
Abstract
The gram-positive bacterium Lactococcus lactis is a useful host for extracellular protein production. A main advantage of L. lactis over other bacterial expression systems is that lactococcal cells display low levels of autolysis and proteolysis. Previously, we developed a set of vectors for nisin-inducible extracellular production of N- or C-terminally hexa-histidine (His6)-tagged proteins. The present study was aimed at expanding our portfolio of L. lactis expression vectors for protein purification and site-specific labeling. Specifically, we present two new groups of vectors allowing N- or C-terminal provision of proteins with a Strep-tag II or AVI-tag. Vectors for AVI-tagging encode an additional His6-tag for protein purification. Another set of vectors allows removal of N-terminal Strep- or His6-tags from expressed proteins with the tobacco etch virus protease. Two possible applications of the developed vectors are presented. First, we show that Strep-tagged LytM of Staphylococcus aureus in the growth medium of L. lactis can be directly bound to microtiter plates coated with an affinity reagent and used for enzyme-linked immunosorbent assays. Second, we show that the AVI-tagged Sle1 protein from S. aureus produced in L. lactis can be directly biotinylated and fluorescently labeled. The fluorescently labeled Sle1 was successfully applied for S. aureus re-binding studies, allowing subcellular localization by fluorescence microscopy. In conclusion, we have developed a set of expression vectors that enhances the versatility of L. lactis as a system for production of proteins with tags that can be used for affinity purification and site-specific protein labeling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The gram-positive bacterium Lactococcus lactis is known to be a suitable host for the expression and secretion of heterologous proteins (Pontes et al. 2011). In most L. lactis expression systems, the production of proteins is induced using the nisin-inducible (NICE) system. Here, the expression of a target gene is directed by the nisA promoter, which is activated in the presence of the food-grade lantibiotic nisin that activates the NisRK two-component regulatory system (Ruyter et al. 1996; Kuipers et al. 1998). Different vectors using the NICE system have been constructed for both cytoplasmic and secreted production of (heterologous) proteins (Mierau and Kleerebezem 2005). For extracellular production, proteins were secreted via the Sec secretion machinery using the signal peptide of the lactococcal protein Usp45 (Borrero et al. 2011; Ng and Sarkar 2013). Recently, a set of vectors suitable for inducible extracellular protein production of N- or C-terminally hexa-histidine (His6)-tagged proteins was published (Neef et al. 2015).
The His6-tag is one of the most widely used tags as it allows efficient one-step purification of tagged proteins by metal affinity chromatography (Jones et al. 1995). However, this tag can have several drawbacks. For example, there may be many contaminating proteins (Lichty et al. 2005) and the His6-tag may lead to protein dimerization (Wu and Filutowicz 1999), instability, or degradation of tagged proteins (Rosales and Lee 2000). Also, His6-tags may interfere with ligand or substrate binding (Fonda et al. 2002). Therefore, the use of alternative protein tags could increase the chances of obtaining efficient protein production and purification and, at the same time, provide opportunities for direct labeling applications. For the isolation or labeling of expressed proteins, several tags have been used in L. lactis, such as the Strep-tag (Lubelski et al. 2006; Frelet-Barrand et al. 2010; Bernaudat et al. 2011), Flag-tag (Diep et al. 2007), Myc-tag (Bosma et al. 2006; Dieye et al. 2010; Visweswaran et al. 2013), and AVI-tag (Seeger et al. 2012; Neef et al. 2014). Only for the last two tags, secretion of heterologous tagged proteins was demonstrated in L. lactis.
The Strep-tag II system is based on an 8-amino-acid peptide tag (WSHPQFEK) with reversible high affinity to Strep-Tactin (an engineered form of streptavidin). It is derived from the well-known extremely high-affinity binding of biotin to streptavidin (Skerra and Schmidt 2000). The short, biologically inert, and proteolytically stable peptide tag allows purification of biologically active Strep-tagged fusion proteins under mild conditions (Schmidt and Skerra 2007). Cytoplasmic expression of a C-terminal Strep-tag fusion in L. lactis using the NICE system was shown for the LmrR protein (Lubelski et al. 2006).
The AVI-tag system involves a 15-amino-acid peptide (GLNDIFEAQKIEWHE) recognized by the biotin ligase BirA that catalyzes the amide linkage between biotin and the lysine residue in the AVI-tag peptide (Cull and Schatz 2000). Production in L. lactis of secreted staphylococcal proteins with an N-terminal AVI-tag for site-specific labeling with biotin has been reported recently (Neef et al. 2014).
The present study was aimed at expanding our portfolio of L. lactis expression vectors. Specifically, we constructed two vector sets by introducing sequences encoding N- or C-terminal AVI- or Strep-tags. The functionality of these vectors was demonstrated by expressing and secreting the tagged staphylococcal reporter proteins LytM and Sle1. The produced and exported proteins were used for rapid immune screening, and direct labeling for detection of localized binding on staphylococcal cells, respectively.
Materials and methods
Bacterial strains and growth conditions
Strains and plasmids are listed in Table 1. L. lactis strains were grown at 30 °C in M17 broth (Oxoid Limited, Hampshire, UK) supplemented with 0.5 or 2% glucose (w/v) (GM17). Standing cultures were supplemented with 0.5% glucose. For optimal production of reporter proteins, the L. lactis cells were grown in medium supplemented with 2% glucose with shaking (250 rpm). When necessary, the medium was supplemented with chloramphenicol (5 μg/ml). The Staphylococcus aureus strains USA300 and NCTC8325 were grown overnight at 37 °C, 250 rpm in Tryptone Soy Broth (TSB; Oxoid Limited).
General molecular biology
Enzymes and buffers were obtained from New England Biolabs (NEB, Ipswich, USA). Genomic DNA of S. aureus USA300, used as template for all PCR reactions, was isolated with the Genelute bacterial genomic DNA kit (Sigma-Aldrich, Zwijndrecht, The Netherlands) according to the manufacturer’s protocol with minor modifications as described before (Neef et al. 2014). PCR reactions were performed with a Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories, Richmond, CA). Primers used in this study, shown in Table 2, were obtained from Eurogentec (Maastricht, The Netherlands). The Taq (Life Technologies, Bleiswijk, The Netherlands) and Phusion Hot Start II (Thermo Fisher Scientific, Wilmington, Delaware, USA) polymerases were used according to the manufacturer’s protocols. PCR products were purified using the High Pure PCR purification kit (Analytic Jena, Jena, Germany). Ligations with T4 DNA ligase and DNA restriction endonuclease digestions were performed following the manufacturer’s protocols (NEB). Plasmids from L. lactis cells were extracted using the innuPREP Plasmid Mini Kit (Analytik Jena) with the following modifications: cell pellets were resuspended in solution A with lysozyme (2 mg/ml, Sigma-Aldrich) and incubated for 10 min at 55 °C. Further DNA purification and concentration were performed with the DNA Clean & Concentrator-5 (Zymo Research, Irvine, CA, USA). The Silica Bead DNA Gel Extraction Kit (Thermo Fisher Scientific) was used for DNA isolation from agarose gels. Nucleotide sequence analyses were performed by Eurofins DNA (Ebersberg, Germany). Electrotransformation of L. lactis was performed using a Gene pulser (Bio-Rad Laboratories) as described before (Leenhouts and Venema 1993).
Construction of expression vectors
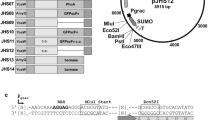
An overall schematic representation of constructed vectors is shown in Fig. 1. The His6-tag present in plasmids pNG4110, pNG4111, and pNG4210 was replaced by the Strep-tag II resulting in the plasmids pNG4110S, pNG4111S, and pNG4210S, respectively. For the construction of plasmid pNG4110S, a PCR fragment was generated using the primers StrepTag.For and Strep110.Rev using plasmid pNG4110 as a template. The PCR product was digested with BstBI and BamHI and ligated to linearized plasmid pNG4110, digested with the same enzymes. The same approach was used to replace the His6-tag to obtain plasmids pNG4111S and pNG4210S using specific primers indicated in Table 2, but in this case, the PCR fragments and vector were digested with BstBI and HindIII. Insertion of the AVI-tag in plasmids pNG4110, pNG4111, and pNG4210 resulted in plasmids pNG4110A, pNG4111A, and pNG4210A, respectively. For the construction of plasmids pNG4110A and pNG4111A, primers AVI-tagNotI.fw and AVI-tagNotI.rev were annealed to obtain a double-stranded DNA fragment with NotI-compatible sticky ends, which was ligated to the NotI-linearized plasmids pNG4110 and pNG4111. In the same manner, primers AVI-tagBamHI.fw and AVI-tagBamHI.rev were annealed and ligated to the BamHI-linearized plasmid pNG4210, resulting in plasmid pNG4210A. To construct lytM and sle1-expressing plasmids, PCR products amplified from S. aureus USA300 genomic DNA with primers indicated in Table 2 were digested with BamHI and NotI and ligated to BamHI/NotI-linearized vectors. Primer combinations using a reverse primer with a stop codon to prevent read-through translation into the plasmid sequences (F1/R2) were used to amplify fragments for ligation to plasmids pNG4110, pNG4111, pNG4110S, and pNG4111S, and primer combinations without a stop codon in the reverse primer (F1/R1) were used to amplify fragments for ligation into plasmids pNG4210, pNG4210S, pNG4110A, pNG4111A, and pNG4210A.

Schematic representation of the expression cassettes present in the different pNG vectors used in this study. a Representation of the secretion signal peptides (ss) and mature regions (26–316 and 26–334) of the S. aureus proteins LytM and Sle1, respectively. b Expression cassettes of the L. lactis pNG-vectors, encoding an N-terminal His6-tag (pNG4110), a C-terminal His6-tag (pNG4210), or a TEV-removable (TEV) N-terminal His6-tag (pNG4111). c Expression cassettes of the L. lactis pNG-vectors, encoding an N-terminal Strep-tag II (pNG4110S), a C-terminal Strep-tag II (pNG4210S), or a TEV-removable (TEV) N-terminal Strep-tag II (pNG4111S). d Expression cassettes of the L. lactis pNG-vectors, encoding an N-terminal His6-tag and a C-terminal AVI-tag (pNG4110A), a C-terminal His6-tag and a N-terminal AVI-tag (pNG4210A), or a TEV-removable (TEV) N-terminal His6-tag (pNG4111A) and a C-terminal AVI-tag. Positions of the restriction enzyme cleavage sites BamHI (B) and NotI (N), the TEV protease cleavage site (TEV), N-terminal or C-terminal His6-tag (h 6 ), Strep-tag II (ST), and AVI-tag (AVI) are indicated. ssu signal sequence of the gene for the secreted lactococcal protein Usp45, *stop codon
Protein expression, purification, and detection
For protein expression, lactococcal cultures were induced in the exponential phase of growth at an optical density at 600 nm (OD600) of 0.5 by the addition of nisin (final concentration 3 ng/ml; Sigma-Aldrich, St. Luis, MO) and harvested after overnight incubation. Cells were then separated from the growth medium by centrifugation. To concentrate protein samples, nisin-induced culture supernatants were precipitated with 10% TCA. The precipitated proteins were resuspended in lithium dodecyl sulfate (LDS) gel-loading buffer (Life Technologies, Grand Island, NY, USA) to denature them prior to LDS-polyacrylamide gel electrophoresis (PAGE). The respective cells were disrupted with 0.1 μm glass beads in LDS sample buffer (Biospec Products, Bartlesville, USA) in a Precellys 24 homogenizer (Bertin Technologies, Saint Quentin en Yvelines Cedex, France). To visualize the extent to which proteins were secreted, secreted and cellular proteins were analyzed by LDS-PAGE using NuPAGE gels (Life Technologies). Proteins were either visualized using Simply Blue Safe Staining (Life Technologies) or by Western blotting on Protan nitrocellulose membranes (Whatman, Germany) using mouse anti-His6-tag (Life Technologies), anti-Strep tag II (Iba Lifesciences, Germany), or anti-AVI-tag (Genscript, Piscataway, USA) primary antibodies. Fluorescent secondary antibodies (goat anti-mouse IRDye 800 CW, LI-COR Biosciences, Lincoln, NE, USA) were used for visualization of bound primary antibodies with an Odyssey Infrared Imaging System (LI-COR Biosciences). Expressed Strep-tag II proteins were purified from growth medium fractions (adjusted to pH 8 with NaOH) using a Strep-Tactin Sepharose 50% suspension following the manufacturer’s protocol (Iba Lifesciences). AVI-tagged Sle1 proteins in cell fractions (Sle1-AVI pellets) were washed twice with phosphate-buffered saline (PBS) and labeled directly with biotin using the BirA biotin ligase (Avidity, Aurora, CO, USA). Cell pellets were then washed twice with PBS and incubated with 0.1 mg Cy3-streptavidin (GE Healthcare Europe, Germany) in PBS for 30 min. After two washes with PBS, pellets were incubated in 6 M urea for 10 min and cell-bound Cy3-Sle1 released to the supernatant was collected after centrifugation. To observe possible non-specific binding of Cy3-streptavidin to biotinylated native L. lactis proteins, Sle1-AVI pellets were washed twice with PBS and incubated with 0.1 mg Cy3-streptavidin in 6 M urea for 10 min. After centrifugation, the supernatant was collected and used as Cy3 negative control supernatant.
Enzyme-linked immunosorbent assays
Strep-tactin-coated microtiter plates (8-well strips, Iba Lifesciences) were incubated for 1 h at room temperature with nisin-induced filter-sterilized growth medium samples (100 μl/well, adjusted to pH 8 with NaOH) containing LytM Strep-tag II fusion proteins. Serial dilutions of previously collected human plasma samples (500–2000,000) were made in PBS, 0.05% Tween 20, 5% skim milk. The plasma samples used were obtained from a patient with epidermolysis bullosa (EB01) and a healthy control volunteer (Control 02) as previously described (van der Kooi-Pol et al. 2013). Specific anti-human IgG secondary antibodies coupled to horseradish peroxidase (dilution 1:2000, Southern Biotechnology, Birmingham, AL) were used according to the manufacturer’s recommendations. Horseradish peroxidase activity was quantified by measuring the hydrolysis of the substrate (O-Phenylenediamine, Sigma-Aldrich) at an optical density of 492 nm (OD492) in a plate reader (Biotek Powerwave XS2, USA). Titers were expressed in arbitrary units (AU) obtained by calculating in Excel the extrapolated initial absorbance at serum dilution 1:1 (titer), using a linear regression equation adjusted through the data points of dilutions with measured OD495 readings between 1.0 and 0.1 (SLOPE(sample OD readings data set, serum dilution factor data set) + INTERCEPT(sample OD readings data set, serum dilution factor data set)), with all R 2 (Pearson product moment correlation coefficient) values > 0.98. Estimated titers of duplicates were averaged.
Microscopy
S. aureus NCTC8325 was grown overnight in TSB and used to inoculate fresh medium (1:50) in the morning. This culture was then grown until the mid-exponential phase. When an OD600 of 1.0 was reached, 1 ml of bacterial culture was collected, washed twice with PBS, and incubated in 1.5 ml of PBS containing 1.5 μg (30 μl) of Cy3-Sle1 or Cy3 control supernatant for 1 h at room temperature. After washing three times with PBS, cells were spotted onto polylysine-coated glass slides. Microscopic images were recorded using a Leica DM5500B epifluorescence microscope equipped with Cy3 filter block and a Leica DFC365FX camera using a ×63 objective (Leica Microsystems BV, The Netherlands). The presence of Sle1 was assayed using rabbit anti-Sle1 antibodies (originally referred to as anti-Aaa antibodies) at a 1:500 dilution using an earlier described protocol (Campo et al. 2004). The anti-Sle1/Aaa antibodies were kindly provided by Christine Heilmann (Heilmann et al. 2005). Secondary Oregon Green anti-rabbit antibodies (Molecular Probes) were used at a dilution of 1:850.
Ethics statements
Plasma from an epidermolysis bullosa patient and a healthy volunteer was collected under the approval of the medical ethics committee of the University Medical Center Groningen (approval no. NL27471,042,09) upon written informed consent and with adherence to the Helsinki Guidelines (van der Kooi-Pol et al. 2013). The necessary written informed consent was obtained from both plasma donors.
Results
Development of vectors for the secretion of Strep- or AVI-tagged proteins from L. lactis
In order to express and secrete AVI-tag or Strep-tag fusion proteins from L. lactis, the vector set pNG4110/111/210 was modified as depicted in Fig. 1. In a previous study, these vectors were successfully used for the extra-cytoplasmic production and purification of heterologous His6-tagged proteins (Neef et al. 2015). The His6-tag-encoding sequences in the vectors pNG4110/111/210 were replaced by the Strep-tag-encoding sequence, generating the vectors pNG4110S/111S/210S (Fig. 1c). Respectively, these vectors can be used to express proteins with N-terminal, N-terminal and TEV-cleavable, or C-terminal Strep-tags. Further, the AVI-tag-encoding sequence was added in the vectors pNG4110/111/210, respectively, generating vectors pNG4110A/111A/210A, which contain both the His6-tag- and AVI-tag-encoding sequences (Fig. 1d). Sequencing of the different vectors with inserted Strep- or AVI- tags showed that all had the expected nucleotide sequences and, upon culturing over multiple generations, no deletions were observed (data not shown). With respect to usage of the AVI-tag, it is noteworthy that BLAST analysis with the 15-residue AVI-tag amino acid sequence showed that none of the genes of L. lactis NZ9000 (i.e., the parental strain of PA1001) encodes proteins with similarity to the AVI-tag. This renders false-positive biotinylation of L. lactis proteins by the BirA enzyme unlikely. Lastly, it should be noted that the restriction sites used for cloning will add additional residues to an expressed protein of interest. This may have consequences for particular applications of the expressed protein.
Production of Strep- or AVI-tagged fusions of the staphylococcal proteins LytM and Sle1
To test our newly constructed “third-generation” vector set for protein production in L. lactis, genes encoding the naturally exported S. aureus proteins LytM and Sle1 were cloned into all of these vectors. The resulting plasmids were then introduced and expressed in L. lactis PA1001. The PA1001 strain lacks the gene for the major peptidoglycan hydrolase AcmA due to which cells do not lyse or separate during and after growth (Steen et al. 2005). This lack of separation results in a sedimentation of cells during growth in a standing culture. To obtain maximal protein production, 2% glucose with aeration was used to obtain a higher cell density compared to 0.5% glucose without aeration. Further, the PA1001 strain displays reduced proteolytic activity due to deletion of the htrA gene (Neef et al. 2014). After induced expression, all fusion proteins were produced as demonstrated by LDS-PAGE, where the respective proteins showed a mobility that matched their expected sizes (Fig. 2).

Expression of various tagged derivatives of the S. aureus LytM and Sle1 proteins. Detection of LytM (a) and Sle1 (b) expression in L. lactis by LDS-PAGE and subsequent Simply Blue staining (upper panels) or Western blotting (lower panels) using antibodies against different tags (α-His6-tag, α-Strep-tag, α-Avi-tag). Expression of the AVI-tagged fusion proteins was also verified using α-His6-tag antibodies showing a similar pattern of expression (results not shown). Expression from different vectors is indicated as follows: H0, pNG4110; H1, pNG4111; H2, pNG4210; S0, pNG4110S; S1, pNG4111S; S2, pNG4210S; A0, pNG4110A; A1, pNG4111A; and A2, pNG4210A. Lanes loaded with cell or growth medium fractions are indicated. Urea, supernatant fraction obtained after incubation of cells with 6 M urea and centrifugation. It should be noted that, in case of Sle1 production (b), the cell fractions correspond to cell fractions after urea incubation. Molecular weights of marker proteins are indicated on the left and the positions of LytM and Sle1 fusion proteins or the major secreted protein of L. lactis (Usp45) are indicated on the right
LytM is a peptidoglycan hydrolase with glycyl-glycine endopeptidase activity that is secreted by all S. aureus strains. In previous studies, LytM was shown to be a very immunogenic protein of S. aureus. This was determined using different techniques, such as enzyme-linked immunosorbent assays (ELISA) (van den Berg et al. 2015) and Luminex bead-based flow cytometry (van der Kooi-Pol et al. 2013). The presently constructed LytM fusion proteins were observed mainly in the growth medium fractions (Fig. 2a). While expression and secretion of the three differently His6-tagged LytM proteins were comparable as shown by Simply Blue staining, immunodetection of LytM with the C-terminal His6-tag was most efficient and lowest for LytM with an N-terminal His6-tag and TEV site. An opposite effect was observed for immunodetection of the AVI-tagged variants as expressed from the plasmids pNG4210A and pNG4111A (Fig. 2a). Furthermore, depending on the tag and its position, different amounts of LytM remained attached to the cells. This suggests that the nature of the tag and its N- or C-terminal position influence the expression, secretion, and/or detection efficiency of the fusion products.
Sle1 is a 32-kDa N-acetylmuramyl-l-alanine amidase which is involved in cell separation (Kajimura et al. 2005). The protein consists of a C-terminal CHAP domain (PF05257), which is responsible for peptidoglycan hydrolysis, plus three N-terminal LysM domains (PF01476) that are responsible for non-covalent cell wall-binding (Dreisbach et al. 2011; Visweswaran et al. 2014). Localized cell surface-binding of this protein has been shown using fusions to the fluorescent mCherry protein (Frankel and Schneewind 2012). Accordingly, all Sle1 fusion proteins were detected in the cell fractions upon nisin-induced expression. Release of Sle1 from the collected cells was achieved by incubation with 6 M urea, which disrupts the interaction of the Lys domains with the cell wall and results in the subsequent release of Sle1 (Fig. 2b). The urea-released Sle1 was effectively recovered by centrifugation. Of note, upon LDS-PAGE and Simply Blue staining, no detectable amounts of proteins other than Sle1 were observed in the respective supernatant fraction. As was observed for the LytM fusion proteins, the expression levels of Sle1 fusion proteins varied depending on the nature and position of the tag. In the case of Sle1 with a C-terminal AVI-tag, hybridizing bands with apparently lower molecular weight were detected upon expression from vectors pNG4110A and pNG4111A (Fig. 2b). Most likely, these relate to degradation products of the respective Sle1 fusions. In all other cases, Sle1 was apparently stably produced. These data show that all fusion products could be produced form the newly constructed vector sets.
Application of Strep-tagged LytM in an ELISA
To test the application potential of secreted Strep-tagged staphylococcal proteins for the detection of specific human antibodies, we applied an ELISA approach. To this end, the Strep-tagged LytM fusion protein was bound to Strep-tactin-coated microwell plates. The Strep-tagged LytM expressed from plasmid pNG4110S-lytM was selected for this application because this Strep-tagged LytM was slightly better detectable than the Strep-tagged LytM expressed from plasmids pNG4111S-lytM and pNG4210S-lytM (Fig. 2a). Specifically, nisin-induced culture supernatants of pNG4110S-lytM were added to microtiter plate wells coated with Strep-tactin. The plates were subsequently washed and ELISA was performed using plasma from an epidermolysis bullosa patient (EB01) and an age-matched healthy control individual (Control 2), both of which had been previously described (van der Kooi-Pol et al. 2013). The results as presented in Fig. 3 show that the EB01 plasma contained a substantially higher level of anti-LytM IgG (2177.5 AU) than the Control 2 plasma (237 AU), which is in full agreement with our previous observations. This shows that Strep-tagged fusions of S. aureus proteins expressed and secreted in L. lactis can be directly recovered from growth medium fractions and used for ELISA.

ELISA of Strep-tagged LytM using human plasma. Strep-tagged LytM was bound to a Strep-tactin 96-well microtiter plate by applying growth medium fractions of L. lactis pNG4110S-lytM. Upon washing of the plate, ELISA was performed using the human plasma samples EB01 and Control 2 as indicated. Regression equations, trend lines (black), and R 2 errors are indicated
Use of the AVI-tag to assess localized Sle1-binding to S. aureus cells
A Sle1 fusion protein with an N-terminal His6-tag and a C-terminal AVI-tag was obtained upon expression from pNG4110A-sle1. Of note, this fusion protein fractionated with the expressing cells, which is in accordance with the presence of three cell wall-binding LysM domains in this protein (Fig. 2b). To assess whether the cell-associated AVI-tagged Sle1 can be directly labeled with biotin, the producing L. lactis cells were collected and incubated with biotin and the BirA ligase. Subsequently, the cells were washed and incubated with Cy3-streptavidin. The resulting Cy3-Sle1 was released from the L. lactis cells by incubation with 6 M urea. Lastly, upon centrifugation, the supernatant fraction containing Cy3-Sle1 was diluted 50-fold and added to S. aureus NCTC8325 cells. Fluorescence microscopy showed that Cy3-labeled Sle1 bound locally on the surface of S. aureus NCTC8325 cells with a preference for the septal region (Fig. 4a). Similar hotspots for binding were observed when using Sle1-specific antibodies in combination with a secondary Oregon Green-labeled antibody (Fig. 4b). In this case, a S. aureus Δspa Δsbi double-mutant strain was used for the immunodetection in order to avoid Fc-specific IgG-binding by the staphylococcal protein A (Spa) and the Sbi protein of S. aureus. Of note, a possible interference by protein A and Sbi is not an issue when using the Cy3-labeled AVI-tagged Sle1 fusion protein. To control for possible Cy3-labeling of biotinylated native L. lactis proteins, L. lactis cells expressing the AVI-tagged Sle1 fusion protein were incubated as described above but in the absence of the BirA ligase. Upon extraction of the AVI-tagged Sle1 from the L. lactis cells and subsequent incubation with S. aureus NCTC8325 cells, no labeling of the staphylococcal cells was observed (Fig. 4c). This shows that an AVI-tagged protein produced in L. lactis can be effectively labeled with biotin and Cy3-streptavidin for further applications.

Binding of AVI-tagged Cy3-labeled Sle1 to S. aureus NCTC8325 cells. a Fluorescence microscopy of S. aureus NCTC8325 cells upon incubation with AVI-tagged Cy3-labeled Sle1. b S. aureus spa sbi double-mutant cells incubated with Sle1-specific rabbit antibodies and secondary Oregon Green anti-rabbit antibodies. c Negative control of cells incubated with non-biotinylated and therefore not fluorescently labeled AVI-tagged Sle1
Discussion
In this study, we describe a third-generation set of cloning vectors for the expression and secretion of differently tagged heterologous proteins from L. lactis. These vectors enable isolation of proteins under mild conditions using Strep-tag fusions or site-specific labeling with biotin using AVI-tag fusions. For both types of tags, N-terminal or C-terminal fusion proteins can be produced as was shown with the reporter proteins LytM and Sle1 from S. aureus. A secreted Strep-tag fusion of the staphylococcal protein LytM was successfully used for rapid immune screening using human sera. LytM was selected for this purpose because it was previously shown to be highly immunogenic (van der Kooi-Pol et al. 2013; van den Berg et al. 2015). An AVI-tagged variant of the staphylococcal Sle1 protein was site-specifically labeled and used for detection of localized binding on staphylococcal cells because it was previously shown to display a particular localization pattern on the S. aureus cell surface (Frankel and Schneewind 2012).
The combination of nisin-inducible expression using the L. lactis PA1001 strain allowed for controlled stable overnight expression of all fusion proteins. As expected based on the autolysin- and protease-deficiency of the PA1001 strain, no autolysis was detectable for any of the fusion protein-expressing derivative strains, and product degradation appears to be negligible. Only a minor possible degradation product of AVI-tagged Sle1 proteins was detectable upon overnight expression. This is in agreement with earlier reports on the usage of this strain, where a possible involvement of cytoplasmic or intramembrane proteases was invoked in residual degradation of the protein ClfB (Neef et al. 2015). These results led to the conclusion that L. lactis might have an additional proteolytric acitvity wich has a substrate specificity and thus does not degrade all secreted proteins. Possibly, Sle1 is like ClB, one of the substrates for this yet unknown protease. Because of this variation in expression, secretion, and stability is protein dependent, we generated a vector set for possible fusion of tags at either end of a protein to determine after detection of expression which fusion can be used best for its purpose.
Measurements of human immune responses against S. aureus LytM were previously performed by ELISA (van den Berg et al. 2015) and Luminex bead-based flow cytometry (van der Kooi-Pol et al. 2013). In both cases, the analyses involved the purification of His6-tagged LytM and subsequent binding of the purified protein to ELISA plates or Luminex beads. As shown in our present study, expression of LytM with a Strep-tag obviates the purification step as Strep-tagged LytM secreted by L. lactis can be directly applied to Strep-tactin-coated microplate wells for LytM immobilization on the plates. Importantly, the recorded human IgG-binding by immobilized LytM matched well with the previously published data, where it was shown that plasma samples from S. aureus-colonized epidermolysis bullosa patients contained significantly higher levels of anti-LytM IgGs than plasma samples from healthy control individuals (van der Kooi-Pol et al. 2013; Swierstra et al. 2015).
Previously, the microscopic detection of non-covalently cell wall-bound S. aureus proteins has been performed in various different ways, including immunofluorescence with labeled antibodies (Steen et al. 2003), in-frame fusions to fluorescent proteins like mCherry (Frankel and Schneewind 2012) or GFP (Visweswaran et al. 2013), or re-binding of purified proteins that had been randomly labeled with fluorophores (Zoll et al. 2012). Of note, such approaches may have certain drawbacks. In particular, in immunofluorescence microscopy, the presence of the IgG-binding proteins Spa (protein A) and Sbi results in an Fc-dependent off-target signal. This can be overcome by using spa sbi mutant strains but it precludes analyses with clinical S. aureus isolates. The use of in-frame fusions between a protein of interest and fluorescent proteins may be hampered by low signal intensities due to low expression levels, possible misfolding of the fluorescent protein, or incompatibilities with the protein secretion machinery (Chudakov et al. 2010). Further, random labeling of proteins used in re-binding studies can potentially interfere with the binding function of the protein. Our present results show that the AVI-tag allows for direct labeling of AVI-tagged Sle1 protein with Cy3-streptavidin. The resulting fluorescently marked Sle1 protein was then used for re-binding studies with staphylococcal cells. Due to the presence of the three LysM peptidoglycan-binding domains, the Sle1 protein binds to the cell wall upon interaction. Previously, Frankel and Schneewind 2012 used mCherry fused to Sle1 for S. aureus cell wall-binding studies to demonstrate the septal binding of Sle1. In our present approach, the previously observed preferential septal binding of Sle1 was clearly reproduced, indicating that the AVI-tag can be used as an alternative to in-frame fusions with fluorescent proteins. Of note, with the AVI-tagged Sle1, we also obtained possible evidence for additional loci where Sle1 may bind to the S. aureus cell surface besides the septal region (Fig. 4a). The latter observation deserves further investigations to identify the specific nature of the respective interactions and their possible biological relevance.
In conclusion, we have developed a set of third-generation expression vectors that enhances the versatility of L. lactis as a system for the production of proteins carrying tags that can be used for affinity purification and site-specific protein labeling.
References
Bernaudat F, Frelet-Barrand A, Pochon N, Dementin S, Hivin P, Boutigny S, Rioux J-B, Salvi D, Seigneurin-Berny D, Richaud P, Joyard J, Pignol D, Sabaty M, Desnos T, Pebay-Peyroula E, Darrouzet E, Vernet T, Rolland N (2011) Heterologous expression of membrane proteins: choosing the appropriate host. PLoS One 6:e29191. https://doi.org/10.1371/journal.pone.0029191
Borrero J, Jiménez JJ, Gútiez L, Herranz C, Cintas LM, Hernández PE (2011) Use of the usp45 lactococcal secretion signal sequence to drive the secretion and functional expression of enterococcal bacteriocins in Lactococcus lactis. Appl Microbiol Biotechnol 89:131–143. https://doi.org/10.1007/s00253-010-2849-z
Bosma T, Kanninga R, Neef J, Audouy SAL, van Roosmalen ML, Steen A, Buist G, Kok J, Kuipers OP, Robillard G, Leenhouts K (2006) Novel surface display system for proteins on non-genetically modified gram-positive bacteria. Appl Environ Microbiol 72:880–889. https://doi.org/10.1128/AEM.72.1.880-889.2006
Campo N, Tjalsma H, Buist G, Stepniak D, Meijer M, Veenhuis M, Westermann M, Müller JP, Bron S, Kok J, Kuipers OP, Jongbloed JDH (2004) Subcellular sites for bacterial protein export. Mol Microbiol 53:1583–1599. https://doi.org/10.1111/j.1365-2958.2004.04278.x
Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA (2010) Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90:1103–1163. https://doi.org/10.1152/physrev.00038.2009
Cull MG, Schatz PJ (2000) Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol 326:430–440
de Ruyter PG, Kuipers OP, de Vos WM (1996) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62:3662–3667
Diep DB, Skaugen M, Salehian Z, Holo H, Nes IF (2007) Common mechanisms of target cell recognition and immunity for class II bacteriocins. Proc Natl Acad Sci 104:2384–2389. https://doi.org/10.1073/pnas.0608775104
Dieye Y, Oxaran V, Ledue-Clier F, Alkhalaf W, Buist G, Juillard V, Lee CW, Piard J-C (2010) Functionality of sortase A in Lactococcus lactis. Appl Environ Microbiol 76:7332–7337. https://doi.org/10.1128/AEM.00928-10
Dreisbach A, van Dijl JM, Buist G (2011) The cell surface proteome of Staphylococcus aureus. Proteomics 11:3154–3168. https://doi.org/10.1002/pmic.201000823
Fonda I, Kenig M, Gaberc-Porekar V, Pristovaek P, Menart V (2002) Attachment of histidine tags to recombinant tumor necrosis factor-alpha drastically changes its properties. Sci World J 2:1312–1325. https://doi.org/10.1100/tsw.2002.215
Frankel MB, Schneewind O (2012) Determinants of Murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J Biol Chem 287:10460–10471. https://doi.org/10.1074/jbc.M111.336404
Frelet-Barrand A, Boutigny S, Moyet L, Deniaud A, Seigneurin-Berny D, Salvi D, Bernaudat F, Richaud P, Pebay-Peyroula E, Joyard J, Rolland N (2010) Lactococcus lactis, an alternative system for functional expression of peripheral and intrinsic Arabidopsis membrane proteins. PLoS One 5:e8746. https://doi.org/10.1371/journal.pone.0008746
Heilmann C, Hartleib J, Hussain MS, Peters G (2005) The multifunctional Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen and fibronectin. Infect Immun 73:4793–4802. https://doi.org/10.1128/IAI.73.8.4793-4802.2005
Jones C, Patel A, Griffin S, Martin J, Young P, O’Donnell K, Silverman C, Porter T, Chaiken I (1995) Current trends in molecular recognition and bioseparation. J Chromatogr A 707:3–22. https://doi.org/10.1016/0021-9673(95)00466-Z
Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, Hayashi I, Yamagishi J, Komatsuzawa H, Sugai M (2005) Identification and molecular characterization of an N-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation of Staphylococcus aureus. Mol Microbiol 58:1087–1101. https://doi.org/10.1111/j.1365-2958.2005.04881.x
Kreiswirth BN, Löfdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, Novick RP (1983) The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. https://doi.org/10.1038/305709a0
Kuipers OP, de Ruyter PGGA, Kleerebezem M, de Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21. https://doi.org/10.1016/S0168-1656(98)00100-X
Leenhouts K, Venema G (1993) Lactococcal plasmid vectors. Plasmids Pract Approach Oxf Univ Press, Oxf UK, pp 65–94
Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S (2005) Comparison of affinity tags for protein purification. Protein Expr Purif 41:98–105. https://doi.org/10.1016/j.pep.2005.01.019
Lubelski J, De Jong A, Van Merkerk R, Agustiandari H, Kuipers OP, Kok J, Driessen AJM (2006) LmrCD is a major multidrug resistance transporter in Lactococcus lactis. Mol Microbiol 61:771–781. https://doi.org/10.1111/j.1365-2958.2006.05267.x
McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC (2003) Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113–5120. https://doi.org/10.1128/JCM.41.11.5113-5120.2003
Mierau I, Kleerebezem M (2005) 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol 68:705–717. https://doi.org/10.1007/s00253-005-0107-6
Neef J, Koedijk DGAM, Bosma T, van Dijl JM, Buist G (2014) Efficient production of secreted staphylococcal antigens in a non-lysing and proteolytically reduced Lactococcus lactis strain. Appl Microbiol Biotechnol 98:10131–10141. https://doi.org/10.1007/s00253-014-6030-y
Neef J, Milder FJ, Koedijk DGAM, Klaassens M, Heezius EC, van Strijpm JAG, Otto A, Becher D, van Dijl JM, Buist G (2015) Versatile vector suite for the extracytoplasmic production and purification of heterologous His-tagged proteins in Lactococcus lactis. Appl Microbiol Biotechnol:1–12. https://doi.org/10.1007/s00253-015-6778-8
Ng DTW, Sarkar CA (2013) Engineering signal peptides for enhanced protein secretion from Lactococcus lactis. Appl Environ Microbiol 79:347–356. https://doi.org/10.1128/AEM.02667-12
Pontes DS, de Azevedo MSP, Chatel J-M, Langella P, Azevedo V, Miyoshi A (2011) Lactococcus lactis as a live vector: heterologous protein production and DNA delivery systems. Protein Expr Purif 79:165–175. https://doi.org/10.1016/j.pep.2011.06.005
Rosales JL, Lee K-Y (2000) Purification of dual-tagged intact recombinant proteins. Biochem Biophys Res Commun 273:1058–1062. https://doi.org/10.1006/bbrc.2000.3063
Schmidt TG, Skerra A (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc 2:1528–1535. https://doi.org/10.1038/nprot.2007.209
Seeger MA, Mittal A, Velamakanni S, Hohl M, Schauer S, Salaa I, Grütter MG, van Veen HW (2012) Tuning the drug efflux activity of an ABC transporter in vivo by in vitro selected DARPin binders. PLoS One 7:e37845. https://doi.org/10.1371/journal.pone.0037845
Skerra A, Schmidt TGM (2000) [18] Use of the Strep-tag and streptavidin for detection and purification of recombinant proteins. In: Enzymology B-M in (ed). Academic Press, Cambridge, pp 271–304
Steen A, Buist G, Horsburgh GJ, Venema G, Kuipers OP, Foster SJ, Kok J (2005) AcmA of Lactococcus lactis is an N-acetylglucosaminidase with an optimal number of LysM domains for proper functioning. FEBS J 272:2854–2868. https://doi.org/10.1111/j.1742-4658.2005.04706.x
Steen A, Buist G, Leenhouts KJ, Khattabi ME, Grijpstra F, Zomer AL, Venema G, Kuipers OP, Kok J (2003) Cell wall attachment of a widely distributed peptidoglycan binding domain is hindered by cell wall constituents. J Biol Chem 278:23874–23881. https://doi.org/10.1074/jbc.M211055200
Swierstra J, Debets S, de Vogel C, Toom NL, Verkaik N, Ramdani-Bouguessa N, Jonkman MF, van Dijl JM, Fahal A, van Belkum A, van Wamel W (2015) IgG4 subclass-specific responses to Staphylococcus aureus antigens shed new light on host-pathogen interaction. Infect Immun 83:492–501. https://doi.org/10.1128/IAI.02286-14
van den Berg S, Koedijk DGAM, Back JW, Neef J, Dreisbach A, van Dijl JM, Bakker-Woudenberg IAJM, Buist G (2015) Active immunization with an octa-valent Staphylococcus aureus antigen mixture in models of S. aureus bacteremia and skin infection in mice. PLoS One 10:e0116847. https://doi.org/10.1371/journal.pone.0116847
van der Kooi-Pol MM, de Vogel CP, Westerhout-Pluister GN, Veenstra-Kyuchukova YK, Duipmans JC, Glasner C, Buist G, Elsinga GS, Westra H, Bonarius HPJ, Groen H, van Wamel WJB, Grundmann H, Jonkman MF, van Dijl JM (2013) High anti-staphylococcal antibody titers in patients with epidermolysis bullosa relate to long-term colonization with alternating types of Staphylococcus aureus. J Invest Dermatol 133:847–850. https://doi.org/10.1038/jid.2012.347
Visweswaran GRR, Leenhouts K, van Roosmalen M, Kok J, Buist G (2014) Exploiting the peptidoglycan-binding motif, LysM, for medical and industrial applications. Appl Microbiol Biotechnol 98:4331–4345. https://doi.org/10.1007/s00253-014-5633-7
Visweswaran GRR, Steen A, Leenhouts K, Szeliga M, Ruban B, Hesseling-Meinders A, Dijkstra BW, Kuipers OP, Kok J, Buist G (2013) AcmD, a homolog of the major autolysin AcmA of Lactococcus lactis, binds to the cell wall and contributes to cell separation and autolysis. PLoS One 8:e72167. https://doi.org/10.1371/journal.pone.0072167
Wu J, Filutowicz M (1999) Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Pol 46:591–599
Zoll S, Schlag M, Shkumatov AV, Rautenberg M, Svergun DI, Götz F, Stehle T (2012) Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J Bacteriol 194:3789–3802. https://doi.org/10.1128/JB.00331-12
Acknowledgements
We thank the anonymous patient with EB from the Dutch Epidermolysis Bullosa Registry for blood donation and Magda van der Kooi-Pol, José Duipmans, and Marcel Jonkman for collecting the EB patient sera in a previous study. We thank Christine Heilmann for providing rabbit antibodies against Sle1.
Funding
F. Romero Pastrana received a scholarship from CONACyT (169643) and was supported in parts by the Graduate School of Medical Sciences from the University of Groningen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
All procedures performed in studies involving human blood donations were in accordance with the standards of the medical ethics committee of the University Medical Center Groningen and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Romero Pastrana, F., Neef, J., van Dijl, J.M. et al. A Lactococcus lactis expression vector set with multiple affinity tags to facilitate isolation and direct labeling of heterologous secreted proteins. Appl Microbiol Biotechnol 101, 8139–8149 (2017). https://doi.org/10.1007/s00253-017-8524-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8524-x