
Abstract
It is well known that various inflammatory cells infiltrate cancer cells. Next to TAMs (tumor-associated macrophages), TAFs (tumor-associated fibroblasts) and TANs (tumor-associated neutrophils) also platelets form the tumor microenvironment. Taking into account the role of platelets in the development of cancer, we have decided to introduce a new term: tumor associated platelets—TAPs. To the best of our knowledge, thus far this terminology has not been employed by anyone. Platelets are the first to appear at the site of the inflammatory process that accompanies cancer development. Within the first few hours from the start of the colonization of cancer cells platelet-tumor aggregates are responsible for neutrophils recruitment, and further release a number of factors associated with tumor growth, metastasis and neoangiogenesis. On the other hand, it also has been indicated that factors delivered from platelets can induce a cytotoxic effect on the proliferating neoplastic cells, and even enhance apoptosis. Undoubtedly, TAPs’ role seems to be more complex when compared to tumor associated neutrophils and macrophages, which do not allow for their division into TAP P1 and TAP P2, as in the case of TANs and TAMs. In this review we discuss the role of TAPs as an important element of tumor invasiveness and as a potentially new therapeutic target to prevent cancer development. Nevertheless, better exploring the interactions between platelets and tumor cells could help in the formulation of new therapeutic goals that support or improve the effectiveness of cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background/Introduction
The association between inflammation and cancer has long been the subject of numerous studies. In 1863, Virchow put forward a hypothesis stating that immune cell infiltrations reflect the site of neoplastic lesions within the chronically affected tissue [1]. A decade later, Dvorak reported that carcinogenesis and inflammatory condition share some growth mechanisms, such as cell proliferation, increased survival, migration and enhanced angiogenesis, which are strictly controlled by growth factors, proinflammatory cytokines and proangiogenic factors. Moreover, he observed that cells involved in inflammation infiltrate the neoplastic tissue as well [2, 3].
Transforming neoplastic cells are surrounded by numerous cells: fibroblasts, endothelial cells, pericytes and mesenchymal cells that together form tumor stroma. These cells produce a number of cytokines and chemokines, which attract the subsequent populations of immune cells, including macrophages, neutrophils, mast cells, dendrites and T, B and NK cells which by infiltrating tumor tissues form the tumor microenvironment [4]. Leukocytes may account for up to 50% of total tumor mass. The interaction of cancer cells with macrophages activates them to produce cytokines, e.g. IL-8 which stimulates further inflow of inflammatory cells [5, 6]. In the tumor microenvironment, cells communicate with one another via direct contact or production of various mediators [7]. The activity of inflammatory cells and the type and level of the expression of factors that modulate inflammation affects the balance between their pro- and antineoplastic activity. In developed tumors, the inflammatory cells act in favor of the tumor, increasing the survival and proliferation of the transformed cells [8, 9].
Tumor-associated macrophages (TAMs)
Macrophages are the main cells playing a role in the inflammatory condition accompanying neoplastic disease. They are derived from precursor cells-monocytes, activated by MCP-1/CCL2 (monocyte chemoattractant protein-1), cytokines (IL-6, CSF-1, VEGF) and chemokines (CCL2, CCL5, CCL8) produced by neoplastic cells. Macrophages are diverted from blood circulation to tissues where they differentiate from mature forms [10]. Present in the tumor microenvironment they are defined as tumor associated macrophages (TAMs). TAMs can promote tumor cell death and preclude the formation of new blood vessels. At the same time they produce proangiogenic factors, growth factors and extracellular matrix metalloproteinases, which in turn may stimulate cancer growth [11, 12].
The opposite action of these cells is associated with the presence of two TAM phenotypes, namely M1 and M2 [12] (Suppl. Fig. 1). M1 macrophages are stimulated by IFN-γ, LPS, TNF-α or GM-CSF, and inhibit tumor growth by producing pro-inflammatory cytokines such as: IL-1, -6, -8, -12, -23, TNF-α, small quantities of IL-10 as well as ROS (reactive oxygen species) and RNI (reactive nitrogen intermediates) [13]. These cells affect Th1 activation and enhance antineoplastic response. They can recognize tumors and kill tumor cells by cytotoxic effect (e.g. ROS) [14]. On the contrary, M2 macrophages display pro-tumor activity. M2, activated by IL-4 and -13, produce small amounts of IL-12 and -23 and large quantities of IL-10, which silences an acute inflammatory state and attenuates antineoplastic immune response of the body [8, 12, 15]. M2 macrophages have an effect on the inactivation of T cells, which crucially decreases the body’s ability to resist cancer development and progression [16]. TAMs can also promote tumor angiogenesis and metastatic potential by the synthesis of cytokines such as: IL-6, -17, -23, and inhibit cytotoxic T cell responses [11].
Due to varying locations, certain TAMs produce growth factors (EGF, PDGF, IL-6, IL-8), or generate immunosuppressive factors (IL-10, TGF-β, PGE2) [12], whereas others produce metalloproteinases (MMP7, MMP9, MMP12), urokinase plasminogen activator (uPA) and cathepsin B able to transform the extracellular matrix (ECM) [17]. In this way, these cells promote tumor proliferation and growth, tissue repair and remodeling and angio- and lymphangiogenesis through the production of VEGF, TNF-α and IL-8, and exert an immunosuppressive effect on the inflammatory cells [18]. The tumor microenvironment is formed first of all by TAM M2. A growing tumor uses substantial amounts of nutrients and oxygen, but lack of contact with the network of blood vessels inside it frequently leads to hypoxia. Hence, angiogenesis is an indispensable stage of cancer progression. The development of new blood vessels depends on the recruitment of TAMs which recognize hypoxia-associated signals. The production and release of TAMs as well as other cells of important proangiogenic factors, such as VEGF, IL-8 (CXCL8), EGF, CXCL1 and hypoxia- induced factor 1 alpha (HIF1α) are regulated by NF-kB, STAT3 and AP-1 [19]. Under the hypoxia condition, HIF-1α stimulates the expression of CXCL12, which leads to the activation and recruitment of endothelial cells [12]. Hypoxia decreases the expression of MMP-9 and TIMP-1 by increasing the expression of disintegrin and metalloproteinase domain-containing protein 8 & 9 coding genes (ADAM8 & ADAM9) [16]. HIF-1α is also the major transcription factor affecting the synthesis of VEGF—the major factor in the initial stages of new blood vessels [12].
TAMs, especially TAM M2, have a significant potential as diagnostic biomarkers in many cancers: breast, gastric, prostate and pancreatic cancer [16]. According to the newest knowledge TAMs could also be used as a prognostic biomarker in lung, esophageal squamous cell and bladder cancer [20].
Tumor-associated fibroblasts (TAFs)
During tumor development, its stromal fibroblasts also undergo some phenotypic changes [21]. Tumor associated fibroblasts (TAFs) do not have a direct effect on cancer growth but they affect healthy fibroblasts in the tumor microenvironment, changing their phenotype to the proneoplastic one. The origin and role of TAFs are poorly understood but TAFs in the tumor environment become activated and secrete a multitude of factors involved in tumorigenesis. TAFs produce extracellular matrix proteins (collagen and fibronectin), basement membrane proteins and metalloproteinases (MMP-1, -7, -9). Other soluble factors secreted by TAFs: IL-10, -13 possess an immunosuppressive effect, and inhibit synthesis of pro-inflammatory cytokines (e.g. IFN-γ, TNF-α, IL-2, -3) and TGF-β, can promote cancer invasiveness and metastasis [22]. TAFs can also promote neoangiogenesis by synthesis of VEGF [12].
Tumor-associated neutrophils (TANs)
Neoplastic cells producing chemokines (e.g. CXCL1), cytokines (TNF-α and IFN-γ) and adhesion proteins can also recruit neutrophilic granulocytes to the tumor microenvironment. Tumor associated neutrophils (TANs) have both pro- and anti-neoplastic functions. Similarly to macrophages, they have two phenotypes: N1 can inhibit tumor growth by increasing the expression of MMP-8, inducing apoptosis via FAS pathway activation and as a result of triggering antibody-dependent cellular cytotoxicity [23]. The N2 phenotype shows a proneoplastic action through the enhancement of MMP-9 expression and release of CXCL-1 and -6, which recruit new proinflammatory cells to the tumor microenvironment. As a consequence of tumor growth and derived cytokines (IL-8 and G-CSF), also NETs (neutrophil extracellular traps) are formed, possibly as a side-effect of the cytokine environment [24]. Its role in cancer is still unclear and the first description of tumor-induced NET formation was published in 2012 [25]. Formation of NETs may seem to be beneficial in fighting infection, as more NETs have a prothrombotic effect inducing intrinsic coagulation pathway, by binding and activation factor XII [26]. It seems that NETs in tumor-bearing mice were associated with the formation of thrombi in the lungs. Experimental research in cancer-bearing mice indicates that NETs also accumulate in the peripheral circulation, cause systemic inflammation and significantly reduce vascular function in the organs that are not sites for either primary or metastatic tumor growth [27].
Tumor-associated platelets (TAPs)
However the phrase “tumor associated platelets” can be found in an earlier publication [28], we are the first to postulate that this term and its abbreviation (TAPs) should be commonly used in the scientific literature, along with TAMs, TAFs, and TANs. Taking into account the role of platelets in the development of cancer, it is justified to introduce the term TAPs. To the best of our knowledge, so far nobody has used this abbreviation, but its use seems to be justified. However, it is difficult to make such a strict division into TAP P1 and P2 as in the case of neutrophils and macrophages, because TAPs’ role is undoubtedly more complex. One example might be the role of CD40 ligand (CD40L) secreted from the granules of active platelets [29]. On the one hand CD40L can induce apoptosis of cancer cells, while on the other hand it can enhance tumor growth and progression [30]. The effect induced by CD40L appears to depend not only on the type of cells that show the receptors’ expression but also on the strength of the signal transmitted by the ligand. The strong signal (high number of CD molecules)—induces apoptosis of cancer cells, whereas the weak signal (small number of CD receptors for the ligand) promotes tumor growth [31].
Tumor-associated platelets (TAPs) like other cells infiltrate the tumor environment, although reports on the effect of blood platelets on tumor growth are inexplicit. It has been indicated that factors released from platelets can induce a cytotoxic effect on the proliferating neoplastic cells, cause mitotic arrest in the G0/G1 phase, and even enhance apoptosis. However, many researchers seem to suggest an active role for platelets in tumor growth and metastasis formation [32].
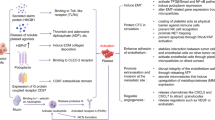
The interactions of blood platelets with cancer cells are complex. The latter induce platelet activation and aggregation both directly and indirectly. Cancer-dependent platelet activation involves both substances secreted by tumor cells and surface molecules of tumor cells [33] (Fig. 1).

Platelet activity in cancer. ADP adenosine diphosphate, ANG-1 angiopoietin 1, CLEC-2 C-type lectin receptor 2, CP cancer procoagulant, EGF epidermal growth factor, LPA lysophosphatidic acid, MMPs matrix metalloproteinases, P2Y12 chemoreceptor for adenosine diphosphate, PAR -1, -4 protease-activated receptor -1, -4, PDGF platelet-derived growth factor, TCIPA tumor cell-induced platelet aggregation, TF tissue factor, TGF-β transforming growth factor β, TXA2 Thromboxane A2, VEGF vascular endothelial growth factor
Tumor cells-platelet binding is possible due to platelet receptors and molecules present on the platelet surface, e.g. GP Ib-IX-V, GP IIb-IIIa, GP V, P-selectin and CLEC-2 (C-type lectin-like receptor 2) (Fig. 2). Newly discovered CLEC-2 and its activator, podoplanin (PDPN) – expressed on multiple tumor cells (e.g. colorectal, lung and bladder carcinomas), are key in platelet aggregation and seem to be important in platelet-cancer cell interaction [34][35][36]. It is possible that CLEC-2 inhibitors could be the new potential antitumor therapeutic target. In an experimental study with several monoclonal antibodies against PDPN-CLEC-2 interactions revealed a decrease in the hematogenous metastasis rate without significantly increasing risk of bleeding [37][38].

Binding of platelet receptors to ligands on cancer cells. CD cluster of differentiation, CLEC-2 C-type lectin receptor 2, GPIb-V-IX glycoprotein Ib-V-IX complex, GPIIbIIIa glycoprotein IIb/IIIa, integrin αIIbβ3, GPVI glycoprotein VI, PAR protease-activated receptor, PDGF platelet-derived growth factor, PDGF-R platelet-derived growth factor receptor, PSGL-1 P-selectin glycoprotein ligand-1, TCIPA tumour cell-induced platelet aggregation, TGF-β transforming growth factor β, TGF-β-R transforming growth factor β receptor, α2β1 integrin also known as VLA-2, GPIa-IIa, CD49b
Platelets are indirectly activated by tumor cells via coagulation activation induced mainly by TF present on tumor cells [39]. Thus, on the one hand platelets can affect the development of the tumor microenvironment, vascular neoangiogenesis and as a consequence metastasis formation. On the other hand, however, cancer cells themselves stimulate blood platelets by secreting TF, CP and collagen, and by using them to initiate processes that warrant survival and proliferation [40]. Adhesion and tumor cell-induced platelet aggregation are very important processes contributing to cancer progression regulated by platelets and their granular contents. Platelet aggregation in response to tumor cell stimulation is known as tumor cell-induced platelet aggregation (TCIPA) [41]. Blood platelets are also a source of TGF-β (transforming growth factor-β), which as an activator of the NF-κB signaling pathway in transformed cells stimulates cancer progression e.g. by promoting metastasis formation [42]. It also stimulates differentiation of endothelial cells into mesenchymal cells able to migrate and proliferate. The epithelial-mesenchymal transition (EMT) is an indispensable process promoting cancer metastases [38]. Platelet molecules such as P-selectin and GP IIb/IIIa have also been implicated in platelet-induced NET formation [40][27]. It is believed that NETs constitute a scaffold for platelet activation and thrombus formation.
Platelets in repressing immune response
To survive in the circulatory system, tumor cells need to finagle immune cells. Therefore they turn to subvert the immune system by hijacking its antitumor properties or attracting immunosuppressive cells. For example, tumor cells may bear a resemblance to platelets by presenting several platelets’ receptors [28]. Circulating tumor cells can also bind to platelets, which protects them from NK cells. This is achieved by the translocation of the major histocompatibility complex class I (MHC class I) from platelets to the tumor cells, resulting in the antitumor properties of NK cells. Moreover, platelets release TGF-β, which reduces the expression of natural-killer group 2, member D on NK cells (NKG2D), and thus leads to the impairment of interferon-γ production and NK cell cytotoxicity. TGF-β also inhibits NK cells activation/function via the suppression of mTOR activity [28, 43, 44].
Platelets can also exert antitumor immunity activity through the debilitation of tumor-infiltrating lymphocytes (TILs). TGF-β released from platelets converts CD4 + (Tcony) cells into induced regulatory T (iTreg) cells, which kills activated T cells in a granzyme B (GzmB)-dependent fashion [44]. Moreover, platelets constitutively express the non-signaling TGF-β-docking receptor glycoprotein A repetitions predominant (GARP) [28]. The role of GARP relies on increases of the functional TGFβ activation in the neighborhood of GARP-expressing cells, which encompass regulatory T (Treg) cells. Rachidi et al. [45] show that the major source of TGFβ in the circulatory system and in the tumor microenvironment are platelets. This is achieved by a constitutive expression of GARP rather than secretion of TGFβ per se. GARP-TGFβ complex on platelets can be formed intracellularly, via the de novo biogenesis, or extracellularly where latent TGFβ (LTGFβ) is snatched by and bound to GARP in the extracellular matrix from non-platelet sources [45]. Besides TGFβ, also other soluble factors released from platelets, mostly lactate, may suppress: T cell proliferation, blastogenesis, and IFNγ production [44, 45].
The discovery of platelet-mediated T cell suppression undoubtedly creates perspectives for the combination of immunotherapy and anti-platelet agents as a therapeutic strategy against cancer [44, 45]. The effectiveness of such therapy has been proven recently by Rachidi et al. [45] in mouse models.
Platelets in tumor angiogenesis
Neoplastic tumors, bigger than 2 mm in diameter, need nutrients and oxygen, as well as suitable cytokines, chemokines or enzymes to grow and metastasize. Platelets are implicated in the early stages of angiogenesis e.g. in the stabilization of newly formed vessels [46]. They stimulate the activation of endothelial cells (ECs) and induce angiogenesis in vivo. Moreover, platelets store in alpha-granules several molecules which exhibit proangiogenic properties like: VEGF (vascular endothelial growth factor), PDGF (platelet derived growth factor), bFGF (basic fibroblast growth factor), EGF (epidermal growth factor) [47]. It is suggested that blood platelets can selectively release VEGF via activation of PAR-1 receptors, and that at the same time the expression of endostatin inhibiting angiogenesis is decreased [48]. The stimulation of the PAR-4 receptor triggers the completely opposite process, i.e. endostatin secretion [49]. It is believed that over 80% of circulating VEGF originates from blood platelets and megakaryocytes, which in physiological conditions promote the repair of damaged tissues and vessels, and its action is inhibited by adequate inhibitors [50].
In many cancers (e.g. lung, breast, colon, and kidney) the platelet count was found to significantly correlate with plasma or serum VEGF concentration [51,52,53]. High VEGF level is associated with shorter overall survival in patients with carcinoma [54, 55]. As shown in our earlier report, blood platelets form complexes with tumor cells in the blood stream. At the site of tumor cell adhesion to ECs, platelets can release their α-granule content, e.g. VEGF, which induces permeability of ECs, facilitates extravasation of cancer cells, and stimulates new vessel formation at the sites of distant metastases. The action of VEGF in neoplastic disease leads to enhanced TF expression by endothelial cells and increased release of vWF. In consequence, platelets adhere to the wall of neoplastic vessels. Moreover, abnormally developed vessels within tumors cause turbulent blood flow, which in turn is associated with the activation and adhesion of platelets and their degranulation [39]. Blood platelets can also stimulate proliferation of endothelial cells and affect the integrity of newly formed or inflamed endothelial cells. This is associated with hemorrhage prevention by releasing substances that affect the adhesion properties of cells [46]. In the course of chronic inflammation and in neoplastic disease, metalloproteinases, serine proteases and other substances that damage the vascular basement membrane are released, leading to hemorrhages. In this case, the role of blood platelets is associated with modulation of the activity of inflammatory cells or inhibition of certain substances by the secretion of such inhibitors as TIMP-1 or serpin [56].
Moreover blood platelets also synthesize and secrete a variety of proteolytic enzymes, such as metalloproteinases (MMP-1,-2, -3, -9 and -14) that degrade the vascular basement membrane and cellular matrix of tissues, which may play a role in the process of extravasation, i.e. migration of cancer cells to the extravascular space [57]. This effect can be enhanced by activation and accumulation of inflammatory cells. MMPs can regulate sCD40L shedding from platelets and pulmonary recruitment of neutrophils [58].
Enhanced proliferation and survival of transformed cells is induced by thrombin, which in turn enhances VEGF expression, increases the adhesion properties of cells and promotes formation of cancer-platelet aggregates [59]. Through the activation of the PAR-1 receptor, thrombin stimulates cancer growth and metastases [60]. Thrombin is released by active blood platelets but it can also be produced by cancer cells.
Platelets in cancer metastasis
Metastasis formation is a most important stage of tumor progression. Transformed cells leave the tumor microenvironment and migrate in the blood to even the most distant organs and their survival before extravasation is a crucial step in metastasis [34]. Therefore, cancer cells activate blood platelets by stimulating various mediators such as cathepsin G or thrombin and by constant expression of tissue factor on their surface. The activated platelets quickly bind to the surface of cancer cells and create their coating [61]. TCIPA is considered crucial for cancer dissemination and resembles the formation of the platelet-monocytes complexes (MPAs). Thus, it can be said that TCIPA increases survival of circulating tumor cells due to the presence of integrins and the GP IIb/IIIa receptor on the platelet surface, which via fibrinogen or vWF interact with tumor integrins. The interaction of platelets with cancer cells is also mediated by P-selectin, which binds to appropriate ligands, e.g. CD24 and CD44 on cancers [62]. Platelet-covered tumor cells can migrate freely, avoiding the immune response of the host, and above all the uptake and damage by NK cells [63]. So it can indicate that platelets may provide some physical barrier to NK cells contact.
Some antiplatelet drugs can have an antimetastatic effect by TCIPA inhibition, e.g. ADP-scavenging agents, apyrase and creatine phosphokinase and also GP IIb/IIA inhibitors [64].
An experimental study, in several mouse models, indicate that antibody-induced or genetic depletion of platelet inhibits metastasis, whereas platelet reconstitution restores metastatic activity [65,66,67]. What’s more, platelets arrest cancer cells in capillaries at the vascular wall via P-selectin and its ligand and facilitate tumor cell extravasation to the subendothelial matrix of the distant organs by activation of the endothelial P2Y2 receptor [68, 69]. Platelet dense granules secrete ATP, which in turn bind to activated endothelial receptors. It leads to the opening of the endothelial barrier and tumor cells can transmigrate and extravasate to form metastasis loci [70], and provide a survival signal, which in turn allows a chemotactic gradient of CCL2 chemokine, which is crucial for monocytes recruitment [71, 72]. The monocytes then differentiate into metastasis-associated macrophages (MAMs) that promote extravasation [73]. Platelets together with leukocytes and tumor cells, induced another chemokine—CCL5/RANTES from EC in an experimental model of colorectal cancer, which enhanced metastatic seeding due to recruitment of monocytes [74].
Besides VEGF, platelets release growth factors to the tumor microenvironment, like TGF-β1 and PDGF, enhancing tumor metastatic potential [38] and instigate tumor cell proliferation, vessel formation and invasiveness [27, 68]. TGF-β1 derived from platelets diminishes NK granule mobilization, cytotoxicity and INF-γ secretion [75] and induced an EMT (epithelial to mesenchymal transition) in carcinoma cells [68] TGF-β1 activity is associated with cancer stages, and microenvironment, and can have the opposite effect. In early stages, it can act as a tumor suppressor and potentially inhibit cancer cell proliferation and tumor growth [76, 77]. However, TGF-β1 released by platelets into the microenvironment can support tumor growth and metastasis formation [76]. In many cancers inhibition of TGF-β signaling in cancer cells strongly reduced intravasation and metastasis [78]. PDGF is involved in the regulation of proteolytic enzyme activity and its elevated concentration in breast cancer reflects a more aggressive and advanced tumor phenotype [79]. Its expression has been identified in various types of tumors, e.g. prostate [66] and colorectal cancer in which it has been proposed as a prognostic marker [80].
As shown in the latest reports, blood platelets are also involved in the formation of the tumor microenvironment during metastasizing [81]. It is believed that the platelet-tumor aggregates are already responsible for neutrophil recruitment within the first two hours after cancer cells have colonized a new site. In this early phase of metastatic formation, no inflow of monocytes, lymphocytes, dendritic cells or NK cells is observed. It appears that the attraction of neutrophils to the tumor microenvironment depends on platelet activation. When functioning of these cells is impaired or in thrombocytopenia this process does not occur. The chemokine CXCL5/7 which is released from blood platelets and then binds to CXCR2 on the surface of neutrophils, is responsible for the activation and migration of granulocytes [82].
It seems that the ability of platelets to form aggregates between tumor cells, platelets and leukocytes may be a crucial step in determining tumor cell survival within the microvasculature of the target organs of metastasis [71].
Platelets count in malignancy
The development of neoplastic disease affects not only platelet activation but also their morphological parameters, i.e. platelet count (PLT) and mean platelet volume (MPV) [83]. A significant drop in PLT—thrombocytopenia—can be associated with intensive chemotherapy and the presence of metastases in bone marrow exerting a suppressive effect on the precursor cells. Cancer patients show increased platelet turnover [84] which improves after anticancer treatment [85, 86].
A systematic drop in platelet count leads to slight bleeding from tiny vessels, whereas considerable thrombocytopenia triggers massive hemorrhages from large blood vessels [81]. Thrombocytopenia experimentally induced by a variety of mechanisms is associated with a reduction in the number of cancer metastases [87].
During the course of cancer, reactive thrombocytosis (more than 400 × 109/L) is much more common and observed in 10–57% of patients, depending on the tumor type [39]. It has long been known that the increased platelet count is associated with more advanced neoplastic disease and is an unfavorable prognostic factor in various cancers [71, 43]. Thrombocytosis is also positively correlated with shortened survival and poor prognosis [88]. However, the underlying mechanisms of thrombocytosis are not completely clear. It is suggested that enhanced production of blood platelets is stimulated by cytokines IL-1, IL-6, and growth factors: GM-CSF and G-CSF released from leukocytes during inflammation and by cancer cells themselves [89]. It is well documented that several tumor cells can produce TPO (thrombopoietin), a main cytokine directly and indirectly regulating megakaryopoiesis and thrombocytopoiesis [90]. These factors stimulate thrombocytopoiesis through binding to the receptors present on megakaryocytes Mpl (TPO-R) [77]. They act synergistically with TPO. In turn, the action of IL-6 is associated with enhanced production of TPO in the liver and a direct effect on megakaryocytes via the membrane receptor IL-6R. This means that platelet count may be markedly increased in the course of inflammation and neoplastic disease. It should also be mentioned that megakaryocytes are able to produce proinflammatory cytokines, e.g. IL-1, -3, -6, GM-CSF, stimulating endothelial cells of bone marrow vessels to produce factors that maintain maturation and differentiation of precursor cells and in consequence platelet production [91]. Moreover, it is known that bone marrow endothelial cells (BMECs) can support megakaryocytopoiesis. BMECs support proliferation and differentiation of megakaryocytic progenitor cells in vitro as well as facilitating the growth and maturation of megakaryocytes in vivo, and release some cytokines, such as IL-6 and TPO [77].
The increased platelet count in the course of neoplastic disease is detrimental and favors the risk of thrombosis which can be the first manifestation of neoplastic disease [92]. Hypercoagulability and excessive thrombin generation, which are commonly observed in the course of malignancy, may contribute to thrombosis in cancer patients, which in fact can be the first symptoms among cancer patients [93].
Targeting platelets in cancer treatment
A wealth of data in the available literature provides information that targeting platelets in cancer treatment would be beneficial [94,95,96,97,98]. Only single studies indicate contrary properties to such an approach [99, 100]. The multitude of reports on this topic highlights the fact that this issue could benefit from being subjected to a separate review. However, Table 1, specially prepared for the purposes of this review, summarizes the main targets of antiplatelet therapy, their best known inhibitors, along with their mechanisms of action and the effect they have on cancer (Table 1). In the clinic, the targeting of only cyclooxygenase molecules is well-characterized [94,95,96, 101,102,103]. Studies concerning other platelet targets (P2Y12, glycoproteins, integrins, L-, P-selectins, PAR-1,-4, thrombin activity, podoplanin-CLEC-2) have so far been utilized mainly on cell lines or animal models [64, 104,105,106,107,108,109]. The targeting of cancer cell-platelet interaction reduces both metastasis and thrombosis [97, 110,111,112,113]. However, this interaction is still not completely understood, thus future studies are required to explicitly explain the role of anti-platelet therapy in cancer prevention and treatment.
Conclusions
There is growing evidence for a close relationship between platelets and cancer development. This specific cross-talk between tumor cells and platelets has been noticed by many authors. Taking into the account the role of platelets in the development of cancer, we have decided to introduce a new term: tumor associated platelets—TAPs. To the best of our knowledge, so far nobody has used this term. Platelets are indirectly activated by tumor cells via coagulation activation induced mainly by TF present on tumor cells. Activated platelets protect tumor cells against cytotoxic activity of NK cells, surrounding them and forming tumor cell-platelet aggregates. Activated platelets also release a number of factors from their granules that can enhance the development of the tumor microenvironment, vascular neoangiogenesis and in consequence metastasis formation. On the other hand, it also has been indicated that factors released from platelets can induce a cytotoxic effect on the proliferating neoplastic cells, and even enhance apoptosis. In respect to the above mentioned information it should be noted that TAPs’ role seems to be more complex as compared to tumor associated neutrophils and macrophages, which does not allow for the easy division of TAPs into TAP P1 and TAP P2. Nevertheless, better exploring the interactions between platelets and tumor cells could help to propose new therapeutic goals that support or improve the effectiveness of cancer treatment.
Data availability
The datasets generated and analyzed during the current study are not publicly available but all are kept at the Medical University of Bialystok and available from the corresponding author (VD-P) on reasonable request.
References
Virchow R (1972) Cellular pathology as based upon physiological and pathological histology. Ann Intern Med 76:157. https://doi.org/10.7326/0003-4819-76-1-157_2
Flier JS, Underhill LH, Dvorak HF (1986) Tumors: wounds that do not heal. N Engl J Med 315:1650–1659. https://doi.org/10.1056/NEJM198612253152606
Trinchieri G (2011) Inflammation in cancer: a therapeutic target? Oncology 25:418–420
de Visser KE, Eichten A, Coussens LM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6:24–37. https://doi.org/10.1038/nrc1782
Balkwill F (2004) Cancer and the chemokine network. Nat Rev Cancer 4:540–550. https://doi.org/10.1038/nrc1388
Koper OM, Kamińska J, Sawicki K et al (2018) Cerebrospinal fluid and serum IL-8, CCL2, and ICAM-1 concentrations in astrocytic brain tumor patients. Ir J Med Sci 187:767–775. https://doi.org/10.1007/s11845-017-1695-8
Piskor BM, Pryczynicz A, Lubowicka E et al (2018) Immunohistochemical expression of Fascin-1 in colorectal cancer in relation to clinical and pathological parameters. Folia Histochem Cytobiol. https://doi.org/10.5603/FHC.a2018.0011
Lin W-W, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117:1175–1183. https://doi.org/10.1172/JCI31537
Zińczuk J, Maciejczyk M, Zaręba K et al (2020) Pro-oxidant enzymes, redox balance and oxidative damage to proteins, lipids and DNA in colorectal cancer tissue is oxidative stress dependent on tumour budding and inflammatory infiltration? Cancers (Basel). https://doi.org/10.3390/cancers12061636
Galdiero MR, Garlanda C, Jaillon S et al (2013) Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 228:1404–1412. https://doi.org/10.1002/jcp.24260
Ishigami S, Natsugoe S, Tokuda K et al (2003) Tumor-associated macrophage (TAM) infiltration in gastric cancer. Anticancer Res 23:4079–4083
Sica A, Allavena P, Mantovani A (2008) Cancer related inflammation: the macrophage connection. Cancer Lett 267:204–215. https://doi.org/10.1016/j.canlet.2008.03.028
Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6:1670–1690. https://doi.org/10.3390/cancers6031670
Liu Y-H, Ding Y, Gao C-C et al (2018) Functional macrophages and gastrointestinal disorders. World J Gastroenterol 24:1181–1195. https://doi.org/10.3748/wjg.v24.i11.1181
Dwyer A, Greenland E, Pixley F (2017) Promotion of tumor invasion by tumor-associated macrophages: the role of CSF-1-activated phosphatidylinositol 3 kinase and Src family kinase motility signaling. Cancers (Basel) 9:68. https://doi.org/10.3390/cancers9060068
Räihä MR, Puolakkainen PA (2018) Tumor-associated macrophages (TAMs) as biomarkers for gastric cancer: a review. Chronic Dis Transl Med 4:156–163. https://doi.org/10.1016/j.cdtm.2018.07.001
Brigati C, Noonan DM, Albini A, Benelli R (2002) Tumors and inflammatory infiltrates: friends or foes? Clin Exp Metastasis 19(3):247–258
Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86:1065–1073. https://doi.org/10.1189/jlb.0609385
Ono M (2008) Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci 99:1501–1506. https://doi.org/10.1111/j.1349-7006.2008.00853.x
Yang L, Zhang Y (2017) Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol 10:58. https://doi.org/10.1186/s13045-017-0430-2
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401. https://doi.org/10.1038/nrc1877
Pardali K, Moustakas A (2007) Actions of TGF-β as tumor suppressor and pro-metastatic factor in humans cancer. Biochim Biophys Acta - Rev Cancer 1775:21–62. https://doi.org/10.1016/j.bbcan.2006.06.004
Fridlender ZG, Albelda SM (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis 33:949–955. https://doi.org/10.1093/carcin/bgs123
Kaplan RN, Riba RD, Zacharoulis S et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827. https://doi.org/10.1038/nature04186
Cedervall J, Zhang Y, Huang H et al (2015) Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res 75:2653–2662. https://doi.org/10.1158/0008-5472.CAN-14-3299
von Brühl M-L, Stark K, Steinhart A et al (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209:819–835. https://doi.org/10.1084/jem.20112322
Cedervall J, Hamidi A, Olsson A-K (2018) Platelets, NETs and cancer. Thromb Res 164:S148–S152. https://doi.org/10.1016/j.thromres.2018.01.049
Xu XR, Yousef GM, Ni H (2018) Cancer and platelet crosstalk: opportunities and challenges of aspirin and other antiplatelet agents. Blood 131:1777–1789. https://doi.org/10.1182/blood-2017-05-743187
Kamińska J, Koper OM, Dymicka-Piekarska V et al (2016) Serum soluble CD40L concentration depending on the stage of multiple myeloma and its correlation with selected angiogenic cytokines. Pol Arch Med Wewn 126(5):321–329. https://doi.org/10.20452/pamw.3427
Korniluk A, Kemona H, Dymicka-Piekarska V (2014) Multifunctional CD40L: pro- and anti-neoplastic activity. Tumor Biol 35:9447–9457. https://doi.org/10.1007/s13277-014-2407-x
Huang J, Jochems C, Talaie T et al (2012) Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood 120:3030–3038. https://doi.org/10.1182/blood-2012-05-427799
Riedl J, Pabinger I, Ay C (2014) Platelets in cancer and thrombosis. Hamostaseologie 34:54–62. https://doi.org/10.5482/HAMO-13-10-0054
Amo L, Tamayo-Orbegozo E, Maruri N et al (2014) Involvement of platelet–tumor cell interaction in immune evasion. Potential role of podocalyxin-like protein 1. Front Oncol 4:245. https://doi.org/10.3389/fonc.2014.00245
Lowe KL, Navarro-Nunez L, Watson SP (2012) Platelet CLEC-2 and podoplanin in cancer metastasis. Thromb Res 129:S30–S37. https://doi.org/10.1016/S0049-3848(12)70013-0
Shirai T, Inoue O, Tamura S et al (2017) C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J Thromb Haemost 15:513–525. https://doi.org/10.1111/jth.13604
Suzuki-Inoue K (2011) Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J Biochem 150:127–132. https://doi.org/10.1093/jb/mvr079
Wojtukiewicz MZ, Hempel D, Sierko E et al (2017) Antiplatelet agents for cancer treatment: a real perspective or just an echo from the past? Cancer Metastasis Rev 36:305–329. https://doi.org/10.1007/s10555-017-9683-z
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20:576–590. https://doi.org/10.1016/j.ccr.2011.09.009
Sierko E, Wojtukiewicz MZ (2004) Platelets and angiogenesis in malignancy. Semin Thromb Hemost 30:95–108. https://doi.org/10.1055/s-2004-822974
Olsson AK, Cedervall J (2018) The pro-inflammatory role of platelets in cancer. Platelets 29:569–573. https://doi.org/10.1080/09537104.2018.1453059
Wojtukiewicz MZ, Hempel D, Sierko E et al (2016) Thrombin—unique coagulation system protein with multifaceted impacts on cancer and metastasis. Cancer Metastasis Rev 35:213–233. https://doi.org/10.1007/s10555-016-9626-0
Sierko E, Wojtukiewicz M (2007) Inhibition of platelet function: does it offer a chance of better cancer progression control? Semin Thromb Hemost 33:712–721. https://doi.org/10.1055/s-2007-991540
Wang Y-H, Deng S-J, Yang Y-D et al (2017) The pretreatment thrombocytosis may predict prognosis of patients with colorectal cancer: a systematic review and meta-analysis. Biomark Med 11:195–210. https://doi.org/10.2217/bmm-2016-0214
Wang S, Li Z, Xu R (2018) Humans cancer and platelet interaction, a potential therapeutic target. Int J Mol Sci 19:1–15. https://doi.org/10.3390/ijms19041246
Rachidi S, Metelli A, Riesenberg B et al (2017) Platelets subvert T cell immunity against cancer via GARP-TGF axis. Sci Immunol. https://doi.org/10.1126/sciimmunol.aai7911
Kisucka J, Butterfield CE, Duda DG et al (2006) Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci 103:855–860. https://doi.org/10.1073/pnas.0510412103
Wojtukiewicz MZ, Sierko E, Hempel D et al (2017) Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev 36:249–262. https://doi.org/10.1007/s10555-017-9673-1
Ma L, Perini R, McKnight W et al (2005) Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from humans platelets. Proc Natl Acad Sci 102:216–220. https://doi.org/10.1073/pnas.0406682102
Italiano JE, Richardson JL, Patel-Hett S et al (2008) Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet α granules and differentially released. Blood 111:1227–1233. https://doi.org/10.1182/blood-2007-09-113837
Holmes CE, Huang JC, Pace TR et al (2008) Tamoxifen and aromatase inhibitors differentially affect vascular endothelial growth factor and endostatin levels in women with breast cancer. Clin Cancer Res 14:3070–3076. https://doi.org/10.1158/1078-0432.CCR-07-4640
Benoy I, Salgado R, Colpaert C et al (2002) Serum interleukin 6, plasma VEGF, serum VEGF, and VEGF platelet load in breast cancer patients. Clin Breast Cancer 2:311–315. https://doi.org/10.3816/CBC.2002.n.008
George ML, Eccles SA, Tutton MG et al (2000) Correlation of plasma and serum vascular endothelial growth factor levels with platelet count in colorectal cancer: clinical evidence of platelet scavenging? Clin Cancer Res 6(8):3147–3152
Dymicka-Piekarska V, Guzinska-Ustymowicz K, Kuklinski A, Kemona H (2012) Prognostic significance of adhesion molecules (sICAM-1, sVCAM-1) and VEGF in colorectal cancer patients. Thromb Res 129:e47–e50. https://doi.org/10.1016/j.thromres.2011.12.004
Kim SJ (2004) Serum vascular endothelial growth factor per platelet count in hepatocellular carcinoma: correlations with clinical parameters and survival. Jpn J Clin Oncol 34:184–190. https://doi.org/10.1093/jjco/hyh039
Ferroni P, Palmirotta R, Spila A et al (2006) Prognostic value of carcinoembryonic antigen and vascular endothelial growth factor tumor tissue content in colorectal cancer. Oncology 71:176–184. https://doi.org/10.1159/000106072
Radomski A, Jurasz P, Sanders EJ et al (2002) Identification, regulation and role of tissue inhibitor of metalloproteinases-4 (TIMP-4) in humans platelets. Br J Pharmacol 137:1330–1338. https://doi.org/10.1038/sj.bjp.0704936
Seizer P, May AE (2013) Platelets and matrix metalloproteinases. Thromb Haemost 110:903–909. https://doi.org/10.1160/TH13-02-0113
Rahman M, Roller J, Zhang S et al (2012) Metalloproteinases regulate CD40L shedding from platelets and pulmonary recruitment of neutrophils in abdominal sepsis. Inflamm Res 61:571–579. https://doi.org/10.1007/s00011-012-0446-6
Yamahata H, Takeshima H, Kuratsu J-I et al (2002) The role of thrombin in the neo-vascularization of malignant gliomas: an intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int J Oncol 20:921–928. https://doi.org/10.3892/ijo.20.5.921
Choi N, Pluthero F, Licht C (2009) Intracellular localization and release of platelet vascular endothelial growth factor. J Undergrad Life Sci
McNicol A, Israels S (2008) Beyond hemostasis: the role of platelets in inflammation, malignancy and infection. Cardiovasc Hematol Disord Targets 8:99–117. https://doi.org/10.2174/187152908784533739
Cheung LS-L, Raman PS, Balzer EM et al (2011) Biophysics of selectin–ligand interactions in inflammation and cancer. Phys Biol 8:015013. https://doi.org/10.1088/1478-3975/8/1/015013
Raab S, Kropp KN, Steinle A et al (2013) Platelets impair NK cell immunosurveillance of metastasizing tumor cells by altering surface expression and shedding of ligands for the activating immunoreceptor NKG2D. Blood 122:3488–3488. https://doi.org/10.1182/blood.V122.21.3488.3488
Lonsdorf AS, Krämer BF, Fahrleitner M et al (2012) Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets. J Biol Chem 287:2168–2178. https://doi.org/10.1074/jbc.M111.269811
Camerer E, Qazi AA, Duong DN et al (2004) Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104:397–401. https://doi.org/10.1182/blood-2004-02-0434
George D (2003) Targeting Pdgf Receptors in Cancer Rationales and Proof of Concept Clinical Trials. In: Advances in Experimental Medicine and Biology. pp 141–151
Bambace NM, Holmes CE (2011) The platelet contribution to cancer progression. J Thromb Haemost 9:237–249. https://doi.org/10.1111/j.1538-7836.2010.04131.x
Schlesinger M (2018) Role of platelets and platelet receptors in cancer metastasis. J Hematol Oncol 11:125. https://doi.org/10.1186/s13045-018-0669-2
McCarty OJT, Mousa SA, Bray PF, Konstantopoulos K (2000) Immobilized platelets support humans colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood 96:1789–1797. https://doi.org/10.1182/blood.V96.5.1789.h8001789_1789_1797
Schumacher D, Strilic B, Sivaraj KK et al (2013) Platelet-Derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24:130–137. https://doi.org/10.1016/j.ccr.2013.05.008
Karachaliou N, Pilotto S, Bria E, Rosell R (2015) Platelets and their role in cancer evolution and immune system. Transl Lung Cancer Res 4(6):713–720
Qian B-Z, Li J, Zhang H et al (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475:222–225. https://doi.org/10.1038/nature10138
Gil-Bernabé AM, Ferjančič Š, Tlalka M et al (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 119:3164–3175. https://doi.org/10.1182/blood-2011-08-376426
Läubli H, Spanaus K-S, Borsig L (2009) Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood 114:4583–4591. https://doi.org/10.1182/blood-2008-10-186585
Kopp H-G, Placke T, Salih HR (2009) Platelet-derived transforming growth factor- down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res 69:7775–7783. https://doi.org/10.1158/0008-5472.CAN-09-2123
Plantureux L, Mège D, Crescence L et al (2020) The interaction of platelets with colorectal cancer cells inhibits tumor growth but promotes metastasis. Cancer Res 80:291–303. https://doi.org/10.1158/0008-5472.CAN-19-1181
Plantureux L, Mège D, Crescence L et al (2018) Impacts of cancer on platelet production, activation and education and mechanisms of cancer-associated thrombosis. Cancers (Basel) 10:1–23. https://doi.org/10.3390/cancers10110441
Biswas S, Guix M, Rinehart C et al (2007) Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest 117:1305–1313. https://doi.org/10.1172/JCI30740
Jabeen S, Espinoza JA, Torland LA et al (2019) Noninvasive profiling of serum cytokines in breast cancer patients and clinicopathological characteristics. Oncoimmunology 8:1–11. https://doi.org/10.1080/2162402X.2018.1537691
Nakamura Y, Tanaka F, Yoshikawa Y et al (2008) PDGF-BB is a novel prognostic factor in colorectal cancer. Ann Surg Oncol 15:2129–2136. https://doi.org/10.1245/s10434-008-9943-9
Labelle M, Begum S, Hynes RO (2014) Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci 111:E3053–E3061. https://doi.org/10.1073/pnas.1411082111
Kawamura M, Toiyama Y, Tanaka K et al (2012) CXCL5, a promoter of cell proliferation, migration and invasion, is a novel serum prognostic marker in patients with colorectal cancer. Eur J Cancer 48:2244–2251. https://doi.org/10.1016/j.ejca.2011.11.032
Korniluk A, Koper-Lenkiewicz OM, Kamińska J et al (2019) Mean platelet volume (MPV): new perspectives for an old marker in the course and prognosis of inflammatory conditions. Mediators Inflamm 2019:1–14. https://doi.org/10.1155/2019/9213074
Donati MB, Falanga A (2001) Pathogenetic mechanisms of thrombosis in malignancy. Acta Haematol 106:18–24. https://doi.org/10.1159/000046585
Hoffmann JJML (2014) Reticulated platelets: analytical aspects and clinical utility. Clin Chem Lab Med 52:1107–1117. https://doi.org/10.1515/cclm-2014-0165
Wang C, Smith BR, Ault KA, Rinder HM (2002) Reticulated platelets predict platelet count recovery following chemotherapy. Transfusion. https://doi.org/10.1046/j.1537-2995.2002.00040.x
Haemmerle M, Stone RL, Menter DG et al (2018) The platelet lifeline to cancer: challenges and opportunities. Cancer Cell 33:965–983. https://doi.org/10.1016/j.ccell.2018.03.002
Ji Y, Sheng L, Du X et al (2015) Elevated platelet count is a strong predictor of poor prognosis in stage I non-small cell lung cancer patients. Platelets 26:138–142. https://doi.org/10.3109/09537104.2014.888547
Kaser A, Brandacher G, Steurer W et al (2001) Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood 98:2720–2725. https://doi.org/10.1182/blood.V98.9.2720
Stone RL, Nick AM, McNeish IA et al (2012) Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med 366:610–618. https://doi.org/10.1056/NEJMoa1110352
Deutsch VR, Tomer A (2006) Megakaryocyte development and platelet production. Br J Haematol 134:453–466. https://doi.org/10.1111/j.1365-2141.2006.06215.x
Dymicka-Piekarska V, Kemona H (2009) Does colorectal cancer clinical advancement affect adhesion molecules (sP- selectin, sE- selectin and ICAM-1) concentration? Thromb Res 124:80–83. https://doi.org/10.1016/j.thromres.2008.11.021
Harrison CN (2005) Platelets and thrombosis in myeloproliferative diseases. Hematology 2005:409–415. https://doi.org/10.1182/asheducation-2005.1.409
Gallicchio L, McSorley MA, Newschaffer CJ et al (2006) Nonsteroidal antiinflammatory drugs, cyclooxygenase polymorphisms, and the risk of developing breast carcinoma among women with benign breast disease. Cancer 106:1443–1452. https://doi.org/10.1002/cncr.21763
Slatore CG, Au DH, Littman AJ et al (2009) Association of nonsteroidal anti-inflammatory drugs with lung cancer: results from a large cohort study. Cancer Epidemiol Biomarkers Prev. https://doi.org/10.1158/1055-9965.EPI-08-1110
Jacobs CD, Chun SG, Yan J et al (2014) Aspirin improves outcome in high risk prostate cancer patients treated with radiation therapy. Cancer Biol Ther 15:699–706. https://doi.org/10.4161/cbt.28554
Shiao J, Thomas KM, Rahimi AS et al (2017) Aspirin/antiplatelet agent use improves disease-free survival and reduces the risk of distant metastases in stage II and III triple-negative breast cancer patients. Breast Cancer Res Treat 161:463–471. https://doi.org/10.1007/s10549-016-4081-8
Lee PC, Yeh CM, Hu YW et al (2016) Antiplatelet therapy is associated with a better prognosis for patients with hepatitis b virus-related hepatocellular carcinoma after liver resection. Ann Surg Oncol 23:874–883. https://doi.org/10.1245/s10434-016-5520-9
Erpenbeck L, Nieswandt B, Schön M et al (2010) Inhibition of platelet GPIbα and promotion of melanoma metastasis. J Invest Dermatol 130:576–586. https://doi.org/10.1038/jid.2009.278
Serebruany VL, Cherepanov V, Cabrera-Fuentes HA, Kim MH (2015) Solid cancers after antiplatelet therapy: confirmations, controversies, and challenges. Thromb Haemost. https://doi.org/10.1160/TH15-01-0077
Sandler RS, Halabi S, Baron JA et al (2003) A randomized trial of aspirin to prevent colorectal adenomas in patients with previous. Eur J Gastroenterol Hepatol 15:579–580. https://doi.org/10.1097/00042737-200305000-00032
Bertagnolli MM, Eagle CJ, Zauber AG et al (2006) Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. https://doi.org/10.1056/NEJMoa061355
Pruthi RS, Derksen JE, Moore D et al (2006) Phase II trial of celecoxib in prostate-specific antigen recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-05-2067
Gareau AJ, Brien C, Gebremeskel S et al (2018) Ticagrelor inhibits platelet–tumor cell interactions and metastasis in humans and murine breast cancer. Clin Exp Metastasis 35:25–35. https://doi.org/10.1007/s10585-018-9874-1
Wang Y, Sun Y, Li D et al (2013) Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE 8:1–12. https://doi.org/10.1371/journal.pone.0080780
Villares GJ, Zigler M, Wang H et al (2008) Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-08-2397
Goertz L, Schneider SW, Desch A et al (2016) Heparins that block VEGF-A-mediated von Willebrand factor fiber generation are potent inhibitors of hematogenous but not lymphatic metastasis. Oncotarget 7(42):68527–68545. https://doi.org/10.1863/ONCOTARGET.11832
Xu M, Wang X, Pan Y et al (2019) Blocking podoplanin suppresses growth and pulmonary metastasis of humans malignant melanoma. BMC Cancer 19:1–11. https://doi.org/10.1186/s12885-019-5808-9
Yu Y, Zhou XD, Liu YK et al (2002) Platelets promote the adhesion of humans hepatoma cells with a highly metastatic potential to extracellular matrix protein: Involvement of platelet P-selectin and GP IIb-IIIa. J Cancer Res Clin Oncol. https://doi.org/10.1007/s00432-002-0325-6
Menter DG, Schilsky RL, DuBois RN (2010) Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clin Cancer Res 16:1384–1390. https://doi.org/10.1158/1078-0432.CCR-09-0788
Menter DG, Tucker SC, Kopetz S et al (2014) Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev 33:231–269. https://doi.org/10.1007/s10555-014-9498-0
Rothwell PM, Wilson M, Price JF et al (2012) Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379:1591–1601. https://doi.org/10.1016/S0140-6736(12)60209-8
Holmes CE, Levis JE, Schneider DJ et al (2016) Platelet phenotype changes associated with breast cancer and its treatment. Platelets. https://doi.org/10.3109/09537104.2016.1171302
Zhao L, Zhang W, Chen M et al (2013) Aspirin induces platelet apoptosis. Platelets 24:637–642. https://doi.org/10.3109/09537104.2012.754417
Mitrugno A, Sylman JL, Ngo ATP et al (2017) Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: Implications for the oncoprotein c-MYC. Am J Physiol - Cell Physiol 312:C176–C189. https://doi.org/10.1152/ajpcell.00196.2016
García Rodríguez LA, Soriano-Gabarró M, Bromley S et al (2017) New use of low-dose aspirin and risk of colorectal cancer by stage at diagnosis: a nested case-control study in UK general practice. BMC Cancer 17:1–11. https://doi.org/10.1186/s12885-017-3594-9
Baron JA, Cole BF, Sandler RS et al (2003) A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. https://doi.org/10.1056/NEJMoa021735
Guillem-Llobat P, Dovizio M, Bruno A et al (2016) Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 7(22):32462–32477. https://doi.org/10.1863/oncotarget.8655
Vad NM, Kudugunti SK, Wang H et al (2014) Efficacy of acetylsalicylic acid (aspirin) in skin B16–F0 melanoma tumor-bearing C57BL/6 mice. Tumor Biol 35:4967–4976. https://doi.org/10.1007/s13277-014-1654-1
Drew DA, Chin SM, Gilpin KK et al (2017) ASPirin intervention for the REDuction of colorectal cancer risk (ASPIRED): a study protocol for a randomized controlled trial. Trials 18:1–11. https://doi.org/10.1186/s13063-016-1744-z
Benamouzig R, Uzzan B, Deyra J et al (2012) Prevention by daily soluble aspirin of colorectal adenoma recurrence: 4-year results of the APACC randomised trial. Gut. https://doi.org/10.1136/gutjnl-2011-300113
Holmes MD, Chen WY, Li L et al (2010) Aspirin intake and survival after breast cancer. J Clin Oncol 28:1467–1472. https://doi.org/10.1200/JCO.2009.22.7918
Rothwell PM, Wilson M, Elwin CE et al (2010) Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 376:1741–1750. https://doi.org/10.1016/S0140-6736(10)61543-7
Rothwell PM, Fowkes FGR, Belch JF et al (2011) Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377:31–41. https://doi.org/10.1016/S0140-6736(10)62110-1
Choe KS, Cowan JE, Chan JM et al (2012) Aspirin use and the risk of prostate cancer mortality in men treated with prostatectomy or radiotherapy. J Clin Oncol 30:3540–3544. https://doi.org/10.1200/JCO.2011.41.0308
Cho MS, Noh K, Haemmerle M et al (2017) Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 130:1235–1242. https://doi.org/10.1182/blood-2017-02-769893
Zhang R, Guo H, Xu J et al (2016) Activated platelets inhibit hepatocellular carcinoma cell differentiation and promote tumor progression via platelet-tumor cell binding. Oncotarget 7(37):60609–60622. https://doi.org/10.1863/oncotarget.11300
Herbert JM, Savi P (2003) P2Y12, a new platelet ADP receptor, target of clopidogrel. Semin Vasc Med. https://doi.org/10.1055/s-2003-40669
Mezouar S, Darbousset R, Dignat-George F et al (2015) Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer 136:462–475. https://doi.org/10.1002/ijc.28997
Jin J, Quinton TM, Zhang J et al (2002) Adenosine diphosphate (ADP)-induced thromboxane A2 generation in humans platelets requires coordinated signaling through integrin αIIbβ3 and ADP receptors. Blood 99:193–198. https://doi.org/10.1182/blood.V99.1.193
Roop RP, Naughton MJ, Van Poznak C et al (2013) A randomized phase ii trial investigating the effect of platelet function inhibition on circulating tumor cells in patients with metastatic breast cancer. Clin Breast Cancer. https://doi.org/10.1016/j.clbc.2013.08.006
Gebremeskel S, Levatte T, Liwski RS et al (2015) The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int J Cancer 136:234–240. https://doi.org/10.1002/ijc.28947
Ballerini P, Dovizio M, Bruno A et al (2018) P2Y12 receptors in tumorigenesis and metastasis. Front Pharmacol 9:1–8. https://doi.org/10.3389/fphar.2018.00066
Uluçkan Ö, Eagleton MC, Floyd DH et al (2008) APT102, a novel ADPase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem. https://doi.org/10.1002/jcb.21709
Bambace NM, Levis JE, Holmes CE (2010) The effect of P2Y-mediated platelet activation on the release of VEGF and endostatin from platelets. Platelets. https://doi.org/10.3109/09537100903470298
Mah-Becherel MCM, Céraline J, Deplanque G et al (2002) Anti-angiogenic effects of the thienopyridine SR 25989 in vitro and in vivo in a murine pulmonary metastasis model. Br J Cancer 86:803–810. https://doi.org/10.1038/sj.bjc.6600142
Cooke NM, Spillane CD, Sheils O et al (2015) Aspirin and P2Y12 inhibition attenuate platelet-induced ovarian cancer cell invasion. BMC Cancer 15:1–10. https://doi.org/10.1186/s12885-015-1634-x
Damiano BP, Derian CK, Maryanoff BE et al (2003) RWJ-58259: a selective antagonist of protease activated receptor-1. Cardiovasc Drug Rev 21:313–326. https://doi.org/10.1111/j.1527-3466.2003.tb00124.x
Ottaiano TF, Andrade SS, de Oliveira C et al (2017) Plasma kallikrein enhances platelet aggregation response by subthreshold doses of ADP. Biochimie. https://doi.org/10.1016/j.biochi.2017.01.010
Etulain J, Mena HA, Negrotto S, Schattner M (2015) Stimulation of PAR-1 or PAR-4 promotes similar pattern of VEGF and endostatin release and pro-angiogenic responses mediated by humans platelets. Platelets. https://doi.org/10.3109/09537104.2015.1051953
Melnikova VO, Balasubramanian K, Villares GJ et al (2009) Crosstalk between protease-activated receptor 1 and platelet-activating factor receptor regulates melanoma cell adhesion molecule (MCAM/MUC18) expression and melanoma metastasis. J Biol Chem 284:28845–28855. https://doi.org/10.1074/jbc.M109.042150
Jain S, Russell S, Ware J (2009) Platelet glycoprotein VI facilitates experimental lung metastasis in syngeneic mouse models. J Thromb Haemost 7:1713–1717. https://doi.org/10.1111/j.1538-7836.2009.03559.x
Qi Y, Chen W, Lieng X et al (2018) Novel antibodies against GPIbα inhibit pulmonary metastasis by affecting vWF-GPIbα interaction. J Hematol Oncol 11:1–17. https://doi.org/10.1186/s13045-018-0659-4
Amirkhosravi A, Mousa SA, Amaya M et al (2003) Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost. https://doi.org/10.1160/th03-02-0102
Trikha M, Zhou Z, Timar J et al (2002) Multiple roles for platelet GPIIb/IIIa and αvβ3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res 62:2824–2833
Lu Q, Lv M, Xu E et al (2015) Recombinant hirudin suppresses the viability, adhesion, migration and invasion of Hep-2 humans laryngeal cancer cells. Oncol Rep 33:1358–1364. https://doi.org/10.3892/or.2015.3717
Lavergne M, Janus-Bell E, Schaff M et al (2017) Platelet integrins in tumor metastasis: do they represent a therapeutic target? Cancers (Basel). https://doi.org/10.3390/cancers9100133
Ludwig RJ, Boehme B, Podda M et al (2004) Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res 64:2743–2750. https://doi.org/10.1158/0008-5472.CAN-03-1054
Stevenson JL, Varki A, Borsig L (2007) Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb Res 120:107–111. https://doi.org/10.1016/S0049-3848(07)70138-X
Heinmöller E, Weinel RJ, Heidtmann HH et al (1996) Studies on tumor-cell-induced platelet aggregation in humans lung cancer lines. J Cancer Res Clin Oncol. https://doi.org/10.1007/bf01209121
Gomes AM, Kozlowski EO, Borsig L et al (2015) Antitumor properties of a new non-anticoagulant heparin analog from the mollusk Nodipecten nodosus: effect on P-selectin, heparanase, metastasis and cellular recruitment. Glycobiology 25:386–393. https://doi.org/10.1093/glycob/cwu119
Battinelli EM, Markens BA, Kulenthirarajan RA et al (2014) Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 123:101–112. https://doi.org/10.1182/blood-2013-02-485011
Vianello F, Sambado L, Goss A et al (2016) Dabigatran antagonizes growth, cell-cycle progression, migration, and endothelial tube formation induced by thrombin in breast and glioblastoma cell lines. Cancer Med 5:2886–2898. https://doi.org/10.1002/cam4.857
Niers TMH, Klerk CPW, DiNisio M et al (2007) Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol, Hematol
Asanuma K, Wakabayashi H, Hayashi T et al (2004) Thrombin inhibitor, argatroban, prevents tumor cell migration and bone metastasis. Oncology. https://doi.org/10.1159/000081004
Nieman MT, LaRusch GA, Fang C et al (2010) Oral thrombostatin FM19 inhibits prostate cancer. Thromb Haemost. https://doi.org/10.1160/TH09-08-0570
Asanuma K, Wakabayashi H, Okamoto T et al (2013) The thrombin inhibitor, argatroban, inhibits breast cancer metastasis to bone. Breast Cancer. https://doi.org/10.1007/s12282-012-0334-5
Yuan L, Liu X (2015) Platelets are associated with xenograft tumor growth and the clinical malignancy of ovarian cancer through an angiogenesis-dependent mechanism. Mol Med Rep 11:2449–2458. https://doi.org/10.3892/mmr.2014.3082
Chang YW, Hsieh PW, Chang YT et al (2015) Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget 6(40):42733–42748. https://doi.org/10.1863/oncotarget.5811
Kato Y, Kaneko MK, Kunita A et al (2008) Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci 99:54–61. https://doi.org/10.1111/j.1349-7006.2007.00634.x
Chandramohan V, Bao X, Kato Kaneko M et al (2013) Recombinant anti-podoplanin (NZ-1) immunotoxin for the treatment of malignant brain tumors. Int J Cancer. https://doi.org/10.1002/ijc.27919
Funding
The research did not receive any specific grant from founding agencies in the public, commercial or not for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conceptualization, Violetta Dymicka-Piekarska; data curation, Joanna Kamińska and Justyna Zińczuk; Funding acquisition, Violetta Dymicka-Piekarska; investigation, Violetta Dymicka-Piekarska, Olga M. Koper-Lenkiewicz and Joanna Kaminska; methodology, Violetta Dymicka-Piekarska; project administration, Violetta Dymicka-Piekarska; resources, Violetta Dymicka-Piekarska, Olga M. Koper-Lenkiewicz and Ewa Kratz; supervision, Violetta Dymicka-Piekarska; validation, Violetta Dymicka-Piekarska and Olga M. Koper-Lenkiewicz; visualization, Violetta Dymicka-Piekarska and Joanna Kamińska; writing – original draft, Violetta Dymicka-Piekarska and Olga M. Koper-Lenkiewicz; writing – review & editing, Joanna Kamińska, Justyna Zińczuk and Ewa Kratz.
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dymicka-Piekarska, V., Koper-Lenkiewicz, O.M., Zińczuk, J. et al. Inflammatory cell-associated tumors. Not only macrophages (TAMs), fibroblasts (TAFs) and neutrophils (TANs) can infiltrate the tumor microenvironment. The unique role of tumor associated platelets (TAPs). Cancer Immunol Immunother 70, 1497–1510 (2021). https://doi.org/10.1007/s00262-020-02758-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02758-7