
Abstract
A Gram-positive rubber-degrading bacterium, Actinoplanes sp. strain OR16 (strain NBRC 114529), is able to grow on agar plates containing natural and synthetic rubber as the sole sources of carbon and energy. When this strain was grown on natural rubber latex overlay agar plates, translucent halos around the cells were observed. To identify the natural rubber degradation genes and other features of its metabolism, its complete genome sequence was determined. The genome of OR16 consists of 9,293,892 bp and comprises one circular chromosome (GenBank accession number AP019371.1) with a G + C content of 70.3%. The genome contains 8238 protein-coding and 18 rRNA genes. A homology search of the genome sequence revealed that three genes (lcp1, lcp2, and lcp3) are homologous to an extracellular latex-clearing protein (Lcp) of Streptomyces sp. K30. RT-PCR analysis revealed that lcp1 and lcp2 seem to constitute an operon. Purified lcp gene products have oxygen consumption activity toward natural rubber latex, suggesting that all these genes encode rubber-degrading enzymes in OR16. Quantitative reverse transcription-PCR analysis indicated that the transcription of these genes is induced during the growth of OR16 on natural rubber. The genes located adjacent to lcp1 and lcp3, which code for a TetR/AcrR-type transcriptional regulator, can bind to the promoter regions of these lcp genes. It is suggested that the putative regulators play a role in regulating the transcription of the lcp genes. These results strongly suggested that three lcp genes are required for the utilization of natural rubber in strain OR16.
Key Points • The complete genome sequence of Actinoplanes sp. strain OR16 was determined. • Three lcp genes which are involved in the natural rubber degradation in OR16 were identified. • Transcription of these lcp genes is induced during utilization of rubber in OR16. • Two regulators, which bind to the promoter regions of lcp, were determined. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural rubber (NR) is a biopolymer containing poly(cis-1,4-isoprene) as a main component and is produced by over 2000 plants including Hevea brasiliensis (Backhaus 1985). NR is used globally in industry for tires, seismic isolation rubbers, medical gloves, and many other products. The waste of these products is permanently disposed and treated by combustion or landfill processes, which are hazardous to the environment. It is necessary to find an alternative treatment process for rubber waste. NR-degrading microorganisms, including Gram-positive and Gram-negative bacteria, have been identified and characterized in the past 20 years (Linos et al. 2000a; Linos et al. 2000b). Because of their rubber degradation ability, these bacteria are expected to be used as practical tools for the treatment process of rubber waste.
Microbial degradation of NR has been widely reported in Gram-positive bacteria such as Streptomyces, Gordonia, Rhodococcus, and Nocardia species and in only a few of Gram-negative bacteria including Steroidobacter cummioxidans 35Y and Rhizobacter gummiphilus NS21T (Bröker et al. 2008; Imai et al. 2011; Jendrossek and Reinhardt 2003; Sharma et al. 2018). Two major types of NR-degrading enzymes, rubber oxygenase (RoxA/RoxB) and latex-clearing protein (Lcp), have been characterized in Gram-negative and Gram-positive bacteria, respectively. Exception includes a Gram-negative bacterium, Solimonas fluminis, which has an lcp homologous gene (Birke and Jendrossek 2019). RoxA from strain 35Y is an exo-type diheme oxygenase that oxidizes poly(cis-1,4-isoprene) into the C15 tri-isoprenoid product 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (Braaz et al. 2004). On the other hand, RoxB degrades NR via endo cleavage of poly(cis-1,4-isoprene) (Birke et al. 2017). It has been reported that the two rubber oxygenases are involved in the NR degradation in Gram-negative bacteria including several myxobacteria (Birke et al. 2017). Recently, it has been reported that the functional RoxB and RoxA orthologs, LatA1 and LatA2, respectively, are essential for NR degradation by R. gummiphilus NS21T (Birke et al. 2018; Kasai et al. 2017).
The NR-degrading actinomycete, Streptomyces sp. strain K30 has an Lcp, which is a cofactor-independent oxygenase that catalyzes the oxidative endo cleavage of poly(cis-1,4-isoprene) into low-molecular-weight products containing aldehyde and keto end groups (Fig. 1) (Birke and Jendrossek 2014; Bröker et al. 2008). In contrast to Gram-negative rubber degraders, almost all rubber-degrading actinomycetes, including Streptomyces, Nocardia, and Rhodococcus species, except for Gordonia polyisoprenivorans VH2, have a single lcp homolog (Hiessl et al. 2012; Luo et al. 2014; Oetermann et al. 2018; Watcharakul et al. 2016).

Proposed cleavage reaction for poly(cis-1,4-isoprene) by Lcps. Poly(cis-1,4-isoprene) is cleaved to form the products with aldehyde and keto end groups
Recently, a rubber-utilizing bacterium, strain OR16 (strain NBRC 114529), was isolated from a botanical garden in Japan (Imai et al. 2011). This strain is capable of growing on solid media containing NR or synthetic isoprene rubber (IR) and produces translucent halos on NR latex overlay agar plates. Taxonomic analysis revealed that strain OR16 belongs to the genus Actinoplanes. The genus Actinoplanes contains the rubber-degrading strains A. missouriensis NBRC 102363, A. italicus DSM 43146, and A. utahensis NBRC 13244 (Jendrossek et al. 1997); however, all the genes and their gene products responsible for rubber degradation have not yet been characterized genetically and biochemically. Here, the genome sequence analysis of strain OR16 identified three lcp homologous genes which are responsible for the rubber utilization of this strain. Multiple lcp genes were characterized in G. polyisoprenivorans VH2, but the two lcp genes are located in chromosome and plasmid. On the other hand, three lcp genes of strain OR16 were located on the chromosome. Recently, three lcp homologous genes have been identified in Streptomyces sp. strain CFMR7 by the genome sequence analysis (Nanthini et al. 2017). However, the functions of the genes have not been characterized at the molecular level, and the roles of their gene products are not clear. To gain insight into the rubber degradation system including three lcp homologous genes and the characterization of the functions of multiple lcp genes of Actinoplanes sp. strain OR16 is important. This is the first report of the functional characterization of rubber-utilizing bacterium which has three lcp genes.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Actinoplanes sp. strain OR16 and its mutant derivatives were routinely grown at 30 °C in PYM medium (0.5% Bactopeptone, 0.3% yeast extract, and 0.1% MgSO4•7H2O; pH 7.0) or W minimal salt medium (Araki et al. 2011) containing 10 mM sodium succinate. Growth on NR or IR was examined using rubber overlay agar plates. To prepare the rubber overlay agar plates, W agar medium containing deproteinized NR (Chaikumpollert et al. 2012) or synthetic isoprene rubber at a final concentration of 0.4% (v/v) was overlaid to form a thin layer on a solid medium. Escherichia coli strains were cultivated aerobically at 30 °C or 37 °C in LB medium. If necessary, ampicillin (100 mg/l), kanamycin (25 mg/l), chloramphenicol (25 mg/l), tetracycline (10 mg/l), and apramycin (100 mg/l) were added to the medium.
DNA manipulations, nucleotide sequencing, and sequence analysis
DNA manipulations including total DNA isolation and nucleotide sequencing were performed as previously described (Masai et al. 1995). Analysis of nucleotide sequences was carried out as previously described (Kasai et al. 2009). The genome sequence of OR16 was determined by the combination of MiSeq, PacBio RS II system, and Sanger sequencing. These sequencing data were assembled by CLC genomics workbench (Qiagen). Annotation was performed using NCBI Prokaryotic Genome Annotation Pipeline ver.3.1 (Tatusova et al. 2016) and RAST server (Aziz et al. 2008). Genome sequences of strain OR16 and of related Actinoplanes spp. were compared by calculating average nucleotide identity (ANI) values using the JSpecies Web Service (Richter et al. 2016). Tat signal sequences were predicted using the TatP 1.0 software (http://www.cbs.dtu.dk/services/TatP/) (Bendtsen et al. 2005).
Expression of lcp genes in E. coli
The coding regions of lcp1, lcp2, and lcp3 were independently amplified by PCR using lcp1_F and R, lcp3_F and R, and lcp2_F and R primer pairs (Table 1). Each forward primer contained an NdeI site at the start codon of the corresponding target gene. The amplified fragments were separately cloned in pJET1.2 to generate the NdeI fragment containing the entire target gene. The NdeI fragments containing either of lcp genes were individually cloned in the expression vector, pET23b. The resultant plasmids were independently introduced into E. coli Rosetta-gami B(DE3)pLysS, and the transformants were grown at 30 °C in LB medium containing kanamycin, chloramphenicol, and tetracycline. When the absorbance at 600 nm (A600) of the culture reached 0.5, 1 mM isopropyl-β-D-thiogalactopyranoside was added, and the cultures were further incubated at 20 °C for 16 h. The resulting cells were harvested and resuspended in 50 mM Tris-HCl (pH 7.4). The cell extract was prepared by using French pressure cell press. After centrifugation, the resulting supernatant was applied to a Ni Sepharose 6 Fast Flow column (GE Healthcare, Buckinghamshire, UK) previously equilibrated with buffer A consisting of 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 100 mM imidazole. Proteins were allowed to bind for 1 min at 4 °C while rotating, followed by washing five times in 5 ml of buffer A. His-tagged proteins were eluted with 5 ml of buffer B consisting of 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 500 mM imidazole, and the fractions were pooled and concentrated.
Enzyme assays
The activities of Lcp1, Lcp2, and Lcp3 were assayed by measuring the substrate-dependent oxygen consumption rate. Each 2-ml assay mixture contained 50 mM Tris-HCl buffer, NR (final concentration was 0.2%), and purified enzyme (25 μg of protein). The reaction mixture was incubated at 30 °C, and the oxygen consumption rate was determined with an oxygen electrode (Dual Digital Model 20; Rank Brothers Ltd., Cambridge, England). One unit of enzyme activity was defined as the amount of activity that resulted in consumption of 1 μmol of O2 per 1 min. Specific activity was expressed in units per milligram of protein. The optimal pH and optimal temperature for Lcp1, Lcp2, and Lcp3 were determined at pH and temperature ranges of 6.0 to 8.5 and 25 to 40 °C, respectively, by using 50 mM Tris-HCl buffer.
Quantitative reverse transcription-PCR (qRT-PCR) analysis
The cells of OR16 were grown on a NR-overlay agar medium or W agar medium containing 10 mM sodium succinate at 30 °C for 3 days. They were collected using centrifugation and washed with 0.9% NaCl. The total RNA was extracted from the cells with ISOGEN II (Nippon Gene Co., Ltd., Tokyo, Japan) according to the manufacturer’s instructions and was treated with RNase-free DNase I (Roche). Single-stranded cDNA was synthesized from 1 μg of total RNA with 100 U of PrimeScript II reverse transcriptase (Takara Bio Inc., Otsu, Japan) and random hexamer primers in a 30 μl reaction mixture. The cDNA was subjected to RT-PCR and qRT-PCR analyses. These PCR analyses were carried out using the specific primers (Table 1) according to the previous reports (Kasai et al. 2017).
Electrophoretic mobility shift assays (EMSAs)
DNA fragments containing the upstream region from lcp1 and lcp3 were prepared by PCR with tetR_lcp1_F/R and lcp3_UP_ F/R primer pairs, respectively (Table 1). The 3′ ends of the probe fragments were labeled with DIG-11-ddUTP using the 2nd generation DIG gel shift kit (Roche), according to the manufacturer’s instructions. Binding reaction was performed at 20 °C for 20 min in a 10-μl reaction mixture containing purified his-tagged ACTI_59620 (ACTI_59620-his) or ACTI_69510 (ACTI_69510-his), 10 nM DIG-labeled probe, 0.1 μg of poly-L-lysine, 5 μg of salmon sperm DNA, 20 mM HEPES (pH 7.6), 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM dithiothreitol, 0.2% (w/v) Tween 20, and 30 mM KCl. The mixtures were then incubated at 20 °C for 20 min, loaded onto 10% polyacrylamide gels in 0.5× Tris-borate-EDTA buffer. Gel electrophoresis was performed as described previously (Kasai et al. 2010). The labeled DNA was detected using a CSPD detection system (Roche) with a C-DiGit image analyzer (LI-Cor Biosciences).
Results
Determination of the genome sequence of strain OR16
To identify the rubber-degrading genes in strain OR16, the genome sequence of this strain was determined. The genomic DNA was sequenced using the MiSeq platform, and the draft genome sequences were de novo assembled to produce 64 scaffolds. Then, the gaps among all contigs were closed by whole-genome sequencing using the PacBio RS II system and a combination of PCR plus Sanger sequencing. The final complete sequence of the OR16 genome consisted of a 9,293,892 bp chromosome with an average GC content of 70.3%. The genome contained 8558 protein-coding sequences (CDSs) and 18 and 65 copies of rRNA and tRNA genes, respectively. This genome was deposited in GenBank under accession number AP019371.1. The 16S rRNA gene sequence of strain OR16 has 96.70% to 95.86% identity with those of the type strains, i.e., A. utahensis IFO 13244T (AB037012), A. philippinensis DSM 43019 T (X93187), A. teichomyceticus DSM 43866T (AJ865472), A. rectilineatus IFO 13941T (AB037010), A. liguriensis DSM 43865T (AJ865471), A. cyaneus DSM 46137T (X93186), A. italicus JCM 3165T (AB048217), A. palleronii JCM 7626T (AB048216), and A. regularis DSM 43151T (X93188). The whole-genome average nucleotide identity (ANI) values between strain OR16 and A. missouriensis 431, Actinoplanes sp. SE50/110, Actinoplanes sp. N902-109, and A. friuliensis DSM 7358 were 87.35%, 85.48%, 84.77%, and 84.36%, respectively. Since the values were less than the same-species threshold (95.0%) (Tong et al. 2015), strain OR16 seemed to be a species distinct from the Actinoplanes spp.
Identification of the lcp orthologs in strain OR16
A tBLASTn homology search of the genome sequence of OR16 was performed using the amino acid sequence of Lcp (AAR25849) of Streptomyces sp. strain K30 as the query, and three homologous genes, ACTI_59630, ACTI_59640, and ACTI_69520, were identified as the most closely related gene sequences; these genes were designated lcp1, lcp2, and lcp3, respectively (Fig. 2a). The deduced amino acid sequences of the lcp1, lcp2, and lcp3 gene products shared overall identifies of 56.6%, 53.9%, and 47.6% with that of Lcp from strain K30, respectively. The amino acid sequence identities among biochemically characterized Lcps from actinomycetes are summarized in Table 2. The deduced amino acid sequences encoded by ACTI_59650 and ACTI_59660 exhibited 71.6% and 78.0% identity with those of oxiB and oxiA, respectively; oxiB and oxiA are involved in degradation of low-molecular-weight rubber-degrading compounds in Streptomyces sp. strain K30 (Rose et al. 2005). In contrast, no other known genes, which may be involved in rubber degradation, were detected in the vicinity of the lcp3 gene. ACTI_59620 and ACTI_69510, encoding putative TetR/AcrR-type transcriptional regulators (TATRs), were located adjacent to lcp1 and lcp3, respectively (Fig. 2a). Hence, there is a possibility that TATRs seem to be involved in the transcriptional regulation of the rubber-degrading genes.

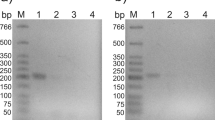
The organization and transcription of the lcp gene clusters. a Open arrows indicate the genes. Locus tags are indicated above the genes. The putative TATRs-coding genes are indicated by R. Double-headed arrows immediately below the gene cluster diagram indicate the locations of the amplified RT-PCR shown in panel B. Boldface bars a and b indicate the positions of DNA probes used in EMSA shown in Fig. 5. b The results of agarose gel electrophoresis of RT-PCR products obtained with primers targeting 1 (expected size 988 bp), 2 (expected size 1215 bp), and 3 (expected size 951 bp) are shown. The amplified regions and the primer sequences are indicated in panel A and Table 1, respectively. M, molecular size markers; + and -, RT-PCR with and without RT, respectively; G, control PCR with the genomic DNA
To define the operon structure of the lcp genes, RT-PCR analysis was performed with total RNA extracted from strain OR16 grown with NR latex. RT-PCR amplification products of the expected size were detected in the intergenic regions of lcp1-lcp2, lcp2-oxiB, and oxiB-oxiA (Fig. 2b). These results suggest that the lcp1, lcp2, oxiB, and oxiA genes are organized in the same transcriptional unit.
Heterologous expression of the lcp genes in E. coli
To examine the roles of Lcp1, Lcp2, and Lcp3, each gene was fused with a 10× histidine-tag and introduced into E. coli Rosetta-gami B(DE3)pLysS cells. When the cell extracts from E. coli expressing each gene were prepared and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), the presence of proteins of corresponding sizes was observed (Fig. 3a–c). To characterize the activity toward NR latex, each of these his-tagged proteins (Lcp1-his, Lcp2-his, and Lcp3-his) was purified by Ni-affinity column chromatography. When the purified proteins were incubated with NR latex, oxygen consumption activity was observed. No consumption of oxygen was observed without protein or NR latex, strongly suggesting that these enzymes are involved in rubber degradation. The optimal temperature and optimal pH for the oxygen consumption activity of Lcp1-his, Lcp2-his, and Lcp3-his with NR latex were determined to be 30 °C and 7.0, 35 °C and 7.5, and 30 °C and 6.5, respectively. The specific activities of Lcp1-his, Lcp2-his, and Lcp3-his were 4.02 ± 0.65 (30 °C, pH 7.0), 1.17 ± 0.07 (35 °C, pH 7.5), and 0.22 ± 0.01 (30 °C, pH 6.5) U/mg protein, respectively (see Fig. S1 in the supplemental material).

SDS-PAGE analysis of protein fractions. Proteins were separated on a SDS 12% polyacrylamide gel and stained with Coomassie brilliant blue. Lcp1-his, Lcp2-his, and Lcp3-his, ACTI_59620-his, and ACTI_69510-his are shown in panels (a), (b), (c), (d), and (e), respectively. M, molecular weight markers; 1, crude extract of the E. coli cells containing lcp1, lcp2, lcp3, ACTI_59620, or ACTI_69510; 2, purified proteins. Molecular masses are given on the left
Transcriptional induction of the lcp genes
To determine whether transcription of the lcp genes is induced in response to NR, the mRNA levels of lcp1, lcp2, and lcp3 were measured by quantitative RT-PCR (qRT-PCR) analysis using the total RNA harvested from cells grown on succinate with or without NR latex. Cells grown on succinate exhibited extremely low transcription levels of these three genes. In contrast, the transcription levels of lcp1, lcp2, and lcp3 in cells grown with NR latex were 22.2-fold, 17.1-fold, and 335-fold higher, respectively, than those in cells grown without NR latex (P < 0.05 by Student’s t test) (Fig. 4). These results strongly suggested that the transcription of these lcp genes is induced during NR utilization in OR16.

Quantification of the expression levels of the lcp genes. Total RNA was isolated from the OR16 cells grown in W medium containing 10 mM succinate without NR (open bars) or with NR (gray bars). The mRNA expression levels were calculated as a ratio of 16S rRNA gene expression. The data are mean values ± standard deviations for four independent experiments. Statistical analysis was performed using Student’s t test. *, P < 0.05; **, P < 0.005
Binding ability of the regulators of the lcp genes
To confirm the role of the ACTI_59620 and ACTI_69510 gene products in regulating the transcription of the lcp genes, these genes were expressed in E. coli BL21(DE3) cells as histidine-tagged proteins (ACTI_59620-his and ACTI_69510-his). To characterize the DNA binding ability of ACTI_59620-his and ACTI_69510-his, these proteins were purified to near homogeneity (Fig. 3d and e). Purified ACTI_59620-his and ACTI_69510-his were used in electrophoretic mobility shift assays (EMSAs) with 199-bp and 203-bp DNA probes containing the upstream regions from lcp1 or lcp3, respectively (Fig. 5). As shown in Fig. 5a, ACTI_59620-his was able to bind to the upstream region from lcp1. Furthermore, a weakly shifted band was observed when the upstream region from lcp3 was used as probe (Fig. 5b). The results suggested that ACTI_59620 mainly bound to the promoter of lcp1 to regulate transcription. In contrast, ACTI_69510-his and DNA complexes were formed when the regions upstream from lcp1 and lcp3 were used as probes (Fig. 5d and e). This indicated that ACTI_69510 is able to interact with both promoters of lcp1 and lcp3. Based on these results, the transcriptional induction of the lcp1 operon seemed to be regulated by ACTI_59620 and ACTI_69510. The regulation of lcp3 transcription mainly requires the binding of ACTI_69510 to the promoter.

Binding of ACTI_59620-his and ACTI_69510-his to the upstream regions from lcp1 and lcp3. DIG labeled DNA probes (10 nM) were incubated in the presence of purified ACTI_59620-his (a–c) and ACTI_69510-his (d–f). DNA fragments including the upstream regions from lcp1 (probe a; A and D) and lcp3 (probe b; B and E) and internal region of 16S rRNA gene of OR16 (negative control; C and F) were used as a probe for EMSA. The size and position of DNA probes a and b are shown in Fig. 2. Arrows with F and C indicate the positions of the unbound probe (free DNA) and protein-DNA complex, respectively
Discussion
Multiple lcp homologs have rarely been identified in rubber degraders such as G. polyisoprenivorans VH2. The lcp1 and lcp2 genes are located on the chromosome and on the plasmid of VH2, respectively (Hiessl et al. 2012). On the other hand, lcp1, lcp2, and lcp3 are all located on the chromosome in OR16. In particular, the lcp1 and lcp2 genes are tandemly located in the same orientation (Fig. 2a). The organization of the lcp genes is similar to that of the lcp genes in Streptomyces sp. strain CFMR7 (Nanthini et al. 2017). The deduced amino acid sequences of the lcp1, lcp2, and lcp3 gene products of strain OR16 have 63.3%, 60.0%, and 59.2% identity, respectively, with those of the corresponding lcp genes from strain CFMR7. Hence, the lcp genes of strain CFMR7 might be involved in the rubber utilization as well as those of strain OR16. Because Lcp was predicted to be secreted via the twin arginine translocation (Tat) pathway (Hiessl et al. 2012; Yikmis et al. 2008), it is critical to identify the Tat signal sequences in Lcp1, Lcp2, and Lcp3. In OR16, the RRxxLx motifs of Lcp1, Lcp2, and Lcp3 were identified between the 6th and 10th, 3rd and 7th, and 6th and 10th residues, respectively. The putative cleavage sites of the Tat signal sequences, which contain the AxA motifs of Lcp1, Lcp2, and Lcp3, were located between 30th and 31st, 26th and 27th, and 33rd to 34th residues, respectively. Therefore, the lcp gene products might be secreted via the Tat pathway in strain OR16.
When NR was incubated with purified Lcps, the oxygen consumption activity was observed. The enzymatic activities are comparable with those of other reported Lcps in strain K30 (4.6 U/mg), strain VH2 (1.3 U/mg), and R. rhodochrous RPK1 (3.1 U/mg) (Birke et al. 2015; Hiessl et al. 2014; Watcharakul et al. 2016). Therefore, these Lcps might be involved in the rubber degradation by OR16 (Fig. 1). The activity of Lcp1 was approximately 7 times and 18 times higher than those of Lcp2 and Lcp3, respectively, suggesting that Lcp1 is mainly involved in rubber degradation in strain OR16. Unlike the enzymatic activities of the Lcp enzymes, the transcription level of lcp3 is significantly higher than those of lcp1 and lcp2 (P < 0.005 by Student’s t test). According to their transcription levels, the role of lcp3 is thought to also be important for the rubber degradation.
RT-PCR analysis revealed that oxiB and oxiA homologous genes (ACTI_59650, ACTI_59660) tandemly located in the downstream region from lcp2 were cotranscribed with the lcp1 and lcp2 genes during rubber utilization. It has been reported that rubber degradation intermediate, low-molecular-weight aldehyde compounds are further metabolized by oxiAB, encoding oxidoreductase, in Streptomyces sp. strain K30 (Rose et al. 2005). It was suggested that ACTI_59650 and ACTI_59660 are involved in the catabolism of aldehyde intermediates for the utilization of rubber by strain OR16.
The ACTI_59620 gene encodes a putative TATR and is located upstream from lcp1 in the opposite direction (Fig. 2a). The deduced amino acid sequence of this gene product is similar to those of TATRs from Rhodococcus sp. Q15 (AAK97457) and R. opacus B-4 (BAH50508). It has been reported that TATRs regulate the transcription of genes involved in antibiotic resistance, antibiotic biosynthesis, catabolic pathways, and biofilm formation in Gram-negative and Gram-positive bacteria as a repressor or an activator (Pompeani et al. 2008; Ramos et al. 2005). Based on these facts, ACTI_59620 seemed to be involved in the regulation of lcp transcription. Furthermore, ACTI_69510, located adjacent to lcp3 and encoding a putative TATR, was found (Fig. 2a). ACTI_69510 seemed to be involved in lcp3 transcription. Interestingly, the amino acid sequences of the N-terminal (residues 22 to 196) and C-terminal (residues 201 to 401) regions of ACTI_69510 exhibited 25.0% identity with each other. In addition, a TATR superfamily-specific domain was found in each region, suggesting that ACTI_69510 was formed by duplication of the regions.
LcpRBA3(2) and LcpRVH2 were identified as transcriptional regulators for putative lcp gene expression in S. coelicolor A3(2) and G. polyisoprenivorans VH2, respectively (Coenen et al. 2019; Oetermann et al. 2019). These promoter regions of lcp1VH2 and lcp2VH2, which interacted with LcpRVH2, contained palindromic sequences, 5′-GATGTTACAACGTTACTCGCGTTGTTACATC-3′ and 5′-GATACAGAGAAGCATAGACTGTAACTCGGTTTC-3′, respectively. In the binding region of ACTI_59620, a palindromic sequence (5′-GATGCGAATTTGTAACAGCGTATCAGCAATC-3′), which is similar to the sequence found in the lcp1VH2 promoter region, was identified. Due to the amino acid sequence similarity (48.7% identity) between LcpRVH2 and ACTI_59620, the formation of a protein-DNA complex by ACTI_59620 requires the palindromic sequence, similar to LcpRVH2.
The amino acid sequence of ACTI_69510 shared 72.3% identity with that of LcpRBA3(2), which binds to the promoter region of lcpA3(2) in strain A3(2) (Coenen et al. 2019). It has been reported that the LcpRBA3(2)-binding sequence contains a palindromic sequence (5′-TATGTTAATGAAAAATCACA-3′). In the upstream region from lcp3, a similar sequence (5′-TATGTTAATGGAAAATCACA-3′) was found. Therefore, the palindromic sequence seemed to be involved in the binding of ACTI_69510. Surprisingly, ACTI_69510 bound not only to the lcp3 promoter but also to the lcp1 promoter. However, a palindromic sequence similar to the palindromic sequence in the lcp3 promoter was not detected in the upstream region from lcp1. Because several palindromic sequences were identified in the lcp1 promoter region, the other sequence seemed to be involved in ACTI_69510 binding. Because ACTI_69510 has two helix-turn-helix DNA-binding motifs that are similar to each other (31.9% identity), ACTI_69510 might be able to recognize several palindromic sequences, including the lcp1 and lcp3 promoters, for its binding. The broad binding specificity of ACTI_69510 is thought to be effective in inducing the transcription of the lcp genes and reasonable for the efficient utilization of rubber in strain OR16.
In this study, we identified the rubber-degrading genes of strain OR16. This strain possesses three lcp genes which might be required for the utilization of natural rubber. Furthermore, two TATRs that have the ability to bind to the promoter regions of the lcp genes were identified. Further analysis including deletion of the TATR-coding genes and the lcp promoter assay will enable us to clarify the actual function and contribution of these TATRs to the transcriptional regulation of rubber-degrading genes.
References
Araki N, Suzuki T, Miyauchi K, Kasai D, Masai E, Fukuda M (2011) Identification and characterization of uptake systems for glucose and fructose in Rhodococcus jostii RHA1. J Mol Microbiol Biotechnol 20(3):125–136. https://doi.org/10.1159/000324330
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. https://doi.org/10.1186/1471-2164-9-75
Backhaus RA (1985) Rubber formation in plants - a minireview. Israel J Bot 34(2–4):283–293
Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S (2005) Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. https://doi.org/10.1186/1471-2105-6-167
Birke J, Jendrossek D (2014) Rubber oxygenase and latex clearing protein cleave rubber to different products and use different cleavage mechanisms. Appl Environ Microbiol 80(16):5012–5020. https://doi.org/10.1128/AEM.01271-14
Birke J, Jendrossek D (2019) Solimonas fluminis has an active latex-clearing protein. Appl Microbiol Biotechnol 103(19):8229–8239. https://doi.org/10.1007/s00253-019-10085-w
Birke J, Rother W, Jendrossek D (2015) Latex clearing protein (Lcp) of Streptomyces sp. strain K30 is a b-type cytochrome and differs from rubber oxygenase a (RoxA) in its biophysical properties. Appl Environ Microbiol 81(11):3793–3799. https://doi.org/10.1128/AEM.00275-15
Birke J, Rother W, Jendrossek D (2018) Rhizobacter gummiphilus NS21 has two rubber oxygenases (RoxA and RoxB) acting synergistically in rubber utilisation. Appl Microbiol Biotechnol 102(23):10245–10257. https://doi.org/10.1007/s00253-018-9341-6
Birke J, Röther W, Jendrossek D (2017) RoxB is a novel type of rubber oxygenase that combines properties of rubber oxygenase RoxA and latex clearing protein (Lcp). Appl Environ Microbiol 83(14). https://doi.org/10.1128/AEM.00721-17
Braaz R, Fischer P, Jendrossek D (2004) Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1,4-isoprene). Appl Environ Microbiol 70(12):7388–7395. https://doi.org/10.1128/AEM.70.12.7388-7395.2004
Bröker D, Dietz D, Arenskötter M, Steinbüchel A (2008) The genomes of the non-clearing-zone-forming and natural-rubber-degrading species Gordonia polyisoprenivorans and Gordonia westfalica harbor genes expressing Lcp activity in Streptomyces strains. Appl Environ Microbiol 74(8):2288–2297. https://doi.org/10.1128/AEM.02145-07
Chaikumpollert O, Yamamoto Y, Suchiva K, Kawahara S (2012) Protein-free natural rubber. Colloid Polym Sci 290:331–338
Coenen A, Oetermann S, Steinbüchel A (2019) Identification of LcpRBA3(2), a novel regulator of lcp expression in Streptomyces coelicolor A3(2). Appl Microbiol Biotechnol 103(14):5715–5726. https://doi.org/10.1007/s00253-019-09896-8
Hiessl S, Böse D, Oetermann S, Eggers J, Pietruszka J, Steinbüchel A (2014) Latex clearing protein-an oxygenase cleaving poly(cis-1,4-isoprene) rubber at the cis double bonds. Appl Environ Microbiol 80(17):5231–5240. https://doi.org/10.1128/AEM.01502-14
Hiessl S, Schuldes J, Thürmer A, Halbsguth T, Bröker D, Angelov A, Liebl W, Daniel R, Steinbüchel A (2012) Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl Environ Microbiol 78(8):2874–2887. https://doi.org/10.1128/AEM.07969-11
Imai S, Ichikawa K, Muramatsu Y, Kasai D, Masai E, Fukuda M (2011) Isolation and characterization of Streptomyces, Actinoplanes, and Methylibium strains that are involved in degradation of natural rubber and synthetic poly(cis-1,4-isoprene). Enzym Microb Technol 49(6–7):526–531. https://doi.org/10.1016/j.enzmictec.2011.05.014
Jendrossek D, Reinhardt S (2003) Sequence analysis of a gene product synthesized by Xanthomonas sp. during growth on natural rubber latex. FEMS Microbiol Lett 224(1):61–65
Jendrossek D, Tomasi G, Kroppenstedt RM (1997) Bacterial degradation of natural rubber: a privilege of actinomycetes? FEMS Microbiol Lett 150(2):179–188
Kasai D, Fujinami T, Abe T, Mase K, Katayama Y, Fukuda M, Masai E (2009) Uncovering the protocatechuate 2,3-cleavage pathway genes. J Bacteriol 191(21):6758–6768. https://doi.org/10.1128/JB.00840-09
Kasai D, Imai S, Asano S, Tabata M, Iijima S, Kamimura N, Masai E, Fukuda M (2017) Identification of natural rubber degradation gene in Rhizobacter gummiphilus NS21. Biosci Biotechnol Biochem 81(3):614–620. https://doi.org/10.1080/09168451.2016.1263147
Kasai D, Kitajima M, Fukuda M, Masai E (2010) Transcriptional regulation of the terephthalate catabolism operon in Comamonas sp. strain E6. Appl Environ Microbiol 76(18):6047–6055. https://doi.org/10.1128/AEM.00742-10
Linos A, Berekaa MM, Reichelt R, Keller U, Schmitt J, Flemming HC, Kroppenstedt RM, Steinbüchel A (2000a) Biodegradation of cis-1,4-polyisoprene rubbers by distinct actinomycetes: microbial strategies and detailed surface analysis. Appl Environ Microbiol 66(4):1639–1645
Linos A, Reichelt R, Keller U, Steinbüchel A (2000b) A gram-negative bacterium, identified as Pseudomonas aeruginosa AL98, is a potent degrader of natural rubber and synthetic cis-1,4-polyisoprene. FEMS Microbiol Lett 182(1):155–161
Luo Q, Hiessl S, Poehlein A, Daniel R, Steinbüchel A (2014) Insights into the microbial degradation of rubber and gutta-percha by analysis of the complete genome of Nocardia nova SH22a. Appl Environ Microbiol 80(13):3895–3907. https://doi.org/10.1128/AEM.00473-14
Masai E, Yamada A, Healy JM, Hatta T, Kimbara K, Fukuda M, Yano K (1995) Characterization of biphenyl catabolic genes of gram-positive polychlorinated biphenyl degrader Rhodococcus sp. strain RHA1. Appl Environ Microbiol 61(6):2079–2085
Nanthini J, Ong SY, Sudesh K (2017) Identification of three homologous latex-clearing protein (lcp) genes from the genome of Streptomyces sp. strain CFMR 7. Gene 628:146–155. https://doi.org/10.1016/j.gene.2017.07.039
Oetermann S, Jongsma R, Coenen A, Keller J, Steinbüchel A (2019) LcpRVH2 - regulating the expression of latex-clearing proteins in Gordonia polyisoprenivorans VH2. Microbiology (SGM) 165(3):343–354. https://doi.org/10.1099/mic.0.000755
Oetermann S, Vivod R, Hiessl S, Hogeback J, Holtkamp M, Karst U, Steinbüchel A (2018) Histidine at position 195 is essential for association of Heme-b in Lcp1VH2. Earth Systems Environ 2(1):5–14. https://doi.org/10.1007/s41748-018-0041-2
Pompeani AJ, Irgon JJ, Berger MF, Bulyk ML, Wingreen NS, Bassler BL (2008) The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol Microbiol 70(1):76–88. https://doi.org/10.1111/j.1365-2958.2008.06389.x
Ramos JL, Martínez-Bueno M, Molina-Henares AJ, Terán W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R (2005) The TetR family of transcriptional repressors. Microbiol Mol Biol Rev 69(2):326–356. https://doi.org/10.1128/MMBR.69.2.326-356.2005
Richter M, Rossello-Mora R, Oliver Glockner F, Peplies J (2016) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32(6):929–931. https://doi.org/10.1093/bioinformatics/btv681
Rose K, Tenberge KB, Steinbüchel A (2005) Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6(1):180–188. https://doi.org/10.1021/bm0496110
Sharma V, Siedenburg G, Birke J, Mobeen F, Jendrossek D, Prakash T (2018) Metabolic and taxonomic insights into the gram-negative natural rubber degrading bacterium Steroidobacter cummioxidans sp. nov., strain 35Y. PLoS One 13(5):e0197448. https://doi.org/10.1371/journal.pone.0197448
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14):6614–6624. https://doi.org/10.1093/nar/gkw569
Tong SY, Schaumburg F, Ellington MJ, Corander J, Pichon B, Leendertz F, Bentley SD, Parkhill J, Holt DC, Peters G, Giffard PM (2015) Novel staphylococcal species that form part of a Staphylococcus aureus-related complex: the non-pigmented Staphylococcus argenteus sp. nov. and the non-human primate-associated Staphylococcus schweitzeri sp. nov. Int J Syst Evol Microbiol 65(Pt 1):15–22. https://doi.org/10.1099/ijs.0.062752-0
Watcharakul S, Rother W, Birke J, Umsakul K, Hodgson B, Jendrossek D (2016) Biochemical and spectroscopic characterization of purified latex clearing protein (Lcp) from newly isolated rubber degrading Rhodococcus rhodochrous strain RPK1 reveals novel properties of Lcp. BMC Microbiol 16:92. https://doi.org/10.1186/s12866-016-0703-x
Yikmis M, Arenskötter M, Rose K, Lange N, Wernsmann H, Wiefel L, Steinbüchel A (2008) Secretion and transcriptional regulation of the latex-clearing protein, Lcp, by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl Environ Microbiol 74(17):5373–5382. https://doi.org/10.1128/AEM.01001-08
Funding
This work was supported by KAKENHI Grant Numbers 16 K12631 and JP15H05639 from Japan Society for the Promotion of Science, Japan. Part of this work was also supported by JST-Mirai Program Grant Number JPMJMI19E6, Japan.
Author information
Authors and Affiliations
Contributions
DK, AS, and MF conceived the project. NG and DK wrote the manuscript. TA, SK, and DVL generated all the physiologic data. NG sequenced the genome and assembled the genome presented here. NG and TA annotated and analyzed the genome of this strain. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 503 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gibu, N., Arata, T., Kuboki, S. et al. Characterization of the genes responsible for rubber degradation in Actinoplanes sp. strain OR16. Appl Microbiol Biotechnol 104, 7367–7376 (2020). https://doi.org/10.1007/s00253-020-10700-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10700-1