
Abstract
There is accumulating evidence that the transforming growth factor beta (TGF-β) and nuclear factor kappa-B (NFκB) pathways are tightly connected and play a key role in malignant transformation in cancer. Immune infiltration by regulatory T- and B-lymphocytes (Tregs, Bregs) has recently gained increased attention for being an important source of TGF-β. There is a plethora of studies examining the pro-tumorigenic functions of carcinoembryonic antigen (CEA), but its receptor CEAR is far less studied. So far, there is a single connecting report that TGF-β also may signal through CEAR. The crosstalk between cancer tissues is further complicated by the expression of CEAR and TGF-β receptors in stromal cells, and implications of TGF-β in epithelial–mesenchymal transition. Furthermore, tumor-infiltrating Tregs and Bregs may directly instruct cancer cells by secreting TGF-β binding to their CEAR. Therefore, both TGF-β and CEA may act synergistically in breast cancer and cause disease progression, and NFκB could be a common crossing point between their signaling. CEAR, TGF-β1–3, TGF-β-R types I–III and NFκB class I and II molecules have an outstanding human–canine sequence identity, and only a canine CEA homolog has not yet been identified. For these reasons, the dog may be a valid translational model patient for investigating the crosstalk of the interconnected CEA and TGF-β networks.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The strategy of comparative oncology is to find homologous molecules, homologous signaling cascades and homologous immune mechanisms to cure cancer in both humans and pets according to the “One Health” principle [1]. Similar to humans, dogs spontaneously develop malignancies with comparable incidence and prevalence and hence represent a natural model for human cancer. For instance, a Swedish study on 80,000 insured female dogs reported that, dependent on higher age and breed, up to 13 % of female dogs had at least one mammary tumor, with an overall-case fatality of 6 % [2]. In humans, females in more highly developed areas have a cumulative risk of 7.1 % of developing mammary cancer by the age of 75, with a mortality rate of 1.7 % [3]. Mammary carcinoma, among others, is thus a burden in both human and veterinary medicines.
The rationale for favouring this tumor entity for comparative studies derives from the fact that it is wise to have access to primary lesions for monitoring tumor progression by caliper measurements. This facilitates the clinical investigations and also takes into consideration that only few centers have access to imaging facilities. Often more than one mamilla are affected in canine cancer patients and may be compared side by side.
It can further be expected that results from comparative oncology studies, investigating naturally occurring cancers due to distinct risk factors in distinct breeds, have a higher translational potential than studies with genetically highly homologous mouse strains [4]. For example, the epidermal growth factor receptor (EGFR) family members EGFR (ErbB1) and human epidermal growth factor receptor 2 (HER-2 (ErbB2)) are molecules of outstanding homology between humans and dogs, and targeting of these molecules results in the same effects on signaling and cancer biology in both species [5, 6].
A more intricate situation was observed for the carcinoembryonic antigen [CEA, also termed carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5)], which represents a classical soluble as well as membrane-expressed tumor marker in human clinical oncology. Serum levels of soluble human CEA correlate with disease progression [7], and its assessment is recommended in monitoring the treatment course of colorectal cancer in combination with other prognostic markers [8, 9]. However, CEA molecules are structurally and evolutionarily diverse between humans and canines [10, 11]. A direct CEA homolog in dogs has not yet been defined and represents “a missing link” (Table 1). In contrast, overexpression of CEA in humans has been known for over 20 years to play an important role in metastasis and cell motility [12] by acting as a ligand for E- and L-selectins [13] and might have a signaling function probably by interacting with the Wnt pathway [14]. Furthermore, vaccination with an adeno-associated virus (AAV)–CEA vector combined with Toll-like receptor-9 or Toll-like receptor-7 agonists in wild-type mice resulted in enhanced Th1-mediated immunity and protection from challenge with MC38 colon tumor cells expressing CEA, whereas the same CEA vaccines in CEA transgenic animals promoted tumor growth due to tolerance phenomena elicited by dendritic and myeloid cells [15]. Some CEA family members such as CEACAM6 may adhere to and inhibit tumor-infiltrating cytotoxic T cells [16]. CEACAM1, CEACAM5 and CEACAM6 may be released from epithelial tumors in microvesicles, whereas tumor endothelia only contain CEACAM1 which has a receptor function for other CEACAMs, influences T cell behavior [17] and regulates the tumor matrix and microvascularization [18]. Hence, CEA may affect the tumor and its stroma at the same time [19].
CEAR binds TGF-β, a cytokine involved in tolerance induction toward malignant tissue
The scientific history of the carcinoembryonic antigen receptor (CEAR) is much more recent. Interestingly, CEAR showed an outstanding sequence identity of 99 % between the human and canine species [20] (Table 1). The great CEA-receptor homology of humans and dogs on the one hand and the lack of a precise canine CEA equivalent on the other hand are discrepancies and indicate that there could be an alternative ligand. The CEAR was originally described in Kupffer cells and identified as the heterogeneous nuclear ribonucleoprotein M4 (hnRNP M4) [21]. Regarding oncology, it was later also found on colon cancer cells [22]. Moreover, its expression was subsequently also detected in mice in the entire gastrointestinal tract including liver and pancreas [23]. CEAR expression has been connected to inflammation in the liver [24]. In Kupffer cells, a full-length hnRNP M4 (CEARL) and a truncated form (CEARS), generated by alternative splicing, were described [14]. The minimal structural element of human CEACAM5 interacting with hnRNP M4/CEAR was reduced to a peptide of eight amino acids [25].
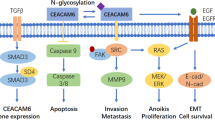
Surprisingly, a recent study has shown that CEA not only signals via its specific receptor, CEAR, but can also bind to the receptor of the important immunomodulatory cytokine transforming growth factor beta (TGF-β, Fig. 1) [26].

Interconnected networks of CEA and TGF-β signaling in cancer. The cancer cell is an autocrine source of CEA as well as of TGF-β which bind to their specific receptors, CEAR or TGF-β-RI:RII, respectively; the latter signaling via the NFκB pathway. Recently, it has been recognized that CEA also signals via TGF-β-R and initiates the same biological effects [26]. Additionally, Tregs and Bregs, as well as stroma cells, participate in this network by secreting TGF-β. It remains open whether the reverse is the case, and TGF-β may also interfere with the CEAR pathway, which is much less defined
TGF-β sources and its function in the tumor
Three high-affinity membrane-bound receptors for TGF-β are known so far: type I, type II and type III. The classical TGF-β signaling, however, occurs via the heterotetrameric complex of 2 TGF-β-receptor (TGF-β-R) type I and 2 TGF-β-receptor type II transmembrane receptors with serine/threonine kinase activity [27–29]. In the tumor microenvironment, TGF-β is most typically derived from human and canine Foxp3+ regulatory T cells (Tregs). It is well known that Tregs can thereby critically dampen anti-tumor immunity and tolerize cytotoxic T cells [30–34]. More recently, intratumoral regulatory B cells (Bregs) have gained attention in human oncology [35, 36]. According to Olkhanud et al. [37], tumor-evoked Bregs should phenotypically resemble activated mature B2 cells (CD19+ CD25hi CD69hi). Lindner et al. [36] reported that intratumoral Bregs also express granzyme-B (stimulated by IL-21 from Tregs) and a signature of CD19+CD38+CD1d+IgM+CD147+, as well as including IL-10, CD25 and indoleamine-2,3-dioxygenase. This population seems interesting as a source of TGF-β and for their capacity to suppress intratumoral CD8+ and CD4+ effector T cells. Bregs can even convert naïve CD4+CD25− T cells to Foxp3+ Tregs [37]. TGF-β, however, may also be derived from tumor stroma cells [19, 38], where it shapes the microenvironment by interacting with growth factors (epidermal growth factor (EGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), insulin-like growth factor (IGF) [39]), cytokines or chemokines, crosstalking to fibroblasts [40] and supporting the enrichment of endothelial cells, which again shape the extracellular matrix [41]. TGF-β promotes the loss of epithelial markers such as E-cadherin and the accumulation of the mesenchymal marker vimentin in the process of epithelial–mesenchymal transition (EMT) [42]. Importantly, in this case tumor stem cells themselves show an enrichment of mesenchymal markers and are a source of TGF-β. Most studies on EMT are done in mouse or human cancer models [43], but there are reports that EMT transition can be achieved by TGF-β in (normal) Madin–Darby canine kidney (MDCK) cells [44].
Physiologically, TGF-β acts as a tumor suppressor, negatively regulating cellular proliferation, but this is changed in the cancer microenvironment toward a tumor promoter function, where it mediates proliferation, migration, invasion, EMT and metastasis, associated with high miR-181a expression, and altogether termed the TGF-β-paradox [45]. In this context, it is important to note that canines are much closer to the human species than murine animal models. The appearance of Tregs also negatively correlates with prognosis in dog cancer patients [46].
For instance, naive CD4+CD25−Foxp3− T cells can be converted to Foxp3+ Tregs when adoptively transferred into Rag−/− mice only in the presence of TGF-β-positive tumors [47]. Thus, the intratumoral milieu amplifies the cellular sources for even more immunosuppressive cytokines. It has been recently shown that elevated levels of TGF-β and IL-6 in the tumor microenvironment support Th17 cells and that the resulting inflammation was supporting the clinical development and progression of gastric cancer [48]. Although Li et al. have shown that CEA binds to TGF-β-R [26], it has not yet been investigated whether the reverse is true, and TGF-β (besides acting via its own TGF-β-R) may crosstalk via CEAR, thereby imitating the tumor-progressive properties of CEA. CEA modulates effector–target interaction by binding to lymphocytes [49]. Only CEACAM1 expression was previously described in T cells [50], whereas the expression of CEACAM5 on T cells was excluded. Regarding this, we are not aware of investigations on the expression of CEAR on T- or B-lymphocytes.
TGF-β signaling
In contrast to CEAR, the cellular signaling function of which has to the best of our knowledge not yet been reported, the signaling cascade for the TGF-β-R is well known. The nuclear factor kappa-B (NFκB) is a key master regulator in growth and survival [51, 52]. In normal cells, TGF-β leads to growth inhibition; in short: TGF-β binds to TGF-β-RII, activating TGF-β-RI and then phosphorylating the SMA and MAD homologs SMAD2 and SMAD3, which associate with SMAD4 and together translocate to the nucleus for transcription of genes. All of this is inhibited by SMAD7 [53]. Interestingly, the TGF-β-R-initiated SMAD pathway was shown to target CEACAM5 (and CEACAM6) genes leading to CEA secretion as a mechanism for proliferation in gastric cancer cells [54]. It will be interesting in the future to investigate whether a synergistic crosstalk between the CEA and TGF-β signaling cascades in cancer cells exists.
In human head and neck squamous cell carcinoma cell lines, Freudlsperger et al. [53] could further demonstrate that TGF-β signaling resulted in a sequential phosphorylation of the transforming growth factor-activated kinase-1 (TAK1), inhibitor of nuclear factor kappa-B kinase (IKK), inhibitor of kappa-B subunit alpha (IκBα) and the v-rel avian reticuloendotheliosis viral oncogene homolog A (RelA); however, the crosstalk to CEA was not addressed in this study. Nor did this study address the consecutive activation of TAK1/mitogen-activated protein kinase kinase (MEK)/protein kinase B (AKT)/NFκB and SMAD pathways upon TGF-β stimulation as Gingery et al. [55] did in osteoclasts.
In human cancers, mutations in the TGF-β pathways (e.g., TGF-β-RII or SMAD4) are frequently observed [56]. A recent study has indicated that, although most tested colorectal cancer cells displayed an inactivated TGF-β signaling pathway, they actively secreted TGF-β acting on stromal cells and were thus driving metastasis [57]. In other cancer cell types, TGF-β signaling is intact, but aberrant NFκB activation and NFκB/RelA stimulate proliferation. In this respect, it should be emphasized that NFκB is constitutively activated in a number of hematologic and solid tumors and is one of the major transcription factors associated with cancer progression, inhibition of apoptosis, limitless replicative potential, tissue invasion and metastasis [58].
The TGF-β-R and NFκB pathways are connected via the TAK-1, which (independently, but parallel to SMAD activation) by phosphorylating IKK can directly stimulate the nuclear factor-κB (NFκB) pathway [55]. It is tempting to speculate that CEA may induce similar signals by interacting with TGF-β-R [26]. TAK1 was expressed in head and neck cancers, where nuclear activation of RelA of the NFκB family also took place. TGF-β induced sequential phosphorylation of several targets including TAK1, IKK, IκBα and RelA; additionally, TAK1 again enhanced TGF-β induced NFκB activation [53]. In human neutrophils, a constitutive association of TAK1 and inhibitor of kappa-B (IκB) was recently reported, indicating a close association of these pathways in inflammatory cells [59]. Neil et al. could show that the TAK1-binding protein 1 (TAB 1) forms complexes with IκB kinase b (IKKb) resulting in stimulation of the TAK1:IKKb:RelA pathway. The authors concluded that this axis, including the NFκB elements, is pivotal in the oncogenic transformation of breast cancer [60]. The fact that NFκB plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human breast cancer cells may additionally complicate the situation [61].
Conclusion
Generally, the dog represents an optimal model organism to study cancer biology in a comparative setting, as many genes represent a great degree of homology to their human counterparts [62]. Even with respect to noncoding RNAs, the significance of similarities between human and dog has recently been acknowledged [63]. Furthermore, the intriguing amino acid homogeneity among human and canine CEAR, TGF-β and TGF-β-R isoforms, NFκB and RelA are given in Table 1, indicating again an advantage of the dog patient in comparative oncology.
We propose that understanding of the crosstalk between CEA and TGF-β signaling toward NFκB as a key cancer regulator, as well as understanding of the Treg and Breg action in tumor tissue, should be extended, possibly with prognostic value. The dog may be a relevant translational model to study these interactions, in line with the comparative oncology strategy [64]. In the future, novel drugs may target the Achilles heel of both obviously interconnected networks.
Abbreviations
- AAV:
-
Adeno-associated virus
- Akt:
-
Protein kinase B
- Breg:
-
Regulatory B-lymphocyte
- CEA:
-
Carcinoembryonic antigen (CEACAM5)
- CEACAM:
-
Carcinoembryonic antigen-related cell adhesion molecule
- CEAR:
-
Carcinoembryonic antigen receptor
- CEARL:
-
Carcinoembryonic antigen receptor, long isoform
- CEARS:
-
Carcinoembryonic antigen receptor, short isoform
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor (ErbB1)
- EMT:
-
Epithelial-to-mesenchymal transition
- FGF:
-
Fibroblast growth factor
- HER-2:
-
Human epidermal growth factor receptor 2 (ErbB2)
- HGF:
-
Hepatocyte growth factor
- hnRNP M4:
-
Heterogeneous nuclear ribonucleoprotein M4 (CEAR)
- IGFs:
-
Insulin-like growth factors
- IKK:
-
Inhibitor of nuclear factor kappa-B kinase
- IKKb:
-
Inhibitor of nuclear factor kappa-B kinase subunit beta
- IκB:
-
Inhibitor of kappa-B
- IκBα:
-
Inhibitor of kappa-B subunit alpha
- MDCK:
-
Madin–Darby canine kidney cell line
- MEK:
-
Mitogen-activated protein kinase kinase
- NFκB:
-
Nuclear factor kappa-B
- PDGF:
-
Platelet-derived growth factor
- RelA:
-
v-Rel avian reticuloendotheliosis viral oncogene homolog A
- SMAD:
-
SMA and MAD homolog
- TAB1:
-
TAK1-binding protein 1
- TAK1:
-
Transforming growth factor-activated kinase-1
- TGF-β:
-
Transforming growth factor beta
- TGF-β-R:
-
Transforming growth factor beta receptor
- Treg:
-
Regulatory T-lymphocyte
References
(The American Veterinary Medical Association) One health initiative task force. One health: a new professional imperative. https://www.avma.org/KB/Resources/Reports/Documents/onehealth_final.pdf. Accessed Sept 27 2013
Egenvall A, Bonnett BN, Ohagen P, Olson P, Hedhammar A, von Euler H (2005) Incidence of and survival after mammary tumors in a population of over 80,000 insured female dogs in Sweden from 1995 to 2002. Prev Vet Med 69:109–127. doi:10.1016/j.prevetmed.2005.01.014
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90. doi:10.3322/caac.20107
Richter SH, Garner JP, Auer C, Kunert J, Wurbel H (2010) Systematic variation improves reproducibility of animal experiments. Nat Methods 7:167–168. doi:10.1038/nmeth0310-167
Singer J, Weichselbaumer M, Stockner T et al (2012) Comparative oncology: ErbB-1 and ErbB-2 homologues in canine cancer are susceptible to cetuximab and trastuzumab targeting. Mol Immunol 50:200–209. doi:10.1016/j.molimm.2012.01.002
Singer J, Fazekas J, Wang W et al (2014) Generation of a canine anti-EGFR (ErbB-1) antibody for passive immunotherapy in dog cancer patients. Mol Cancer Ther 13:1777–1790. doi:10.1158/1535-7163.MCT-13-0288
Iwanicki-Caron I, Di Fiore F, Roque I et al (2008) Usefulness of the serum carcinoembryonic antigen kinetic for chemotherapy monitoring in patients with unresectable metastasis of colorectal cancer. J Clin Oncol 26:3681–3686. doi:10.1200/jco.2007.15.0904
Bast RC, Ravdin P, Hayes DF et al (2001) 2000 Update of recommendations for the use of tumor markers in breast and colorectal cancer: clinical practice guidelines of the american society of clinical oncology*. J Clin Oncol 19:1865–1878
Ogoshi K, Miyaji M, Nakamura K, Kondoh Y, Makuuchi H, Tajima T (1998) Immunotherapy and combined assay of serum levels of carcinoembryonic antigen and acute-phase reactants. Cancer Immunol Immunother : CII 46:14–20
Zebhauser R, Kammerer R, Eisenried A, McLellan A, Moore T, Zimmermann W (2005) Identification of a novel group of evolutionarily conserved members within the rapidly diverging murine Cea family. Genomics 86:566–580. doi:10.1016/j.ygeno.2005.07.008
Kammerer R, Popp T, Hartle S, Singer BB, Zimmermann W (2007) Species-specific evolution of immune receptor tyrosine based activation motif-containing CEACAM1-related immune receptors in the dog. BMC Evol Biol 7:196. doi:10.1186/1471-2148-7-196
Hostetter RB, Augustus LB, Mankarious R, Chi K, Fan D, Toth C, Thomas P, Jessup JM (1990) Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J Natl Cancer Inst 82:380–385. doi:10.1093/jnci/82.5.380
Thomas SN, Zhu F, Schnaar RL, Alves CS, Konstantopoulos K (2008) Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J Biol Chem 283:15647–15655. doi:10.1074/jbc.M800543200
Bajenova O, Chaika N, Tolkunova E, Davydov-Sinitsyn A, Gapon S, Thomas P, O’Brien S (2014) Carcinoembryonic antigen promotes colorectal cancer progression by targeting adherens junction complexes. Exp Cell Res 324:115–123. doi:10.1016/j.yexcr.2014.04.007
Triozzi PL, Aldrich W, Ponnazhagan S (2011) Inhibition and promotion of tumor growth with adeno-associated virus carcinoembryonic antigen vaccine and Toll-like receptor agonists. Cancer Gene Ther 18:850–858. doi:10.1038/cgt.2011.54
Witzens-Harig M, Hose D, Junger S et al (2013) Tumor cells in multiple myeloma patients inhibit myeloma-reactive T cells through carcinoembryonic antigen-related cell adhesion molecule-6. Blood 121:4493–4503. doi:10.1182/blood-2012-05-429415
Muturi HT, Dreesen JD, Nilewski E, Jastrow H, Giebel B, Ergun S, Singer BB (2013) Tumor and endothelial cell-derived microvesicles carry distinct CEACAMs and influence T-cell behavior. PLoS One 8:e74654. doi:10.1371/journal.pone.0074654
Muller MM, Singer BB, Klaile E, Obrink B, Lucka L (2005) Transmembrane CEACAM1 affects integrin-dependent signaling and regulates extracellular matrix protein-specific morphology and migration of endothelial cells. Blood 105:3925–3934. doi:10.1182/blood-2004-09-3618
Vannucci L (2014) Stroma as an active player in the development of the tumor microenvironment. Cancer Microenviron. doi:10.1007/s12307-014-0150-x
Weichselbaumer M, Willmann M, Reifinger M et al (2011) Phylogenetic discordance of human and canine carcinoembryonic antigen (CEA, CEACAM) families, but striking identity of the CEA receptors will impact comparative oncology studies. PLoS Curr 3:RRN1223. doi:10.1371/currents.RRN1223
Bajenova OV, Zimmer R, Stolper E, Salisbury-Rowswell J, Nanji A, Thomas P (2001) Heterogeneous RNA-binding protein M4 is a receptor for carcinoembryonic antigen in Kupffer cells. J Biol Chem 276:31067–31073. doi:10.1074/jbc.M104093200
Laguinge L, Bajenova O, Bowden E, Sayyah J, Thomas P, Juhl H (2005) Surface expression and CEA binding of hnRNP M4 protein in HT29 colon cancer cells. Anticancer Res 25:23–31
Zhao HM, Zhang S, Gao F (2010) Expression of carcinoembryonic antigen receptor in digestive organs. Zhonghua Wei Chang Wai Ke Za Zhi 13:608–611
Thomas P, Forse RA, Bajenova O (2011) Carcinoembryonic antigen (CEA) and its receptor hnRNP M are mediators of metastasis and the inflammatory response in the liver. Clin Exp Metastasis 28:923–932. doi:10.1007/s10585-011-9419-3
Palermo NY, Thomas P, Murphy RF, Lovas S (2012) Hexapeptide fragment of carcinoembryonic antigen which acts as an agonist of heterogeneous ribonucleoprotein M. J Pept Sci 18:252–260. doi:10.1002/psc.2393
Li Y, Cao H, Jiao Z, Pakala SB, Sirigiri DN, Li W, Kumar R, Mishra L (2010) Carcinoembryonic antigen interacts with TGF-{beta} receptor and inhibits TGF-{beta} signaling in colorectal cancers. Cancer Res 70:8159–8168. doi:10.1158/0008-5472.CAN-10-1073
Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP (2002) Crystal structure of the human TbetaR2 ectodomain–TGF-beta3 complex. Nat Struct Biol 9:203–208. doi:10.1038/nsb766
Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J (1992) TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71:1003–1014. doi:10.1016/0092-8674(92)90395-S
Huang T, David L, Mendoza V et al (2011) TGF-β signalling is mediated by two autonomously functioning TβRI: TβRII pairs. EMBO J 30:1263–1276. doi:10.1038/emboj.2011.54
Adeegbe DO, Nishikawa H (2013) Natural and induced T regulatory cells in cancer. Front Immunol 4:190. doi:10.3389/fimmu.2013.00190
Carvalho MI, Pires I, Prada J, Queiroga FL (2014) A role for T-lymphocytes in human breast cancer and in canine mammary tumors. Biomed Res Int 2014:130894. doi:10.1155/2014/130894
O’Neill K, Guth A, Biller B, Elmslie R, Dow S (2009) Changes in regulatory T cells in dogs with cancer and associations with tumor type. J Vet Intern Med 23:875–881. doi:10.1111/j.1939-1676.2009.0333.x
Pinheiro D, Chang YM, Bryant H et al (2014) Dissecting the regulatory microenvironment of a large animal model of non-Hodgkin lymphoma: evidence of a negative prognostic impact of FOXP3 + T cells in canine B cell lymphoma. PLoS One 9:e105027. doi:10.1371/journal.pone.0105027
Whiteside TL (2014) Regulatory T cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother: CII 63:67–72. doi:10.1007/s00262-013-1490-y
Biragyn A, Lee-Chang C, Bodogai M (2014) Generation and identification of tumor-evoked regulatory B cells. Methods Mol Biol 1190:271–289. doi:10.1007/978-1-4939-1161-5_19
Lindner S, Dahlke K, Sontheimer K et al (2013) Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res 73:2468–2479. doi:10.1158/0008-5472.CAN-12-3450
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A (2011) Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res 71:3505–3515. doi:10.1158/0008-5472.CAN-10-4316
Mele V, Muraro MG, Calabrese D et al (2014) Mesenchymal stromal cells induce epithelial-to-mesenchymal transition in human colorectal cancer cells through the expression of surface-bound TGF-beta. Int J Cancer 134:2583–2594. doi:10.1002/ijc.28598
Lee C, Jia Z, Rahmatpanah F, Zhang Q, Zi X, McClelland M, Mercola D (2014) Role of the adjacent stroma cells in prostate cancer development and progression: synergy between TGF-beta and IGF signaling. Biomed Res Int 2014:502093. doi:10.1155/2014/502093
Zhang J, Wang Y, Li D, Jing S (2014) Notch and TGF-beta/Smad3 pathways are involved in the interaction between cancer cells and cancer-associated fibroblasts in papillary thyroid carcinoma. Tumour Biol 35:379–385. doi:10.1007/s13277-013-1053-z
Gupta DK, Singh N, Sahu DK (2014) TGF-beta mediated crosstalk between malignant hepatocyte and tumor microenvironment in hepatocellular carcinoma. Cancer Growth Metastasis 7:1–8. doi:10.4137/CGM.S14205
Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA (2010) Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle 9:4461–4468
Dunning NL, Laversin SA, Miles AK, Rees RC (2011) Immunotherapy of prostate cancer: should we be targeting stem cells and EMT? Cancer Immunol Immunother: CII 60:1181–1193. doi:10.1007/s00262-011-1065-8
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29:4741–4751. doi:10.1038/onc.2010.215
Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP (2013) TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Investig 123:150–163. doi:10.1172/JCI64946
Kim JH, Hur JH, Lee SM, Im KS, Kim NH, Sur JH (2012) Correlation of Foxp3 positive regulatory T cells with prognostic factors in canine mammary carcinomas. Vet J 193:222–227. doi:10.1016/j.tvjl.2011.10.022
Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, Goedegebuure PS, Linehan DC (2009) Tumor-derived TGF-beta mediates conversion of CD4 + Foxp3 + regulatory T cells in a murine model of pancreas cancer. J Immunother 32:12–21. doi:10.1097/CJI.0b013e318189f13c
Li Q, Li Q, Chen J et al (2013) Prevalence of Th17 and Treg cells in gastric cancer patients and its correlation with clinical parameters. Oncol Rep 30(3):1215–1222. doi:10.3892/or.2013.2570
Kammerer R, von Kleist S (1996) The carcinoembryonic antigen (CEA) modulates effector-target cell interaction by binding to activated lymphocytes. Int J Cancer 68:457–463. doi:10.1002/(SICI)1097-0215(19961115)68:4<457:AID-IJC10>3.0.CO;2-2
Chen L, Chen Z, Baker K et al (2012) The short isoform of the CEACAM1 receptor in intestinal T cells regulates mucosal immunity and homeostasis via Tfh cell induction. Immunity 37:930–946. doi:10.1016/j.immuni.2012.07.016
Oeckinghaus A, Hayden MS, Ghosh S (2011) Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12:695–708. doi:10.1038/ni.2065
Pasparakis M, Luedde T, Schmidt-Supprian M (2006) Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ 13:861–872. doi:10.1038/sj.cdd.4401870
Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, Chen Z, Van Waes C (2013) TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 32:1549–1559. doi:10.1038/onc.2012.171
Han SU, Kwak TH, Her KH et al (2008) CEACAM5 and CEACAM6 are major target genes for Smad3-mediated TGF-beta signaling. Oncogene 27:675–683. doi:10.1038/sj.onc.1210686
Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ (2008) TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res 314:2725–2738. doi:10.1016/j.yexcr.2008.06.006
Elliott RL, Blobe GC (2005) Role of transforming growth factor Beta in human cancer. J Clin Oncol 23:2078–2093. doi:10.1200/JCO.2005.02.047
Calon A, Espinet E, Palomo-Ponce S et al (2012) Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22:571–584. doi:10.1016/j.ccr.2012.08.013
Naugler WE, Karin M (2008) NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev 18:19–26. doi:10.1016/j.gde.2008.01.020
Ear T, Fortin CF, Simard FA, McDonald PP (2010) Constitutive association of TGF-beta-activated kinase 1 with the IkappaB kinase complex in the nucleus and cytoplasm of human neutrophils and its impact on downstream processes. J Immunol 184:3897–3906. doi:10.4049/jimmunol.0902958
Neil JR, Schiemann WP (2008) Altered TAB 1: I kappaB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res 68:1462–1470. doi:10.1158/0008-5472.CAN-07-3094
Oida K, Matsuda A, Jung K et al (2014) Nuclear factor-kB plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human breast cancer cells. Sci Rep 4:4057. doi:10.1038/srep04057
Lindblad-Toh K, Wade CM, Mikkelsen TS et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819. doi:10.1038/nature04338
Wagner S, Willenbrock S, Nolte I, Murua Escobar H (2013) Comparison of non-coding RNAs in human and canine cancer. Front Genet 4:46. doi:10.3389/fgene.2013.00046
Riccardo F, Aurisicchio L, Impellizeri JA, Cavallo F (2015) The importance of comparative oncology in translational medicine. Cancer Immunol Immunother: CII 64:137–148. doi:10.1007/s00262-014-1645-5
Acknowledgments
The authors were supported by Project P23398-B11 of the Austrian Science Fund (FWF), Judit Fazekas by the Cell Communication in Health and Disease (CCHD) PhD Program W1205-B09 of the Austrian Science Fund (FWF). We would like to thank Mrs. Amelia Wein for proofreading the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest pertaining to the contents of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Jensen-Jarolim, E., Fazekas, J., Singer, J. et al. Crosstalk of carcinoembryonic antigen and transforming growth factor-β via their receptors: comparing human and canine cancer. Cancer Immunol Immunother 64, 531–537 (2015). https://doi.org/10.1007/s00262-015-1684-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1684-6