]. MELOE-1 antigen has never been targeted in any immunotherapy trial so far, but the very peculiar expression of this antigen led us to believe that the injection of MELOE-1-specific T-cells would be safe. Indeed, MELOE-1 antigen derives from one of the short ORFs of a lncRNA overexpressed in the melanocytic lineage [12, 13]. The IRES-dependent translation of MELOE-1 from this lncRNA is only effective in melanomas but not in melanocytes, conferring to this antigen a strict melanoma expression profile [14, 15]. Thus, the choice of these two antigen-specific T cell repertoires for melanoma immunotherapy appeared a realistic and relevant option. Based on these arguments and thanks to an original technique for sorting these specific T lymphocytes [16], we set up a unique phase I/II clinical trial called MELSORT (NCT02424916, https://clinicaltrials.gov), in which Melan-A and MELOE-1 specific T cells were infused to 6 metastatic melanoma patients. Here we report the characterization of infused T-cells, the characteristics of included patients and their clinical outcome, and the immune follow-up of infused T-cells.
Material and methods
Melanoma patients
Treated patients were HLA-A2 melanoma patients, with unresectable stage III or IV melanoma, with tumors expressing Melan-A and MELOE antigens detected by RT-PCR. This prospective, open, monocentric phase I/II trial, promoted by Nantes University Hospital (France), was designed and conducted in accordance with the Declaration of Helsinki. The clinical protocol was approved by the ethics committee, and the national Health Agency (ANSM), and it was registered with the regulatory state authority (NCT02424916). All patients gave written informed consent before enrollment in the study and met the following inclusion criteria: male or female ≥ 18 and ≤ 75 years, patient expressing the HLA-A*0201 subtype of the human leukocyte antigen (HLA -A2), patient with metastatic melanoma stage IIIc or IV (AJCC 2010) except brain metastases, tumor expressing the antigens Melan-A and MELOE-1 detected by RT-qPCR, ECOG ≤ 1, lymphocyte count ≥ 1500/µL. Prior adjuvant melanoma treatment (before metastatic stage) were authorized (anti- BRAF, anti-CTLA4, IFN, TIL…). The primary objective was the evaluation of clinical and biological safety and the secondary objectives were the clinical efficiency and immune monitoring of infused T cells. The study design is illustrated by Fig. 1. After signed consent, patients were selected and 120 mL of blood were taken from HLA-A*0201 patients with tumor expressing Melan-A and MELOE antigens. One month after patients received intravenously autologous specific-T cells, with subcutaneous injections of IL-2 (6 MU, Chiron) from day 0 to day 4. From month 1 to month 12, patients were clinically (each month) and radiologically (every three months) followed-up and did not receive any other anti-cancer treatment (Fig. 1). The primary objective of safety was assessed by the NCI common toxicity criteria (Version 4.0, May 2009, http://ctep.cancer.gov). Overall tumor response (complete response, partial response, stable disease) was evaluated according to Response Evaluation Criteria in Solid Tumor (RECIST) and immune-related Response Criteria (irRC), every 3 months until M12.

Therapeutic schedule of MELSORT clinical trial. a Antigen expression was validated by qPCR on a tumor biopsy from HLA-A*0201 metastatic melanoma patients, 7 days before the initiation of T cell production process. b Melan-A and MELOE-1 specific T-cells were produced in the Unit of cell Therapy of Nantes University hospital, from a sample of 120 mL of patient’s blood. c One month after the initiation of the production process, specific T cells were infused intravenously to the autologous patients, with subcutaneous injections of IL-2 (6 MU) during 5 days. d Patients were monthly followed clinically and radiologically
Persistence of injected specific T cells evaluated by immune monitoring with HLA-peptide tetramers, at day 1, day 7, Day 30 and Day 90.
Production of Melan-A and MELOE-1 specific T-cells from patient PBMC
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Hypaque gradient centrifugation, washed three times and seeded in 96 well/plates at 2 × 105 cells/well in RPMI 1640 medium supplemented with 8% human serum (HS) (a pool from 20 donors prepared and secured by the EFS of Nantes) with 50U/mL of IL-2. PBMC were immediately stimulated with clinical grade peptides (CSBio, USA), either 1 µg/mL of Melan-AA27L peptide (ELAGIGILTV) or 10 µg/mL of MELOE-136–44 peptide (TLNDECWPA) for 14 days. Following stimulation, each microculture was evaluated for the percentage of specific CD8 T lymphocytes by double staining with the relevant APC-conjugated HLA-peptide tetramer (from the SFR Sante recombinant protein facility) and PE-conjugated anti-CD8 mAb (BD Biosciences, France) using a FACSCanto. Microcultures that contained at least 0.5% of Melan-AA27L or MELOE-136–44 specific T cells were selected, pooled and sorted with the relevant clinimers, as previously described [16]. These Clinimers were produced at GMP grade by the company Px-Therapeutics. Briefly, clinical grade M450-epoxy magnetic beads (Clin Ex-vivo Dynabeads, Life technologies) were covalently coupled to a monoclonal antibody specific for the peptide AviTag™ (Avidity, Aurora, CO, USA) that is fused to the heavy chain of the HLA constructs. Clinical batches of this Ab were produced by PX’Therapeutics from a master cell bank of clinical grade CHO-DG44 cell line (Life Technologies). HLA-A0201/peptide 3-mutated monomers were produced in GMP conditions by PX’Therapeutics. Final assembly of the Clinimer™ reagent was performed right before T cell sorting in the Unit of cell therapy, by incubating Chim-AvT beads with the appropriate HLA/peptide monomers (1 g of monomer for 107 beads). The quality control of the resulting clinical grade multimers of HLA/peptide complexes (Clinimers™) was performed by staining with a PE-conjugated anti-HLA-A2 mAb (BD Biosciences, France).
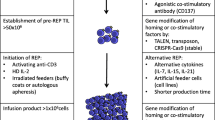
Sorted-specific specific T cells were seeded in 96 well plates for polyclonal amplification as previously described [17, 18] with irradiated feeder cells (PBMC pool from 4 donors and lymphoblastoid B cells-LAZ), clinical grade IL-2 (150 IU/mL) (Novartis Pharma, Rueil Malmaison, France), and PHA-L (1 µg/mL) (Sigma, Lyon, France), in 150 µL of RPMI1640 medium; containing 8% of HS. After a 14-days culture period, the total number of amplified T-cells is estimated by manual counting, and their specificity is assessed by double staining with the relevant APC-conjugated HLA-peptide tetramer (from the SFR Sante recombinant protein facility) and PE-conjugated anti-CD8 mAb (BD Biosciences, France). The purity threshold for cell injection has been set at 90% of cells labelled with the specific tetramer. Concerning the absolute number of infused T-cells, specifications had been fixed between 108 and 5 × 108 of each specificity, with an equal quantity of each T-cell population. Reactivity of amplified T cells was finally evaluated by TNF-α production in response to their cognate epitope. In brief, T cells were stimulated for 5 h in the presence of brefeldin A (10 µg/mL) with the appropriate peptide (10 µM for MELOE-136–44 and 1 µM for Melan-AA27L). T-cells were then stained with APC-conjugated tetramer, fixed with 4% paraformaldehyde (Euromedex), permeabilized with PBS 0.1% saponin, intracellularly labelled with PE-conjugated anti-TNF-α mAb (BD Biosciences) for 30 min at room temperature and analyzed by flow cytometry. The reactivity threshold for cell injection has been set at 50% of cells producing TNF-α among tetramer-positive T-cells, and the cell viability threshold at 90%. The safety of the infused T-cells was controlled by evaluation of the residual magnetic beads by flow cytometry [16] in the cell suspension, the non-proliferation of the irradiated feed cells, the absence of mycoplasma and viruses as HHV6, HHV8, CMV and the absence of bacterial and fungal contamination. This production process and associated controls are summarized in Fig. 2. With the exception of patient P14 (who received only 6 × 107 MELOE-1 specific T-cells), Melan A and MELOE-1 specific T-cells were finally collected in equal amounts (between 100 and 500 × 106 cells for each specificity) up to 109 total T-cells, and conditioned in a volume of 200 ml of 40 mg/ml of human serum albumin (Vialebex 40 mg/ml, LFB, France).

Methodology of melanoma-specific CD8 T cells production from HLA-A2 melanoma patients PBMC and controls in process. a PBMC from HLA-A2 melanoma patients were stimulated with each of the two antigen-derived peptide (Melan-AA27L and MELOE-136–44). b After 2 weeks, the amplification of specific-T cells is assessed by tetramer labelling. c Specific T-cells are sorted with HLA-peptide multimer-coated magentic beads, and amplified on irradiated feeders cells. d After 2 weeks, purity and reactivity of amplified T-cells on peptide-pulsed T2 cells are assessed and T-cells are infused to the patients
TRBV repertoire diversity
Vß diversity of sorted Melan-A and MELOE-1 specific T cell lines was analyzed by labeling with 24 anti-Vß mAbs included in the IOTest Beta Mark TCR V Kit (Beckman-Coulter, IM3497). These cytometric analyses were performed on a Facs Canto II (BD Biosciences). We followed throughout the manuscript the nomenclature of IMGT database [19].
For TCR sequencing of MELOE-1-specific T cells from P5 patient, total RNA was extracted from 5 × 105 specific-T cells using QIAGEN RNeasy Kit. 25 ng of RNA was used to build libraries with the QIAseq Immune Repertoire T-cell Receptor Panel (Catalog 333705—IMHS-001Z), as previously described [9]. FASTQ files were analyzed in the QIAGEN GeneGlobe Data Analysis Center using the Immune Repertoire Application. The clonotype calls are generated using the IMSEQ software [20]. Clonotypes were defined on the basis of unique amino-acid sequences of CDR3 beta regions. In our set of data, the total number of unique TCR sequences was identical to the number of clonotypes.
Cell lines
The human TAP deficient cell line T2 (174 × CEM.T2) used as a presenting cell was purchased from the ATCC (CRL-1992). Melanoma cell lines, registered in the Biocollection PC-U892-NL (CHU Nantes, France), were established from metastatic tumor fragments in the Unit of Cell therapy of Nantes. All these cells were cultured in RPMI1640 medium supplemented with 10% of Fetal Bovine Serum (Eurobio), 2 mM L-glutamine (Gibco), 100 U/mL penicillin (Gibco) and 0.1 mg/mL streptomycin (Gibco).
Functional avidity and melanoma reactivity
Functional avidity of antigen-specific sorted T-lymphocytes was evaluated after co-culture with TAP-deficient T2 cells loaded with a range of either Melan-AA27L (ELAGIGILTV) or MELOE-136–44 peptides at the effector/target ratio 1/2, through the measurement of CD107a mobilization. Reactivity of sorted T-cell populations was also evaluated on 3 HLA-A*0201 melanoma cell lines, at various effector/target ratios, through the measurement of CD107a mobilization. CD107a mobilization was measured after a 3 h. of co-culture at 37 °C in the presence of a CD107a-specific mAb (H4A3 clone, BioLegend). T lymphocytes were then stained with anti-CD8 antibodies (Clone RPA-T8, BioLegend) and analyzed by flow cytometry on a Facs Canto II (BD Biosciences).
RT-qPCR for MLANA and meloe expression in melanoma cell lines
Total RNA was extracted from tumors or melanoma cell lines by RNA purification system NucleoSpin RNA II (Macherey–Nagel, Hoerdt-FRANCE) (RNA integrity number > 7). Retrotranscription was performed using 1 μg of total RNA, oligodT, and SuperScript III reverse transcriptase (Invitrogen-Life-Technologies, Saint-Aubin-FRANCE). Relative quantification of meloe, MLANA, and housekeeping genes (HKG) RPLPO and cyclophilin-A expression was performed using Brilliant SYBR Green qPCR (Stratagene-Agilent Technologies, Les-Ulis-FRANCE). cDNA samples (20 ng) were added to SYBR Green Master Mix with specific primers at 200 nM. RPLPO: 5′-GTGATGTGCAGCTGATCAAGACT-3′ and 5′-GATGACCAGCCCAAAGGAGA-3′; cyclophilin-A: 5′-CCACCGTGTTCTTCGACAT-3′ and 5′-CCAGTGCTCAGAGCACGAAA-3′; meloe: 5′-GTCCTCCCCAGCACCAGAGT-3′ and 5′-AGCCTGCCATCTGCAATCCT-3′; MLANA: 5′-TGCTCATCGGCTGTTGGTATTG-3′ and 5′-GGAGCATTGGGAACCACAGG-3′. For the four genes, thermal cycling was 95 °C for 10′, 40 cycles at 95 °C for 30″, 60 °C for 1′, and 72 °C for 1′. The efficiency of PCR reaction was validated with duplicate series of tenfold-diluted cDNA from the melanoma cell line M113, performed in parallel to plot the standard curves for the three genes. Mean threshold cycle (CT) values from duplicate PCR reactions were normalized to mean CT values for the two HKG from the same cDNA preparations. The relative expression ratio of a target gene was calculated based on the PCR efficiency (E) and the CT deviation between a given tumour or melanoma cell line (x) and a calibrator (M113 melanoma cell line), expressed in comparison with the mean of the HKG: ratio = (E target) ΔCT target (calibrator − x)/mean ((E HKG) ΔCT HKG (calibrator − x)).
Phenotypic characterization of infused T cells
Therapeutic cells were thawed in RPMI (Gibco) containing 8% human serum, washed twice, separated in two tubes and stained in 30 µL of staining buffer (PBS, 0.1% BSA) containing 7AAD (BD Biosciences), a specific tetramer (HLA-A2/Melan-A or HLA-A2/MELOE-1 conjugated to PE) and antibodies from panel 1 and 2 and incubated for 1 h at 4 °C. Cells were then washed twice before acquisition on a BD Symphony A5.2 analyzer. Data were collected and analyzed using the BD FACS Diva Software v8.05.
-
Panel 1 (activation/exhaustion) TIGIT (PE-Cy7), PD-1 (BV421), KLRG1 (APC), CD38 (BV480), CD39 (BUV737), CD62L (FITC), CD103 (BB700).
-
Panel 2 (Chemokine receptors) CCR6 (PE-Cy7), CXCR3 (BV421), CCR9 (PE), CXCR4 (BV711), CXCR5 (BV605), CCR10 (BB515), CD103 (BV480).
Immune follow-up of infused T cells
To minimize non-specific staining, HLA-A2-Melan-A and MELOE-1 tetramers were tittered and used at the lowest concentration that showed a clearly distinguishable positive T-cell population [1]. Total blood (3 mL) from melanoma patients at different time-points were incubated with 10 µg/mL of either Melan-A or MELOE-1/HLA-A*0201 APC-tetramers and with PE-conjugated anti-CD8 (Biologend) and FITC-conjugated anti-CD3 (Biolegend), for 1 h at 4 °C in the dark, with gentle agitation every 15 min. After incubation, the red blood cells are lysed through incubation in 10 volumes of lysis solution (BD Biosciences), during 15 min at room temperature. After washing in PBS-0.1%BSA, cells were resuspended in PBS and analyzed on a FACSCanto. Frequencies of antigen-specific T-cells are calculated based on the number of tetramer positive cells, divided by the total number of CD3+/CD8+ T lymphocytes analyzed.
Results
Patients pre-included and treated by ACT with melanoma-specific sorted T cells
Twenty metastatic melanoma patients were pre-selected for this clinical trial (Table S1), and finally 6 patients were treated by ACT with T-cells specific for the two selected melanoma antigens (Table 1). Indeed, 7 patients did not meet the inclusion criteria, such as improper HLA-A2 allele, absence of antigen expression by the tumor, or severe lymphopenia which hampered the production of sufficient numbers of specific T-lymphocytes.
In five patients, the production process was unsuccessful either because of a too low frequency of antigen-specific circulating T cells (for 3 patients), a mishap in the sorting procedure for Melan-A-specific T cells (one patient), or an insufficient amount and reactivity of MELOE-1-specific T-cells (one patient). Of note, one of the patients exhibiting a too low frequency of MELOE-1-specific T-cells (P4) received 5 × 108 Melan-A-specific CD8 T-cells as a compassionate treatment. Patient P11 could not receive the treatment due to a general status alteration during the production process that required transfer to palliative care and patient P15 could not benefit from the treatment because the regulatory agency was considering an amendment to the protocol, and did not give its authorization within the deadline.
All but one of the 6 patients who received the complete treatment were stage IIIC metastatic patients (Table 1), with tumors expressing the two targeted antigens. Four of these patients received antigen-specific T lymphocytes as a first line of treatment, which was conceivable at that time, considering that recommendations for the use of anti-PD-1 as first-line therapy did not come until May 2016, and considering the progressive capacity of melanoma in these patients. The two other patients had been treated previously either with Nivolumab or a combination of Nivolumab and Ipilimumab.
Production of therapeutic T cells and controls
Melan-A and MELOE-1-specific T-cells were produced from HLA-A*0201 PBMC from melanoma patients, according to a validated procedure [16]. After a peptide stimulation step of PBMC, the frequency of individual microcultures containing specific-T cells was assessed through CD8/tetramer double labelling (Fig. 2 and Table 2). Positive microcultures were grouped and specific-T cells were sorted with magnetic beads coated with each relevant tetramer, called Clinimers™ (Fig. 2).
The number of sorted-T cells was estimated (Table 2) and specific T lymphocytes were further amplified on feeder cells. At day 24 of the process (± 2 days), the absence of residual magnetic beads in the cell cultures was assessed by flow cytometry [16]. Finally, at the end of the amplification process (D30), amplified T cells were enumerated. As shown in Table 2, T lymphocytes underwent between 8 and 16 divisions during this amplification step after stimulation with the cognate peptides. Cell viability was always above 90%, and the purity of the amplified T cells always above 94% (assessed by tetramer labelling). The reactivity of specific T-cells was always above 60% as assessed by the production of TNF-α upon peptide stimulation.
The research for mycoplasma, viral (HHV6, HHV8 and CMV), bacterial and fungal contamination was always negative, as well as the presence of residual magnetic beads in cell suspension.
All 6 patients but one received equal numbers of Melan-A and MELOE-1 specific T lymphocytes (between 3.1 × 108 and 5 × 108 of each specificity). Patient 14 received 3.6 × 108 Melan-A-specific T cells but only 6 × 107 MELOE-1-specific T cells due to a low number of divisions of these T cells during the amplification process (only 8 divisions within 15 days of amplification) (Table 2). This patient was therefore the only patient to have received less than 108 T cells of this specificity. The decision to treat him despite this non-compliance was justified by the rapid progression of his disease and by the availability of Melan-A specific cells in sufficient numbers. Not to treat this patient would have been a loss of opportunity, given the favorable benefit-risk ratio.
Side effects and clinical outcome
Regarding the safety, no serious adverse events related to the treatment were reported during this study. The only observed adverse events were mild to moderate and are summarized in the Figure S1, classified according the system organ class (SOC). Overall, the most represented SOC among the reported events were general manifestations, consistent with the manifestations observed during IL-2 injection, such as asthenia, flu-like syndrome and pain at site injection, followed by gastrointestinal disorders. All the reported adverse events related to IL-2 treatment had resolved by the month following the last cytokine injection, as previously observed with TIL and IL-2 injection [21].
Regarding the clinical efficacy, among the 6 patients who received the treatment, one experienced a partial response according to RECIST criteria (Table 1). The other patients progressed either at month 3 or month 6. Of note, one of the progressing patient received anti-PD-1 treatment 4 months after ACT and experienced a complete response after 9 cures, still ongoing (P13).
Frequency and diversity of therapeutic T lymphocytes
The frequency of microcultures containing antigen-specific T cells after in vitro peptide stimulation was calculated as the ratio between wells containing at least 1% of specific T-cells and the total number of stimulated microcultures. On this basis, the frequency of microcultures containing Melan-A-specific T cells ranged between 17 and 70%, whereas that of those containing MELOE-1 specific T cells ranged between 1.7 and 31% (Fig. 3a). For each individual patient, the frequency of MELOE-1 specific T cells was always lower than that of Melan-A-specific T cells, as previously observed in healthy donors and melanoma patients [5]. Patients P5 and P16 exhibited the highest frequencies of Melan-A and MELOE-1 specific T cells (Fig. 3a). The diversity of antigen-specific sorted T-cell populations based on the frequency of Vß subfamilies was further documented for each patient (Fig. 3b) with a panel of 24 specific Vß-specific antibodies, covering the most frequently expressed TRBV chains. As shown in Fig. 3b and Table 3, Melan-A and MELOE-1-sorted T-cell populations expressed between 2 and 8 TRBV chains and 1–10 TRBV chains, respectively, with a frequent dominant expression of one or two TRBV chains. We could not document complete antigen-specific TRBV repertoire for P5, P14 and P16 patients (less than 50% of TRBV repertoire identified), due to the lack of available antibodies against frequently expressed TRBV chains within Melan-A and MELOE-1 T cell repertoires (especially for TRBV6 and TRBV7 families) [9]. To partially answer this question, we analyzed the cumulated frequencies of TRBV chains within MELOE-1-specific T cells from P5 patient by TCR sequencing (Fig. 3c). This parameter illustrates the relative abundance of CDR3ß clonotypes using a given TRBV gene, within a given repertoire. In this TCR repertoire, we identified 31 CDR3ß clonotypes, and this analysis indeed confirmed that the most abundant clonotypes expressed TRBV chains for which the specific antibodies were not available in the antibody panel (white sections in Fig. 3c).

Frequency and Vbeta diversity of Melan-A and MELOE-1-sorted T cells. a Percentages of microcultures containing antigen-specific T cells after the step of peptide-stimulation. b TRBV diversity of sorted-antigen specific T cells, determined by flow cytometry. Each stack represents a distinct TRBV subfamily, and dominant TRBV chains are indicated within the corresponding stack. ND: Not determined. c Cumulative frequencies (UMI counts) of each TRBV chains in the MELOE-1 specific T cell repertoire from P5 patient. The number of clonotypes using a given TRBV chain is indicated
Functional avidity and reactivity of antigen-specific T lymphocytes
We tested the functional avidity of each antigen-specific T-cell population on T2 cells loaded with a range of each cognate peptide, using double staining with a specific anti-CD8 Ab together with CD107a labeling (Fig. 4a). As illustrated by Fig. 4a (upper panel) and Table 3, the range of functional avidities for Melan-A-specific T-lymphocytes was between 10−11 M and 10−14 M, whereas this range was lower and much narrower for MELOE-1 specific lymphocytes, ranging from 10−10 M and 10−11 M (Fig. 4a, lower panel and Table 3). Interestingly, with the exception of P13 patient, there was a relative proportionality between the EC50 of Melan-A and MELOE-1 specific lymphocytes for a given patient. As an example, Melan-A and MELOE-1 specific T lymphocytes from patient P5 both exhibited high functional avidities (black curves in Fig. 4a), whereas these T-lymphocytes from patient P16 both exhibit the lowest functional avidities (green curves in Fig. 4a). We also tested the functional avidity of the main TRBV subfamilies within a given T-cell repertoire (Figure S2). Globally, there were no drastic differences between the functional avidities of total CD8 T cells and that of dominant TRBV subfamilies. As expected, the reactivity of these T-cell populations, measured on HLA-A*0201 melanoma cell lines (Fig. 4b) were mostly consistent with their functional avidities. Melan-A-specific T-cells from P5 and P13 patients exhibited the highest reactivity against the 3 tested melanoma cell lines, compared to the 4 other populations (Fig. 4b, upper panel and Table 3). This difference was especially marked against M134 cell line, that displayed the lowest level of Melan-A expression, measured by RT-qPCR (Fig. 4c, upper panel). MELOE-1-specific T-lymphocytes from P5 patient were also among the most reactive against the three melanoma cell lines, with T-cells from P17 and P14 patients (Fig. 4b, lower panel). Conversely, MELOE-1 specific-T-cells from P10, P13 and P16 patients barely recognized these cell lines, consistent with their lower functional avidity against the MELOE-1 peptide (Fig. 4a, lower panel).

Functional avidities of Melan-A and MELOE-1-specific CTL lines and PD-1 expression. a CD107a expression by Melan-A -specific CTL lines, in response to TAP-deficient T2 cells loaded with a range of Melan-AA27L (up) or MELOE-136–44 (bottom) peptides. b Anti-tumor reactivity of Melan (left) and MELOE-1 (right)-specific CTL lines was validated through the measurement of CD107a degranulation in response to 3 HLA-A2 melanoma cell lines expressing the two antigens, at 3 different E/T ratios. c mlana (left) and meloe (right) genes relative expression were calculated from the Ct value normalized on the Ct value of of a reference melanoma cell line, and on the Ct value of two HKG (cyclophilin and RPLPO)
In conclusion, these results showed that therapeutic T-cell infused to P5 patient, who exhibited a partial response (Table 1) displayed the best profile, both in terms of functional avidity of Melan-A and MELOE-1 specific T cells and of reactivity against target melanoma cells expressing the two antigens.
Activation/exhaustion markers expressed by therapeutic T-cells
We previously described that functional avidity of Melan-A-specific T cells was associated with the co-expression of PD-1 and TIGIT receptors [22]. This T cell subset thereafter called DPOS subset is an heterogeneous T cell population containing both highly activated T cells and exhausted T cells [23]. We therefore investigated through multiparametric flow cytometry the proportion of DPOS T cells in each antigen-specific T cell population, together with their expression of additional exhaustion markers, CD39, KLRG1 and CD103. The gating strategy is depicted in Figure S3, by a representative example.
Three Melan-A specific T cell populations (from P5, P10 and P13) contained high proportion of DPOS T cells, above 30% (Fig. 5a) and among these 3 populations, those from P10 and P13 patients both expressed high levels of CD39, together with KLRG1 and CD103 for P10 patient and CD103 for P13 patient (Fig. 5b). Conversely, Melan-A-specific T cells from P5 patient almost did not express any of those molecules. In the three other Melan-A specific T cell population, the fraction of DPOS T cells is low (Fig. 5a), and these subpopulations all expressed CD39 with KLRG1 and CD103 for P14 patient (Fig. 5b).

Fraction of PD-1+/TIGIT+ T cells among specific-T cells and exhaustion markers expressed by this subset, analyzed on a BD-Symphony cytometer. a and c Fraction of PD-1+/TIGIT+ (DPOS) T cells among Melan-A and MELOE-1 infused specific T-cells. b and d Percentages of Melan-A and MELOE-1 DPOS T cells expressing CD39, KLRG1 and CD103 markers
For MELOE-1-specific T cells, 2 populations (P5 and P10) contained more than 30% of DPOS T cells (Fig. 5c). Among these two subpopulations, only those from P10 contained a high proportion of CD39 positive T lymphocytes (Fig. 5d). MELOE-1 specific T cells from P14 and P16 patients contained around 20% of DPOS T-cells, enriched with significant fractions of CD39+ and CD103+ T lymphocytes. Finally, MELOE-1-specific T-cells from P17 and especially P13 patients contained low fractions of DPOS T cells. These results, together with the functional avidities and the tumor reactivity of these therapeutic T cells suggested that the presence of a high proportion of DPOS T cells lacking expression of exhaustion markers, especially CD39, could condition therapeutic efficacy.
Immune follow-up of infused antigen-specific-T cells in the blood of patients
Another feature associated with clinical efficacy of ACT is the in vivo persistence of infused T-cells and their ability to migrate to the tumors. In order to evaluate blood persistence of infused cells, we measured the frequency of circulating antigen-specific T cells from day 1 to day 90 after ACT, by labeling 3 ml of whole blood with HLA-peptide-specific tetramers and CD3 and CD8-specific antibodies. Frequencies of specific T-cells were calculated among total CD8 T cells. Before T cell transfer, the frequencies of tetramer positive cells, respectively, ranged from 1.3 × 10–4 to 3.7 × 10–4 for Melan-A-specific T-cells and between 8.9 × 10–5 and 5.5 × 10–4 for MELOE-1-specific T-cells. (Table S2 and Fig. 6a). After infusion, these frequencies increased significantly at day 1 in all treated patients (Table S2 and Fig. 5a), with the corresponding whole blood staining shown in Fig. 6b. However, two groups of patients could be clearly distinguished at D1: patients P10, P13 and P14 had very high frequencies of specific cells in the blood (up to 10–2) while in patients P5, P16 and P17 the majority of the infused cells already disappeared from the blood. We were thus prompted to look for possible differences in homing markers or chemokine receptors that may explain these two different profiles. We performed a multipanel labelling of the infused T cells to analyze the expression of CD62L, CXCR4, CCR10, CCR9 and CCR6 (Figure S4) but we could not identify any differential expression that could account for these different behaviors.

Immune follow-up of infused antigen-specific T lymphocytes. a Frequency of Melan-A and MELOE-1-specific T cells among CD8 T cells detected in patients’ blood across time points post ACT. b Dot plots illustrating the presence of tetramer-positive T-lymphocytes, at day 1 post-injection, through multiple labelling performed on 3 mL of total blood. Frequencies of specific-T cells are indicated in the dot plots and upper numbers within brackets indicate the amount of infused T cells.
Discussion
This study demonstrates the feasibility and safety of adoptive transfer of T lymphocytes specific for two melanoma antigens, selected via their TCR by an original method [16]. An advantage of this method is its relative simplicity of implementation, its speed and safety, with no need for genetic modification, as compared with the production of CAR T-cells or TCR transfer. In addition, selecting therapeutic cells from the endogenous repertoire naturally limits the range of their functional avidity, which may also reduce the severe autoimmune off-target toxicities sometimes observed with engineered T cells with very high affinities [24, 25]. Nevertheless, one of the prerequisites for successful production of antigen-specific T cells is the frequency of the corresponding T cell repertoires in the peripheral blood of patients. As such, Melan-A and MELOE-1 antigen-specific T repertoires are among the most frequent for tumor antigen-specific T repertoires [5, 9]. Indeed, blood frequencies of Melan-A and MELOE-1 specific T cells are, respectively, around 10–4 and 10–5 among CD8 T cells in HLA-A*0201 melanoma patients, allowing their sorting from patient blood after a single step of peptide stimulation. This production method may thus require some minor adjustments for the amplification of less frequent T cell repertoires, such as those specific for some cancer germline antigens or neo-antigens. In this respect, we previously demonstrated that the multimer sorting procedure remained efficient to sort much rarer repertoires, such as gp100 or NA17-A specific T cells [26].
Despite this relatively high peripheral frequency, for 2 of the 11 patients for whom the production process of specific T lymphocytes was initiated, stimulation by the MELOE-136–44 peptide did not allow the amplification of specific lymphocytes, probably due to less frequent T-cell repertoires in these two patients (BM-02 and SE-04). Of note, in one of these two patients (BM-02), the peptide stimulation step also failed to amplify specific Melan-A T lymphocytes, which is more surprising in view of the high frequency of this T-cell repertoire in all HLA-A*0201 patients. Melan-AA27L peptide stimulation also failed to amplify specific T-cells from another patient (SC-01), while the amplification of MELOE-1 specific T-cells was successful for this same patient. For these patients, we can hypothesize an exhaustion of Melan-A specific T-cells, due to chronic restimulation, as observed for virus-specific T lymphocytes in pathological contexts [27].
In addition to the amplitude of their specific T repertoires, Melan-A and MELOE-1 antigens were also selected on the basis of their frequent and shared expression by melanoma tumors, and regarding MELOE-1 antigen, for its strict tumor specificity. Indeed, this antigen, unlike Melan-A, had never been targeted in melanoma immunotherapy trials, and it was crucial to guarantee the safety of injecting T lymphocytes specific for this antigen. This non-toxicity was, if not totally guaranteed, at least strongly supported by the dual expression control of this antigen, both at the transcriptional level, which limits its expression in the melanocyte lineage [12], and at the translational level, with a protein expression controlled by an IRES sequence, active only in melanomas [14, 15]. No side effects were observed for the 6 treated patients, apart from the expected side effects related to IL-2 injections, carried out to promote the survival of the injected T lymphocytes [10]. For all treated patients, we observed increased frequencies of tetramer-specific T cells in blood the day after T-cell infusion, with marked increase for three of them (Fig. 6). These T cells progressively disappeared from blood at day 7 that may reflect their migration to secondary lymphoid organs and tumors, as previously shown after the injection of Melan-A specific T-cells [11].
Another major point for the effectiveness of adoptive transfer is the functional avidity of the injected T-lymphocytes. As the MELOE-1 antigen is not expressed in the thymus, T lymphocytes specific for this antigen are not subject to thymic selection, thus favoring the presence of T lymphocytes with high functional avidity within this T cell repertoire. The same is true for the Melan-A specific T repertoire, despite the fact that Melan-A has long been considered as a self-antigen tolerized in the thymus. Indeed, although Melan-A mRNA can be detected in medullary thymic epithelial cells, it was documented that in the majority of individuals, this mRNA is truncated due to a mis-initiation of gene transcription [28]. In the truncated mRNA transcripts, the immunodominant HLA-A2 Melan-A26-35 epitope is lost thus leading to the evasion of central self- tolerance towards this epitope. Functional avidities of Melan-A and MELOE-1 sorted-T cells were assessed using T2 cells pulsed with the Melan-A analog peptide (A27L) or the MELOE-136–44 peptide. Among Melan-A specific T-cells, T lymphocytes from P5 and P13 patients exhibited the highest functional avidity, followed by T cells from P10 and P14, and from P16 and P17 patients (Fig. 4a and Table 3). The range of functional avidities is much narrower for MELOE-1 specific T cells, with T cells from P5, P14 and P17 exhibiting better EC50 on the natural peptide than T cells from P10, P13 and P16 patients (Fig. 4a and Table 3). Based on these results, it is interesting to observe that only lymphocytes from the single responding patient (P5, black curves in Fig. 4a) displayed high avidities against both Melan-A and MELOE-1 peptides. These results were further confirmed by measuring the reactivity of T cells against HLA-A2 melanoma cell lines, spontaneously expressing the two target antigens (Fig. 4b and c). Again, Melan-A specific T cells from P5 and P13 patients were the most reactive against melanoma cell lines, as well as MELOE-1-specific T cells from P5, P14 and P17 (Fig. 4b). Besides patient P5, lymphocytes of both specificities from patient P14 also exhibited good functional avidity and were reactive against melanoma cells (blue curves and histograms in Figs. 4a and 4b). Nonetheless, P14 patient did not benefit from ACT and the major difference when compared to patient P5 was the poor diversity of its T cell repertoire (Fig. 3b). Indeed, Melan-A-specific T cell repertoire from P14 patient, was dominated by a single Vbeta subfamily (probably mainly composed of a dominant clonotype), and we could not identify the Vbeta subfamilies that constitute MELOE-1-specific T-cell repertoire, also suggesting the dominance of a clonotype not recognized by the available Vbeta-specific antibodies. In contrast, Melan-A and MELOE-1 specific T-cell repertoires from P5 patient were rather diverse, with, respectively, 6 and more than 9 Vbeta subfamilies (Fig. 3b and Table 3). More specifically, through TCR sequencing, we could identify 31 distinct clonotypes within MELOE-1 specific T-cells from this patient (Fig. 3c). The diversity of a given T-cell repertoire increases the probability of success of adoptive cell transfer, in particular by diversifying the expression profiles of the immune check points. Conversely, the injection of a population with a very low polyclonality increases the probability of T-cell exhaustion within tumor microenvironment, despite a high anti-tumor reactivity.
Considering this point, we analyzed the expression of a panel of activation/exhaustion markers through multiparametric flow cytometry on each infused T-cell population. We and others have previously suggested that PD-1 and TIGIT co-expression may define a relevant CD8 T cell population (thereafter called DPOS), of high functional avidity, associated with clinical efficacy of PD-1 blockade [22, 23, 29,30,31]. This DPOS subset is a heterogeneous population containing both activated and exhausted T cells, and the delicate balance between these two differentiation status is probably a key feature of therapeutic efficacy. As expected, specific T cells from P5 contained a significant proportion of DPOS T cells either for Melan-A or MELOE-1-specific T cells (Fig. 5a and b). This was also the case for specific T cells from P10 patient, but contrary to DPOS T cells from P5, these T cells contained a high proportion of CD39+, KLRG1+ and CD103+ T lymphocytes, suggesting an advanced exhaustion status. Indeed, it has been previously shown that CD39 and CD103 co-expression identifies a subpopulation of TILs found in tumor microenvironment, with a TRM phenotype, expressing high levels of exhaustion markers, and enriched in tumor reactive T cells. Nonetheless, repetitive antigen stimulation led to impaired effector function and tumor escape [32]. Furthermore, the co-expression of KLRG1 on these specific T-cells also suggests an engagement into the exhaustion process [33].
In conclusion, we have demonstrated the feasibility of adoptive transfer of T lymphocytes from the endogenous repertoire of patients, produced according to an original method never tested before. We also demonstrated the safety of targeting the non-classical MELOE-1 antigen, never targeted before in immunotherapy protocols. Of the 6 patients treated, one patient developed a clinical response, and we hypothesize that this therapeutic response is related to the diversity and functional avidity of the Melan-A and MELOE-1 specific T lymphocytes that this patient received. This feature is strongly associated with the fraction of DPOS T cells and the activation/exhaustion status of antigen-specific repertoires.
This study demonstrates that the use of endogenous T lymphocytes, in addition to its safety, can lead to therapeutic efficacy, which can be further enhanced by the selection of highly reactive T cells, based on PD-1 and TIGIT co-expression. Such a therapy could be considered for patients refractory to ICI strategies, which would provide these patients directly with a highly tumor-specific T-repertoire, whose activity could be further enhanced by a combination with ICI.
Data availability
The dataset used and/or analyzed during the current study is available from the corresponding authors on reasonable request.
References
Vignard V, Lemercier B, Lim A et al (2005) Adoptive transfer of tumor-reactive Melan-A-specific CTL clones in melanoma patients is followed by increased frequencies of additional Melan-A-specific T cells. J Immunol 175:4797–4805
Khammari A, Labarriere N, Vignard V et al (2009) Treatment of metastatic melanoma with autologous Melan-A/MART-1-specific cytotoxic T lymphocyte clones. J Invest Dermatol 129:2835–2842
Godet Y, Moreau-Aubry A, Guilloux Y et al (2008) MELOE-1 is a new antigen overexpressed in melanomas and involved in adoptive T cell transfer efficiency. J Exp Med 205:2673–2682
Benlalam H, Vignard V, Khammari A et al (2007) Infusion of Melan-A/Mart-1 specific tumor-infiltrating lymphocytes enhanced relapse-free survival of melanoma patients. Cancer Immunol Immunother 56:515–526
Godet Y, Desfrancois J, Vignard V et al (2010) Frequent occurrence of high affinity T cells against MELOE-1 makes this antigen an attractive target for melanoma immunotherapy. Eur J Immunol 40:1786–1794
Pittet MJ, Zippelius A, Valmori D et al (2002) Melan-A/MART-1-specific CD8 T cells: from thymus to tumor. Trends Immunol 23:325–328
Zippelius A, Pittet MJ, Batard P et al (2002) Thymic selection generates a large T cell pool recognizing a self-peptide in humans. J Exp Med 195:485–494
Trautmann L, Labarriere N, Jotereau F et al (2002) Dominant TCR V alpha usage by virus and tumor-reactive T cells with wide affinity ranges for their specific antigens. Eur J Immunol 32:3181–3190
Simon S, Wu Z, Cruard J et al (2018) TCR analyses of two vast and shared melanoma antigen-specific T cell repertoires: common and specific features. Front Immunol 9:1962
Yee C, Thompson JA, Byrd D et al (2002) Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA 99:16168–16173
Meidenbauer N, Marienhagen J, Laumer M et al (2003) Survival and tumor localization of adoptively transferred melan-A-specific T cells in melanoma patients. J Immunol 170:2161–2169
Bobinet M, Vignard V, Florenceau L et al (2013) Overexpression of meloe gene in melanomas is controlled both by specific transcription factors and hypomethylation. PLoS ONE 8:e75421
Chalopin B, Florenceau L, Fradin D et al (2015) A lineage-specific methylation pattern controls the transcription of the polycistronic mRNA coding MELOE melanoma antigens. Melanoma Res 25:279–283
Carbonnelle D, Vignard V, Sehedic D et al (2013) The melanoma antigens MELOE-1 and MELOE-2 are translated from a bona fide polycistronic mRNA containing functional IRES sequences. PLoS ONE 8:e75233
Charpentier M, Croyal M, Carbonnelle D et al (2016) IRES-dependent translation of the long non coding RNA meloe in melanoma cells produces the most immunogenic MELOE antigens. Oncotarget 7:59704–59713
Labarriere N, Fortun A, Bellec A et al (2013) A full GMP process to select and amplify epitope-specific T lymphocytes for adoptive immunotherapy of metastatic melanoma. Clin Dev Immunol 2013:932318
Dreno B, Nguyen J-M, Khammari A et al (2002) Randomized trial of adoptive transfer of melanoma tumor-infiltrating lymphocytes as adjuvant therapy for stage III melanoma. Cancer Immunol Immunother 51:539–546
Khammari A, Nguyen J-M, Pandolfino M-C et al (2007) Long-term follow-up of patients treated by adoptive transfer of melanoma tumor-infiltrating lymphocytes as adjuvant therapy for stage III melanoma. Cancer Immunol Immunother 56:1853–1860
Yousfi Monod M, Giudicelli V, Chaume D, Lefranc M-P (2004) IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinformatics 20(Suppl 1):i379–i385
Kuchenbecker L, Nienen M, Hecht J et al (2015) IMSEQ–a fast and error aware approach to immunogenetic sequence analysis. Bioinformatics 31:2963–2971
Khammari A, Nguyen J-M, Leccia M-T et al (2020) Tumor infiltrating lymphocytes as adjuvant treatment in stage III melanoma patients with only one invaded lymph node after complete resection: results from a multicentre, randomized clinical phase III trial. Cancer Immunol Immunother 69:1663–1672
Simon S, Vignard V, Varey E et al (2017) Emergence of high-avidity melan-A-specific clonotypes as a reflection of anti-PD-1 clinical efficacy. Cancer Res 77:7083–7093
Simon S, Voillet V, Vignard V et al (2020) PD-1 and TIGIT coexpression identifies a circulating CD8 T cell subset predictive of response to anti-PD-1 therapy. J Immunother Cancer 8:e001631
Morgan RA, Chinnasamy N, Abate-Daga D et al (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36:133–151
D’Ippolito E, Schober K, Nauerth M, Busch DH (2019) T cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer Immunol Immunother 68:1701–1712
Bouquié R, Bonnin A, Bernardeau K et al (2009) A fast and efficient HLA multimer-based sorting procedure that induces little apoptosis to isolate clinical grade human tumor specific T lymphocytes. Cancer Immunol Immunother 58:553–566
Youngblood B, Oestreich KJ, Ha S-J et al (2011) Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 35:400–412
Pinto S, Sommermeyer D, Michel C et al (2014) Misinitiation of intrathymic MART-1 transcription and biased TCR usage explain the high frequency of MART-1-specific T cells. Eur J Immunol 44:2811–2821
Chauvin JM, Pagliano O, Fourcade J et al (2015) TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest 125:2046–2058
Inozume T, Yaguchi T, Furuta J et al (2016) Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J Invest Dermatol 136:255–263
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC (2017) TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 276:112–120
Duhen T, Duhen R, Montler R et al (2018) Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9:2724–2813
Chen Z, Ji Z, Ngiow SF et al (2019) TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51:840-855.e5
Acknowledgements
The authors acknowledge the participation of the patients in this clinical trial.
Funding
This study was supported by grants from the INCa/PHRC and promoted by Nantes hospital (Réf. RC12_0261), the LabEx IGO program ANR-11-LABX-0016-01 (funded by the «Investissements d’Avenir» French Government program, managed by the French National Research Agency (ANR)), and the Ligue Contre le Cancer GO.
Author information
Authors and Affiliations
Contributions
FL, NL, and BD contributed to conceptualization; SSa, AF, VV, TB, and SSi contributed to methodology; FL, NL, AK, BD, and NJ were involved in formal analysis and investigation; NL and FL contributed to writing—original draft preparation; NL, FL, SSa, AK, and BD contributed to writing—review and editing; FL, BD, and NL contributed to funding acquisition; FL, NL, and BD contributed to supervision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
The trial complied with all French (Agence Nationale de Sécurité du Médicament et des Produits de Santé, ANSM, « loi n°2012-300, 5 Mars 2012, relative aux recherches impliquant la personne humaine», and all its amendments) and European requirements for the conduct of clinical trials (European Medicine Agency, EMA, CPMP/ICH/377/95, EMEA) and all the principles of Good clinical practices, the 1964 Declaration of Helsinki, received approval for all study sites from the ethics committee of Rennes, France (CPP Ouest V- Rennes; réf CPP: 14/19-935) and is registered at ClincalTrials.gov under the identifier: NCT02424916.
Informed consent
All patients provided written informed consent prior to inclusion.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
262_2021_2961_MOESM1_ESM.pdf
Fig. S1: Distribution of non-serious events by SOC (System Organ Class). The coding of adverse events and reactions used the MedDRA thesaurus.Fig. S2: Functional avidities of the main TRBV subfamilies of Melan-A and MELOE-1-specific CTL lines. T cell functional avidities were evaluated by measuring CD107a mobilization in response to T2 cells loaded with a range of Melan-AA27L (upper panel) or MELOE-136-44 peptides, at an E:T ratio of 1:2. CD107a positive fraction was evaluated by flow cytometry with a specific antibody, gated on the relevant TRBV subtypes or on total CD8 T cells.Fig. S3: Gating strategy for phenotypic characterization of infused T-cells. Tetramer positive cells were first analyzed on viable cells, within singlets among lymphoid cells. Activation/exhaustion markers were then analyzed on tetramer-positive cells.Fig. S4 : Chemokine receptors expression by infused T-cells. The expression of various chemokine receptors was assessed on Melan-A and MELOE-1 specific T lymphocytes, through a multipanel labeling analyzed on a BD-Symphony cytometer. (PDF 466 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dréno, B., Khammari, A., Fortun, A. et al. Phase I/II clinical trial of adoptive cell transfer of sorted specific T cells for metastatic melanoma patients. Cancer Immunol Immunother 70, 3015–3030 (2021). https://doi.org/10.1007/s00262-021-02961-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-02961-0