
Abstract
Fumonisins are carcinogenic mycotoxins that are frequently found as natural contaminants in maize from warm climate regions around the world. The aminotransferase FumI is encoded as part of a gene cluster of Sphingopyxis sp. MTA144, which enables this bacterial strain to degrade fumonisin B1 and related fumonisins. FumI catalyzes the deamination of the first intermediate of the catabolic pathway, hydrolyzed fumonisin B1. We used a preparation of purified, His-tagged FumI, produced recombinantly in Escherichia coli in soluble form, for enzyme characterization. The structure of the reaction product was studied by NMR and identified as 2-keto hydrolyzed fumonisin B1. Pyruvate was found to be the preferred co-substrate and amino group receptor (K M = 490 μM at 10 μM hydrolyzed fumonisin B1) of FumI, but other α-keto acids were also accepted as co-substrates. Addition of the co-enzyme pyridoxal phosphate to the enzyme preparation enhanced activity, and saturation was already reached at the lowest tested concentration of 10 μM. The enzyme showed activity in the range of pH 6 to 10 with an optimum at pH 8.5, and in the range of 6°C to 50°C with an optimum at 35°C. The aminotransferase worked best at low salt concentration. FumI activity could be recovered after preincubation at pH 4.0 or higher, but not lower. The aminotransferase was denatured after preincubation at 60°C for 1 h, and the residual activity was also reduced after preincubation at lower temperatures. At optimum conditions, the kinetic parameters K M = 1.1 μM and k cat = 104/min were determined with 5 mM pyruvate as co-substrate. Based on the enzyme characteristics, a technological application of FumI, in combination with the fumonisin carboxylesterase FumD for hydrolysis of fumonisins, for deamination and detoxification of hydrolyzed fumonisins seems possible, if the enzyme properties are considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fumonisins are a group of structurally related mycotoxins produced by several species of Fusarium (Rheeder et al. 2002) as well as by Alternaria alternata (Chen et al. 1992), Aspergillus niger (Frisvad et al. 2007), and Tolypocladium species (Mogensen et al. 2011). Fusarium verticillioides may be considered the most important fumonisin producer from the aspect of food and feed safety, as it is an important pathogen of maize in warm climate regions around the world. The fumonisins produced by this fungus were also the first to be isolated (Gelderblom et al. 1988; Bezuidenhout et al. 1988) and thoroughly studied (Marasas 2001). Fumonisin B1 (FB1), the most prevalent fumonisin of F. verticillioides, can cause equine leukoencephalomalacia (Marasas et al. 1988; Kellerman et al. 1990), porcine pulmonary edema (Harrison et al. 1990; Haschek et al. 2001), liver cancer in rats (Gelderblom et al. 1991), neural tube disorder in mice (Sadler et al. 2002; Gelineau-van Waes et al. 2005), alteration of the immune response in pigs (Taranu et al. 2005), and shows several other toxic effects in laboratory and domestic animals (Voss et al. 2007). Exposure to fumonisins is also associated with cancer and neural tube disorder in humans (Wild and Gong 2010). At the molecular level, fumonisins interfere with sphingolipid metabolism by inhibiting the enzyme ceramide synthase (Wang et al. 1991). The resulting imbalance of sphingolipids, which also have signaling functions as cellular messenger molecules, is responsible for toxic and carcinogenic effects of fumonisins (Merrill et al. 2001; Riley et al. 2001). However, other mechanisms of fumonisin toxicity and carcinogenicity were also proposed, including activation of mitogen-activated protein kinase (Wattenberg et al. 1996), lipid peroxidation (Abel and Gelderblom 1998), and alteration of the biosynthesis of other lipids in addition to the sphingolipids (Gelderblom et al. 2001).
The known fumonisin biodegradation pathways in black yeast strains (Blackwell et al. 1999) and bacteria (Duvick et al. 2003; Heinl et al. 2010) all start with the hydrolytic release of the two tricarballylic acid side chains, catalyzed by a fumonisin carboxylesterase. The second step is deamination of hydrolyzed fumonisin B1 (HFB1), either by an amine oxidase in black yeast strains (Blackwell et al. 1999) or an aminotransferase in bacterial strains (Heinl et al. 2010, 2011) (Fig. 1). Enzymatic detoxification of fumonisins may be a suitable approach to increase feed and food safety (Karlovsky 1999). However, it is unclear whether toxicity is sufficiently reduced by enzymatic cleavage of the two tricarballylic acid side chains. Hydrolyzed fumonisins were reported not to have cancer-initiating potency (Gelderblom et al. 1993), to be less hepatotoxic (Howard et al. 2002), less disruptive of sphingolipid metabolism, and not inducing neural tube disorder (Voss et al. 2009) in vivo. Hydrolysis of fumonisins by nixtamalization was reported to reduce, but not eliminate, toxicity (Hendrich et al. 1993; Voss et al. 1996). The in vitro effects of hydrolyzed fumonisins were reported to range from less (Flynn et al. 1997; Norred et al. 1997; Schmelz et al. 1998; van der Westhuizen et al. 1998; Seefelder et al. 2003) to more toxic than those of the intact fumonisins (Gelderblom et al. 1993). The 2-amino group of hydrolyzed fumonisins plays a key role for toxicity, since it can be acylated both in vitro (Humpf et al. 1998) and in vivo (Seiferlein et al. 2007), and the N-acyl-metabolites are even more cytotoxic than FB1 (Humpf et al. 1998). In summary, deamination in addition to removal of the side chains may be necessary for complete fumonisin detoxification. Since the bacterial aminotransferases, contrary to the black yeast amine oxidases, do not require molecular oxygen for activity, they may be more suitable for applications in anaerobic environments, for instance as feed or food enzymes for detoxification directly in the intestinal tract of animals or humans. Another possible application would be in ethanol fermentation of maize mash, where fumonisins normally accumulate in the distillers dried grains and solubles, which are subsequently used as animal feed.

Initial steps of fumonisin B1-degradation pathway of Sphingopyxis sp. MTA144: FB1 (2-amino-12,16-dimethyl-3,5,10-trihydroxy-14,15-propan-1,2,3-tricarboxyicosane) is substrate of the fumonisin carboxylesterase FumD, which catalyzes hydrolytic cleavage of both tricarballylic acid (TCA) chains off the core chain to produce HFB1 (2-amino-12,16-dimethylicosane-3,5,10,14,15-pentol) and tricarballylic acid (1,2,3-propanetricarboxylic acid). Aminotransferase FumI transfers the 2-amino group from HFB1 to pyruvate, producing 2-keto-HFB1 (3,5,10,14,15-pentahydroxy-12,16-dimethylicosane-2-one) and alanine
We have previously cloned the fumI gene of Sphingopyxis sp. MTA144, which catalyzes deamination of HFB1 (Heinl et al. 2010), expressed the gene in E. coli and purified the enzyme (Hartinger et al. 2010). Here, we report the characterization of the enzyme together with its kinetic parameters as a starting point for evaluation of the application potential of the enzyme.
Materials and methods
Chemicals
FB1 and 13C-FB1 were obtained from Biopure Referenzsubstanzen GmbH (Tulln, Austria). HFB1 was prepared by complete hydrolysis of FB1 with purified recombinant carboxylesterase FumD (Heinl et al. 2010). All other chemicals were purchased from Sigma (St. Louis, MO, USA) or Sigma–Aldrich (Steinheim, Germany). Water was purified by reverse osmosis in an arium® RO 61316 system (Sartorius Stedim Biotech GmbH, Göttingen, Germany).
Recombinant enzyme production and purification
Construction of the expression vector pET-30a-AT144HIS, gene expression in E. coli ArcticExpress(DE3), and purification of the 6xHis-tagged recombinant aminotransferase FumI by immobilized nickel affinity chromatography were previously described (Hartinger et al. 2010). The recombinant enzyme FumI-HIS comprises the FumI sequence (GenBank ACS27061) with a C-terminal extension of ASSVDKLAAALEHHHHHH. The calculated molecular mass is 48,271.5 Da. Preparations of FumI-HIS were stored at −20°C after addition of glycerol to 25%.
Enzyme activity assays
Possible co-substrates of the aminotransferase were tested in a reaction system containing 94 ng/ml FumI-HIS in 20 mM Tris–HCl pH 7.4, 50 mM NaCl, 2 mM CaCl2, 20 μM pyridoxal phosphate (PLP), and 0.1 mg/ml bovine serum albumin (BSA). HFB1 as the substrate was employed at a concentration of 15 μM, and no co-substrate or 15 μM or 3 mM of the α-keto acids pyruvate, α-ketobutyrate, α-ketoglutarate, glyoxylate, or oxaloacetate, respectively, were added. Samples of 200 μl were taken immediately after addition of the enzyme to the reaction mixture (t = 0 h), as well as after 0.5, 1, and 3 h of incubation in a water bath at 25°C. For correlation of enzyme activity with pyruvate and oxaloacetate concentration, the assay was performed as above except with 10 μM HFB1, and co-substrate concentrations were varied from 0.01 to 100 mM. Aminotransferase activity was determined by calculating the slope of the initial, linear reaction rates in Δ μmol HFB1 per liter per minute. Oxaloacetate was dissolved directly before use to avoid decarboxylation. To test the need for addition of PLP, the assay was performed as above with 15 μM HFB1 and PLP concentrations ranging from 0 to 400 μM. The effect of salt on enzyme activity was tested by using 20 mM Tris–HCl pH 8.0 buffers with 1,000 mM, 100 mM, 10 mM NaCl, or without added NaCl, Teorell–Stenhagen buffer pH 8.0 (Teorell and Stenhagen 1938), or 100 mM potassium phosphate buffer, all with 20 μM PLP, 0.1 mg/ml BSA, 3 mM pyruvate, and 15 μM HFB1.
The optimum pH for enzyme activity was determined by setting Teorell–Stenhagen buffer to pH values ranging from pH 1.8 to 11.5 at 22°C (Teorell and Stenhagen 1938). The assay was performed using 67 ng/ml FumI-HIS, 10 μM HFB1, 20 μM PLP, 0.1 mg/ml BSA, and 5 mM pyruvate at 25°C. Samples were taken before addition of enzyme and after 15, 30, 45, 60, 120, and 240 min. Enzyme activities were calculated from early time points, where reaction rates were linear, and are expressed as μM 2-keto-HFB1 formed per minute. The optimum temperature for enzyme activity was determined by using the same conditions as for the pH dependence, with the buffer set to pH 8.0 at 22°C, and incubation temperatures in water baths ranging from 6°C to 50°C. Reaction mixtures were temperature-equilibrated for 30 min before addition of enzyme.
To determine pH stability of FumI-HIS, the Teorell–Stenhagen buffer was set to pH values between pH 1.1 and 10.7, and 14 μg/ml of enzyme was incubated in these buffers at 35°C for 1 h. The residual enzyme activity was determined in the same buffer at pH 7.8 after dilution of FumI-HIS to 50 ng/ml, using 15 μM HFB1, 20 μM PLP, 0.1 mg/ml BSA, and 5 mM pyruvate. Samples were taken, and reaction rates were determined as for the correlation of activity with pH and temperature. The thermal stability of FumI-HIS was determined by incubating 14 μg/ml of enzyme in Teorell–Stenhagen buffer, set to pH 8.0 at 22°C, for 1 h in water baths at temperatures ranging from 7°C to 60°C. Residual enzyme activity was measured at 35°C after dilution in the same buffer with final concentrations of FumI-HIS, HFB1, PLP, BSA, and pyruvate as described for pH stability. Samples were taken, and reaction rates were calculated as for the determination of optimum pH and temperature.
Enzyme kinetics were determined by incubating 10 ng/ml FumI-HIS at 35°C in 20 mM Tris–HCl pH 8.0, 20 μM PLP, 0.1 mg/ml BSA, and 5 mM pyruvate, while the concentration of HFB1 was varied from 0.1 to 100 μM. Samples were taken, and reaction rates were determined as above.
Liquid chromatography–mass spectrometry
Samples taken from enzyme assays were inactivated at 99°C for 10 min and stored frozen. After thawing, mixing, and centrifugation, aliquots of samples containing nominally 0.2 nmol HFB1, but no more than 100 μl, were dried at 50°C under a stream of nitrogen and dissolved in 200 μl solvent with 13C-FB1 as internal standard. Thus, the final nominal HFB1 concentration for analysis was 1 μM, or less if the original concentration was less than 2 μM. All samples were analyzed using the previously described instrumental setup and method (Heinl et al. 2010), to which 2-keto-HFB1 has been integrated (Hartinger et al. 2010).
Preparative isolation of 2-keto-HFB1 and structure determination by NMR
FumI-HIS was produced in E. coli ArcticExpress (DE3) as previously described (Hartinger et al. 2010). Biomass was resuspended in 1/100 of the culture volume, homogenized in a French Press, and 1.2 ml of the clarified lysate was added to 4.05 mg HFB1 in 25 ml buffer (20 mM Tris–HCl pH 8.0, 6.6 mM pyruvate, 30 μM PLP, and 0.1 mg/ml BSA). After incubation at 30°C, complete deamination of HFB1 was confirmed by liquid chromatography–mass spectrometry (LC–MS). The reaction mixture was centrifuged, lyophilized, and dissolved in 6 ml of 40% acetonitrile in water (v/v). After centrifugation, 600-μl portions were separated on a preparative Agilent 1100 HPLC system (Agilent, Waldbronn, Germany) consisting of a G1361A pump, a G2260A autosampler, an adjustable flow splitter (Analytical Scientific Instruments, CA, USA), an evaporative light-scattering detector (SEDEX LT-ELSD Model 85, Sedere, Alfortville, France), a G1365B multi-wavelength detector, and a G1364B fraction collector. A Gemini-NX C18 column (150 × 21.2 mm, 5-μm particle size, 110-Å pore size; Phenomenex, Aschaffenburg, Germany) protected by a Gemini C18 pre-column (15 × 21.2 mm) was operated at a flow rate of 16 ml/min using gradient elution. Mobile phase A, consisting of acetonitrile/H2O (10:90, v/v), was degassed by membrane filtration. Acetonitrile was used as mobile phase B. The gradient was as follows: 0–0.2 min: 0% B, 0.2–5 min: linear increase to 95% B, 5–6.4 min: isocratic elution at 95% B, 6.4–6.5 min: linear decrease to 0% B, 6.5–9.5 min: re-equilibration at 0% B. The column effluent was split 1:70, one part moving into the evaporative light-scattering detector and the main part passing through the multivariate wavelength detector to the fraction collector. The 2-keto-HFB1 eluted between 4.85 and 5.7 min and fractions were collected in that time window. The combined fractions were evaporated to dryness on a rotary evaporator, dissolved in 7 ml of ethyl acetate, and transferred into an 8-ml screw cap vial; the ethyl acetate solution was evaporated to dryness under a stream of nitrogen at 50°C. The 2-keto-HFB1, a viscous, clear, and colorless oily substance, was stored at 4°C until NMR analysis.
1H- and 13C-NMR spectra were obtained from CD3CN solutions using an Avance DRX-400 FT-NMR spectrometer (Bruker BioSpin, Rheinstetten, Germany), operating at 400.13 MHz for 1H and 100.62 MHz for 13C, at 295 K using a 5-mm inverse broadband Z-gradient probe head. Data were recorded and evaluated using TOPSPIN 1.3 software (Bruker Biospin). All pulse programs were taken from the Bruker software library. Chemical shifts were established on the basis of residual solvent resonances.
Results
Biochemical characterization of FumI-HIS
Clarified cell lysates of E. coli overexpressing the fumI gene were previously used to obtain preliminary information, namely that HFB1 is a substrate and that an α-keto acid such as pyruvate is required as co-substrate for the activity of the aminotransferase FumI (Heinl et al. 2010). The detailed biochemical characterization reported here was made using FumI-HIS, which was prepared and purified to electrophoretic homogeneity as described before (Hartinger et al. 2010). This enzyme preparation was found to be stable for several months when stored in 25% glycerol at −20°C. Clarified cell lysates of E. coli ArcticExpress (DE3) producing FumI and FumI-HIS from the same expression vector pET-30a under identical conditions showed a very similar HFB1 deamination activity, indicating that the His-tag does not affect the enzymatic activity.
The α-keto acids pyruvate, α-ketobutyrate, α-ketoglutarate, glyoxylate, and oxaloacetate were tested as potential co-substrates for aminotransferase FumI (Fig. 2). No deamination of HFB1 was detectable when no co-substrate was added or in the presence of α-ketoglutarate, while pyruvate, oxaloacetate, glyoxylate, and α-ketobutyrate, when included at 3 mM concentration, supported the reaction, with HFB1 deamination efficiency decreasing in this order. When tested with 10 μM HFB1, the enzyme showed maximal activity at 10 mM pyruvate, and the K M for pyruvate was 490 μM. Deamination activity was significantly reduced when the concentration of the co-substrate pyruvate was in the same range as that of HFB1, and substrate inhibition was observed for pyruvate concentrations in the range of 50–100 mM (Fig. 3). With oxaloacetate as co-substrate, reaction rates were lower, and the K M for oxaloacetate was 4,150 μM (Fig. 3). Addition of PLP enhanced the aminotransferase activity of FumI-HIS, and saturation was reached already at the lowest concentration tested (10 μM PLP) since a further increase in PLP concentration to 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 300, and 400 μM in the assay mixture did not result in a further increase in the reaction rate. Without added PLP, FumI-HIS showed about 23% of the maximum activity.

Comparison of HFB1 deamination activity of FumI-HIS with five different α-keto acids as co-substrate. The reactions were incubated in 20 mM Tris–HCl buffer (pH 7.4) at 25°C with 94 ng/ml FumI and 3 mM α-keto acid

Correlation between pyruvate concentration or oxaloacetate concentration and HFB1 deamination activity. The reactions were incubated with 94 ng/ml FumI-HIS and 10 μM HFB1 in 20 mM Tris–HCl (pH 7.4) with 0.1 mg/ml BSA, 20 μM PLP, and the indicated concentration of pyruvate or oxaloacetate at 25°C. Samples were taken and analyzed, and reaction rates were calculated as described in Materials and Methods. The reaction mixtures without co-substrates (negative controls), which are not shown in the semi-logarithmic plot, gave HFB1 deamination rates of zero
Addition of NaCl to the 20 mM Tris–HCl (pH 8.0) reaction buffer reduced the HFB1 deamination rate (Fig. 4). When using 100 mM potassium phosphate or Teorell–Stenhagen buffer, enzyme activity was also lower than in 20-mM Tris–HCl buffer of the same pH. Nevertheless, Teorell–Stenhagen buffer was used to determine the optimum pH for enzyme activity, since this buffer can be adjusted to any pH between 2.0 and 12.0 (Teorell and Stenhagen 1938). Aminotransferase FumI-HIS was active in the range of pH 6.5 to 9.7 and showed a rather sharp optimum at pH 8.0 and 8.5 (Fig. 5a). The enzyme was active over a broad temperature range of 6°C to 45°C with an optimum for the 30 min assay at 35°C (Fig. 5b). Significant and rapid thermal inactivation was observed at 50°C, where traces of 2-keto-HFB1 were formed only at the beginning of the incubation period (data not shown). Chromatograms of samples from the HFB1 deamination reaction of this experiment at 35°C are shown in Fig. 6. Furthermore, FumI-HIS was incubated with intact FB1 instead of hydrolyzed FB1 under identical conditions, but FB1 was not deaminated. Reaction with a hydrolyzed total fumonisin extract from F. verticillioides culture material performed under the same conditions showed that, in addition to HFB1, HFB2, and HFB3 were substrates of FumI-HIS as well. When FumI-HIS was preincubated at an acidic pH and 35°C for 1 h before HFB1 deamination was measured, the activity was reduced after incubation at pH 6.5 or less, and completely abolished after incubation at pH below 4.0 (Fig. 7a). Incubation at pH 8.0 for 1 h at various temperatures resulted in a steady decrease of FumI activity with increasing temperature, and the enzyme was completely inactivated after incubation at 60°C. Even after incubation at 7°C, a 15% loss of enzyme activity compared to non-preincubated FumI was determined (Fig. 7b).

Effect of different buffers and salt concentrations on the HFB1-deamination rate of aminotransferase FumI-HIS. Negative control: 20 mM Tris–HCl (pH 8.0) with buffer instead of enzyme

Determination of optimum pH (a) and temperature (b) for FumI-HIS activity. The reactions were incubated with 67 ng/ml FumI-HIS and 10 μM HFB1 in Teorell–Stenhagen buffer, with supplements as described in Materials and Methods, set to the indicated pH values (a) or to pH 8.0 (b), at 25°C (a) or the indicated temperatures (b)

LC–MS chromatograms of samples from a HFB1 transamination reaction. Samples of 10 μM HFB1 in Teorell–Stenhagen buffer (pH 8.0) at 35°C were taken before (a), 30 min (b), or 240 min (c) after addition of purified aminotransferase FumI-HIS to a final concentration of 67 ng/ml. HFB1 and 2-keto-HFB1 were separated on a C8 reversed phase column in a formate–acetonitrile gradient and detected in SIM mode

Stability of FumI-HIS at various pH values (a) and temperatures (b). Fourteen micrograms per milliliter FumI-HIS was incubated in Teorell–Stenhagen buffer at the indicated pH at 35°C (a) or in the same buffer at pH 8.0 at the indicated temperature (b) for 1 h. Residual enzyme activity was determined as described in Materials and Methods. The measured activities after incubation at pH 8.0 (a) or without preincubation (b) were set to 100%
The apparent steady-state kinetic parameters of FumI-HIS catalyzed HFB1 deamination were determined for a saturating co-substrate concentration of 5 mM pyruvate in 20 mM Tris–HCl buffer (pH 8.0) at 35°C, and the Michaelis–Menten plot is shown in Fig. 8. The apparent V max value of 0.022 μM/min was reached at 5 μM HFB1 when 10 ng/ml of FumI-HIS was present in the reaction mixture. A further increase in substrate (HFB1) concentration resulted in a significant reduction of the reaction rate, indicating substrate inhibition. The k cat value was calculated to be 104 min−1 using the theoretical molecular mass of FumI-HIS, and the Michaelis constant K M for HFB1 was determined to be 1.1 μM. The specific HFB1 deamination activity was 2.2 μmol/min/mg.

Initial HFB1 deamination reaction rate as a function of substrate concentration. The reactions were performed with 10 ng/ml FumI-HIS and 5 mM pyruvate, a saturating co-substrate concentration, at 35°C in 20 mM Tris–HCl pH 8.0, 20 μM PLP, 0.1 mg/ml BSA. HFB1 concentrations ranged from 0.1 to 100 μM
Structure determination of the reaction product by NMR
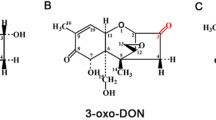
From the 1D 1H- to 13C-APT-NMR spectra, it was obvious that the sample did not contain a single species, but three different forms. In order to analyze the molecular structures of these compounds, 1H1H COSY, 1H13C HSQC, and 1H13C HMBC spectra were recorded. Using these two-dimensional methods, all of the structures could be elucidated unambiguously, and complete assignments of 1H and 13C signals were established (Table 1). Thus, the structures were identified as 2-keto-HFB1 and two cyclic hemiketals formed by ring closure between the 2-keto moiety and the 5-OH. While the hemiketals have already been described (Blackwell et al. 1999), the open-chain keto form has not yet been characterized.
Discussion
Gastrointestinal detoxification of fumonisins by specific enzymes is a promising concept to ameliorate the effects that fumonisins, which are frequently found as natural contaminants of maize in large parts of the world, have on the health and performance of domestic animals. It is important to know the characteristics of an enzyme-catalyzed reaction before a technological application can be considered. We determined some key properties of the aminotransferase FumI of Sphingopyxis sp. MTA144 with respect to its reaction with fumonisins. The enzyme is active with hydrolyzed FB1, the first intermediate in all known pathways of FB1 catabolism, and not with FB1 itself and catalyzes the transfer of the 2-amino group onto a suitable acceptor (Heinl et al. 2010). Since the corresponding amino group of structurally similar sphinganine is acylated by ceramide synthase, which is the molecular target that is inhibited by fumonisins (Wang et al. 1991), the deamination reaction can be considered as important for detoxification of fumonisins. To our knowledge, no reports on the toxicity of 2-keto-HFB1 have been published, and we have not studied its toxicity yet. However, the amino group is thought to play a key role for the toxicity of FB1 (Gelderblom et al. 1993; Lu et al. 1997; Norred et al. 2001; Fernandez-Surumay et al. 2004), and obviously, 2-keto-HFB1 cannot be acylated in a similar way as HFB1 (Humpf et al. 1998; Seiferlein et al. 2007). Therefore, it seems reasonable to speculate that 2-keto-HFB1 may be non-toxic.
Numerous aminotransferases have been identified and studied since the first description of this class of enzymes (Needham 1930), and several are used in industrial applications for the production of a range of natural and non-natural amino acids and amines (Taylor et al. 1998; Hwang et al. 2005; Zhu and Hua 2009). However, this is the first characterization of an aminotransferase that catalyzes deamination of a fumonisin. Our work may lead to the further expansion of the range of aminotransferase applications to mycotoxin detoxification. Since FB1 is not a substrate for FumI, a possible technological application of the enzyme will require its combined use together with a fumonisin carboxylesterase such as FumD, so that HFB1, the substrate of FumI, is generated by hydrolytic cleavage of the two tricarballylic acid side chains of FB1. In this respect, the substrate specificity of FumI is quite remarkable, since the direct vicinity of the 2-amino group is identical in FB1 and HFB1 (Fig. 1). Since HFB2 and HFB3 are also deaminated by the enzyme, the 10-hydroxyl and 5-hydroxyl groups of HFB1, which are lacking in HFB2 and HFB3, respectively, are apparently not essential for substrate recognition. The ability of FumI to use several different α-keto acids as amino group acceptors is also noteworthy. Even though pyruvate was the preferred co-substrate when various α-keto acids were tested at the identical molar concentrations in our study, it is tempting to speculate that the relaxed co-substrate specificity of FumI may have biological significance for its function in Sphingopyxis sp. MTA144. Depending on the available α-keto acids, FumI could thus produce alanine from pyruvate, aspartate from oxaloacetate, or glycine from glyoxylate in vivo. The affinity of FumI for its co-substrate (Fig. 3), which is much lower than the affinity for HFB1 (Fig. 8) when judged by the K M values, may also be important for the enzyme in vivo, since the compatible α-keto acids are also intermediates in energy metabolism. In this context, it is interesting to note that the fum gene cluster of Sphingopyxis sp. MTA144 encodes three genes, the tricarballylate proton symport pump FumG, the tricarballylate dehydrogenase FumE, and the citrate utilization protein B FumF, which supposedly allow utilization of the two tricarballylic acid side chains of FB1 and their conversion to citric acid (Heinl et al. 2010). Oxaloacetate and glyoxylate, which both accept the amino group during the FumI-catalyzed reaction, can both be generated from citric acid in the Krebs cycle or the glyoxylate cycle. For a technological application of FumI, this co-substrate requirement together with the comparatively low K M value implies that pyruvate or one of the other suitable α-keto acids will have to be present or will have to be provided together with the enzyme at millimolar concentration to enable efficient and fast HFB1 deamination. However, if the enzyme is to be used for gastrointestinal HFB1 deamination, this co-substrate requirement might be covered to some extent by the presence of α-keto acids in the chymus. It cannot be excluded that other amino group acceptors that we have not yet identified may enable higher reaction rates even at low concentration.
Blackwell et al. have noted before that 2-keto-HFB1 can undergo cyclization between the 5-hydroxyl group and C-2 of 2-keto-HFB1 to form two isomers of a hemiketal, and that this reaction is favored by the presence of methanol in the solvent (Blackwell et al. 1999). We made the same observation and adapted our procedure for purification and NMR characterization of 2-keto-HFB1 to work without methanol. Nevertheless, the NMR characterization showed that our preparation of 2-keto-HFB1 was not pure, and the signals indicative of the hemiketals were still present. As the C-1–C-5 part of 2-keto-HFB1 resembles a simplified ketose structure, an equilibrium between the open-chain and cyclized forms may be the explanation for this behavior since in carbohydrate chemistry, such equilibria are ubiquitous. This may also be one reason that the 2-keto-HFB1 peaks that eluted from a C8 HPLC column were broader than the HFB1 peaks.
Overexpression of FumI in E. coli and the subsequent purification procedure (Hartinger et al. 2010) result in enzyme preparations that apparently are not saturated with the prosthetic group PLP, as can be concluded from the finding that addition of PLP to such an enzyme preparation enhances activity. Aminotransferases were reported to occur as mixtures of their apo- and holo-forms also when isolated from their natural environment (Moss 1976). Increasing the PLP concentrations above 10 μm in the reconstitution experiments of recombinant FumI-HIS showed no further increase of aminotransferase activity, which is consistent with the tight binding of PLP that has been reported for other aminotransferases (Ford et al. 1980; Eliot and Kirsch 2004). If batches of enzyme need to be prepared for technological application in the future, they can be saturated with low concentrations of PLP before their application to ensure that FumI is predominantly available as a holoenzyme.
FumI-HIS displayed HFB1 deamination activity in all buffers we tested, and these covered a wide range of salt concentrations (Fig. 4). This indicates that the proposed application of FumI is unlikely to be limited by buffer salt requirements. The lack of enzyme activity at acidic pH (Fig. 5a) rules out that FumI is already active in the stomach when provided as an animal feed supplement. However, FumI can be expected to function in sections of the upper intestine where the pH is close to neutral or even slightly higher. The temperature optimum of 35°C and good activity at 40°C found for FumI-HIS (Fig. 5b) imply that the actual body temperature of animals, which e.g. was reported to be 38.8°C for piglets (Ingram and Legge 1970), should be well suited for high enzyme activity. Based on our determination of pH stability (Fig. 7a), it will be necessary to provide FumI in a formulation that enables shuttling through the stomach without inactivation at low pH. Simultaneously, the enzyme could thus be protected from proteolytic degradation. Technologies for microencapsulation and targeted release in the intestine have previously been developed (Champagne and Fustier 2007).
We were surprised to see a reduction of HFB1 deamination activity after preincubation of FumI-HIS at 7°C, because we had observed that a preparation of enzyme that was stored in the fridge apparently retained full activity over several weeks. Maybe handling of the enzyme at low concentration contributed to the observed loss of activity. Likewise, the maximal specific reaction rate measured in the experiment shown in Fig. 8, using 10 ng/ml FumI-HIS, was lower than in the experiment shown in Fig. 3, when 94 ng/ml FumI-HIS was used.
The K M value we determined for HFB1 deamination, 1.1 μM, corresponds to 0.794 mg/kg FB1. Since the fumonisin concentrations frequently found in maize (Binder et al. 2007; Gonzalez Pereyra et al. 2008; Gong et al. 2009; Monbaliu et al. 2010) and known to have toxic effects on animals (Ross et al. 1991; Taranu et al. 2005) are higher, FumI should have sufficient affinity for the substrate HFB1 to enable detoxification in the range of fumonisin concentrations that are relevant in agriculture. The decrease of FumI activity observed at HFB1 concentrations higher than 10 μM, which is equivalent to 7.22 mg/l FB1, can be interpreted as substrate inhibition. Since the guidance value for the maximum acceptable concentration of FB1 and FB2 in e.g. pig feed is 5 mg/kg according to the European Commission Recommendation 2006/576/EC, this observed inhibition of activity at higher HFB1 concentrations may not be relevant from a technological point of view. In case enzymatic detoxification by a combination of FumD and FumI of a highly contaminated commodity is required, substrate inhibition of FumI may be avoided by adapting FumD activity to that of FumI, so that the HFB1 is continuously formed in sub-inhibitory concentrations.
Feed enzymes may be useful for the degradation of antinutritive substances that are naturally contained in agricultural commodities, or to enhance the nutritional value of feed. The use of phytase as a feed enzyme to release phosphate from phytate that is naturally contained in plant material is well established (Lei and Porres 2003; Rao et al. 2009). Based on the enzyme characteristics reported here, FumI may also be suitable for future applications as a feed enzyme to enhance the salubriousness of maize that is naturally contaminated with fumonisins, if the specific properties and requirements of FumI are considered in this application.
References
Abel S, Gelderblom WCA (1998) Oxidative damage and fumonisin B1-induced toxicity in primary rat hepatocytes and rat liver in vivo. Toxicology 131:121–131
Bezuidenhout SC, Gelderblom WCA, Gorst-Allman CP, Horak RM, Marasas WFO, Spiteller G, Vleggaar R (1988) Structure elucidation of the fumonisins, mycotoxins from Fusarium moniliforme. J Chem Soc, Chem Commun 743–745
Binder EM, Tan LM, Chin LJ, Handl J, Richard J (2007) Worldwide occurrence of mycotoxins in commodities, feeds and feed ingredients. Anim Feed Sci Technol 137:265–282
Blackwell BA, Gilliam JT, Savard ME, Miller JD, Duvick JP (1999) Oxidative deamination of hydrolyzed fumonisin B1 (AP1) by cultures of Exophiala spinifera. Nat Toxins 7:31–38
Champagne CP, Fustier P (2007) Microencapsulation for the improved delivery of bioactive compounds into foods. Curr Opin Biotechnol 18:184–190
Chen J, Mirocha CJ, Xie W, Hogge L, Olson D (1992) Production of the mycotoxin fumonisin B1 by Alternaria alternata f. sp. lycopersici. Appl Environ Microbiol 58:3928–3931
Duvick JP, Maddox JR, Gilliam, JT (2003) Compositions and methods for fumonisin detoxification. US Patent 6538177, Pioneer Hi-Bred International
Eliot AC, Kirsch JF (2004) Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem 73:383–415
Fernandez-Surumay G, Osweiler GD, Yaeger MJ, Hauck CC, Hendrich S, Murphy PA (2004) Glucose reaction with fumonisin B1 partially reduces its toxicity in swine. J Agric Food Chem 52:7732–7739
Flynn TJ, Stack ME, Troy AL, Chirtel SJ (1997) Assessment of the embryotoxic potential of the total hydrolysis product of fumonisin B1 using cultured organogenesis-staged rat embryos. Food Chem Toxicol 35:1135–1141
Ford GC, Eichele G, Jansonius JN (1980) Three-dimensional structure of a pyridoxal-phosphate-dependent enzyme, mitochondrial aspartate aminotransferase. Proc Natl Acad Sci USA 77:2559–2563
Frisvad JC, Smedsgaard J, Samson RA, Larsen TO, Thrane U (2007) Fumonisin B2 production by Aspergillus niger. J Agric Food Chem 55:9727–9732
Gelderblom WCA, Abel S, Smuts CM, Marnewick J, Marasas WF, Lemmer ER, Ramljak D (2001) Fumonisin-induced hepatocarcinogenesis: mechanisms related to cancer initiation and promotion. Environ Health Perspect 109(Suppl 2):291–300
Gelderblom WCA, Cawood ME, Snyman SD, Vleggaar R, Marasas WF (1993) Structure-activity relationships of fumonisins in short-term carcinogenesis and cytotoxicity assays. Food Chem Toxicol 31:407–414
Gelderblom WCA, Jaskiewicz K, Marasas WF, Thiel PG, Horak RM, Vleggaar R, Kriek NP (1988) Fumonisins-novel mycotoxins with cancer-promoting activity produced by Fusarium moniliforme. Appl Environ Microbiol 54:1806–1811
Gelderblom WCA, Kriek NP, Marasas WF, Thiel PG (1991) Toxicity and carcinogenicity of the Fusarium moniliforme metabolite, fumonisin B1, in rats. Carcinogenesis 12:1247–1251
Gelineau-van Waes J, Starr L, Maddox J, Aleman F, Voss KA, Wilberding J, Riley RT (2005) Maternal fumonisin exposure and risk for neural tube defects: mechanisms in an in vivo mouse model. Birth Defects Res A Clin Mol Teratol 73:487–497
Gong HZ, Ji R, Li YX, Zhang HY, Li B, Zhao Y, Sun L, Yu F, Yang J (2009) Occurrence of fumonisin B1 in corn from the main corn-producing areas of China. Mycopathologia 167:31–36
Gonzalez Pereyra ML, Pereyra CM, Ramirez ML, Rosa CA, Dalcero AM, Cavaglieri LR (2008) Determination of mycobiota and mycotoxins in pig feed in central Argentina. Lett Appl Microbiol 46:555–561
Harrison LR, Colvin BM, Greene JT, Newman LE, Cole JR (1990) Pulmonary edema and hydrothorax in swine produced by fumonisin B1, a toxic metabolite of Fusarium moniliforme. J Vet Diagn Invest 2:217–221
Hartinger D, Heinl S, Schwartz HE, Grabherr R, Schatzmayr G, Haltrich D, Moll WD (2010) Enhancement of solubility in Escherichia coli and purification of an aminotransferase from Sphingopyxis sp. MTA144 for deamination of hydrolyzed fumonisin B1. Microb Cell Fact 9:62
Haschek WM, Gumprecht LA, Smith G, Tumbleson ME, Constable PD (2001) Fumonisin toxicosis in swine: an overview of porcine pulmonary edema and current perspectives. Environ Health Perspect 109(Suppl 2):251–257
Heinl S, Hartinger D, Thamhesl M, Schatzmayr G, Moll WD, Grabherr R (2011) An aminotransferase from bacterium ATCC 55552 deaminates hydrolyzed fumonisin B1. Biodegradation 22:25–30
Heinl S, Hartinger D, Thamhesl M, Vekiru E, Krska R, Schatzmayr G, Moll WD, Grabherr R (2010) Degradation of fumonisin B1 by the consecutive action of two bacterial enzymes. J Biotechnol 145:120–129
Hendrich S, Miller KA, Wilson TM, Murphy PA (1993) Toxicity of Fusarium proliferatum fermented nixtamalized corn-based diets feed to rats: effect of nutritional status. J Agric Food Chem 41:1649–1654
Howard PC, Couch LH, Patton RE, Eppley RM, Doerge DR, Churchwell MI, Marques MM, Okerberg CV (2002) Comparison of the toxicity of several fumonisin derivatives in a 28-day feeding study with female B6C3F1 mice. Toxicol Appl Pharmacol 185:153–165
Humpf HU, Schmelz EM, Meredith FI, Vesper H, Vales TR, Wang E, Menaldino DS, Liotta DC, Merrill AH (1998) Acylation of naturally occurring and synthetic 1-deoxysphinganines by ceramide synthase. Formation of N-palmitoyl-aminopentol produces a toxic metabolite of hydrolyzed fumonisin, AP1, and a new category of ceramide synthase inhibitor. J Biol Chem 273:19060–19064
Hwang BY, Cho BK, Yun H, Koteshwar K, Kim BG (2005) Revisit of aminotransferase in the genomic era and its application to biocatalysis. J Mol Catal B Enzym 37:47–55
Ingram DL, Legge KF (1970) Variations in deep body temperature in the young unrestrained pig over the 24 h period. J Physiol 210:989–998
Karlovsky P (1999) Biological detoxification of fungal toxins and its use in plant breeding, feed and food production. Nat Toxins 7:1–23
Kellerman TS, Marasas WF, Thiel PG, Gelderblom WCA, Cawood M, Coetzer JA (1990) Leukoencephalomalacia in two horses induced by oral dosing of fumonisin B1. Onderstepoort J Vet Res 57:269–275
Lei XG, Porres JM (2003) Phytase enzymology, applications, and biotechnology. Biotechnol Lett 25:1787–1794
Lu Z, Dantzer WR, Hopmans EC, Prisk V, Cunnick JE, Murphy PA, Hendrich S (1997) Reaction with fructose detoxifies fumonisin B1 while stimulating liver-associated natural killer cell activity in rats. J Agric Food Chem 45:803–809
Marasas WF (2001) Discovery and occurrence of the fumonisins: a historical perspective. Environ Health Perspect 109(Suppl 2):239–243
Marasas WF, Kellerman TS, Gelderblom WCA, Coetzer JA, Thiel PG, van der Lugt JJ (1988) Leukoencephalomalacia in a horse induced by fumonisin B1 isolated from Fusarium moniliforme. Onderstepoort J Vet Res 55:197–203
Merrill AH, Sullards MC, Wang E, Voss KA, Riley RT (2001) Sphingolipid metabolism: roles in signal transduction and disruption by fumonisins. Environ Health Perspect 109(Suppl 2):283–289
Mogensen JM, Moller KA, von Freiesleben P, Labuda R, Varga E, Sulyok M, Kubatova A, Thrane U, Andersen B, Nielsen KF (2011) Production of fumonisins B2 and B4 in Tolypocladium species. J Ind Microbiol Biotechnol. doi:10.1007/s10295-010-0916-1 (in press)
Monbaliu S, Van Poucke C, Detavernier C, Dumoulin F, Van De Velde M, Schoeters E, Van Dyck S, Averkieva O, Van Peteghem C, De Saeger S (2010) Occurrence of mycotoxins in feed as analyzed by a multi-mycotoxin LC-MS/MS method. J Agric Food Chem 58:66–71
Moss DW (1976) Reactivation of the apoenzyme of aspartate aminotransferase in serum. Clin Chim Acta 67:169–174
Needham DM (1930) A quantitative study of succinic acid in muscle: glutamic and aspartic acids as precursors. Biochem J 24:208–227
Norred WP, Riley RT, Meredith FI, Poling SM, Plattner RD (2001) Instability of N-acetylated fumonisin B1 (FA1) and the impact on inhibition of ceramide synthase in rat liver slices. Food Chem Toxicol 39:1071–1078
Norred WP, Plattner RD, Dombrink-Kurtzman MA, Meredith FI, Riley RT (1997) Mycotoxin-induced elevation of free sphingoid bases in precision-cut rat liver slices: specificity of the response and structure-activity relationships. Toxicol Appl Pharmacol 147:63–70
Rao DE, Rao KV, Reddy TP, Reddy VD (2009) Molecular characterization, physicochemical properties, known and potential applications of phytases: an overview. Crit Rev Biotechnol 29:182–198
Rheeder JP, Marasas WF, Vismer HF (2002) Production of fumonisin analogs by Fusarium species. Appl Environ Microbiol 68:2101–2105
Riley RT, Enongene E, Voss KA, Norred WP, Meredith FI, Sharma RP, Spitsbergen J, Williams DE, Carlson DB, Merrill AH (2001) Sphingolipid perturbations as mechanisms for fumonisin carcinogenesis. Environ Health Perspect 109(Suppl 2):301–308
Ross PF, Rice LG, Plattner RD, Osweiler GD, Wilson TM, Owens DL, Nelson HA, Richard JL (1991) Concentrations of fumonisin B1 in feeds associated with animal health problems. Mycopathologia 114:129–135
Sadler TW, Merrill AH, Stevens VL, Sullards MC, Wang E, Wang P (2002) Prevention of fumonisin B1-induced neural tube defects by folic acid. Teratology 66:169–176
Schmelz EM, Dombrink-Kurtzman MA, Roberts PC, Kozutsumi Y, Kawasaki T, Merrill AH (1998) Induction of apoptosis by fumonisin B1 in HT29 cells is mediated by the accumulation of endogenous free sphingoid bases. Toxicol Appl Pharmacol 148:252–260
Seefelder W, Humpf H-U, Schwerdt G, Freudinger R, Gekle M (2003) Induction of apoptosis in cultured human proximal tubule cells by fumonisins and fumonisin metabolites. Toxicol Appl Pharmacol 192:146–153
Seiferlein M, Humpf HU, Voss KA, Sullards MC, Allegood JC, Wang E, Merrill AH (2007) Hydrolyzed fumonisins HFB1 and HFB2 are acylated in vitro and in vivo by ceramide synthase to form cytotoxic N-acyl-metabolites. Mol Nutr Food Res 51:1120–1130
Taranu I, Marin DE, Bouhet S, Pascale F, Bailly JD, Miller JD, Pinton P, Oswald IP (2005) Mycotoxin fumonisin B1 alters the cytokine profile and decreases the vaccinal antibody titer in pigs. Toxicol Sci 84:301–307
Taylor PP, Pantaleone DP, Senkpeil RF, Fotheringham IG (1998) Novel biosynthetic approaches to the production of unnatural amino acids using transaminases. Trends Biotechnol 16:412–418
Teorell T, Stenhagen E (1938) Ein Universalpuffer für den pH-Bereich 2.0 bis 12.0. Biochem Z 299:416–419
van der Westhuizen L, Shephard GS, Snyman SD, Abel S, Swanevelder S, Gelderblom WCA (1998) Inhibition of sphingolipid biosynthesis in rat primary hepatocyte cultures by fumonisin B1 and other structurally related compounds. Food Chem Toxicol 36:497–503
Voss KA, Bacon CW, Meredith FI, Norred WP (1996) Comparative subchronic toxicity studies of nixtamalized and water-extracted Fusarium moniliforme culture material. Food Chem Toxicol 34:623–632
Voss KA, Riley RT, Snook ME, Waes JG (2009) Reproductive and sphingolipid metabolic effects of fumonisin B1 and its alkaline hydrolysis product in LM/Bc mice: hydrolyzed fumonisin B1 did not cause neural tube defects. Toxicol Sci 112:459–467
Voss KA, Smith GW, Haschek WM (2007) Fumonisins: toxicokinetics, mechanism of action and toxicity. Anim Feed Sci Technol 137:299–325
Wattenberg EV, Badria FA, Shier WT (1996) Activation of mitogen-activated protein kinase by the carcinogenic mycotoxin fumonisin B1. Biochem Biophys Res Commun 227:622–627
Wang E, Norred WP, Bacon CW, Riley RT, Merrill AH (1991) Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J Biol Chem 266:14486–14490
Wild CP, Gong YY (2010) Mycotoxins and human disease: a largely ignored global health issue. Carcinogenesis 31:71–82
Zhu D, Hua L (2009) Biocatalytic asymmetric amination of carbonyl functional groups—a synthetic biology approach to organic chemistry. Biotechnol J 4:1420–1431
Acknowledgments
We would like to thank Markus Kainz and Elisabeth Pichler from Quantas Analytics GmbH (Tulln, Austria) for collaboration on LC–MS analysis, and Patricia Fajtl for support with making preparations of FumI-HIS. This work was supported in part by the Austrian Research Promotion Agency FFG and the Christian Doppler Research Association.
Conflict of interest
The authors declare no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hartinger, D., Schwartz, H., Hametner, C. et al. Enzyme characteristics of aminotransferase FumI of Sphingopyxis sp. MTA144 for deamination of hydrolyzed fumonisin B1 . Appl Microbiol Biotechnol 91, 757–768 (2011). https://doi.org/10.1007/s00253-011-3248-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3248-9