
Abstract
A bifunctional hydratase/alcohol dehydrogenase was isolated from the cyclohexanol degrading bacterium Alicycliphilus denitrificans DSMZ 14773. The enzyme catalyzes the addition of water to α,β-unsaturated carbonyl compounds and the subsequent alcohol oxidation. The purified enzyme showed three subunits in SDS gel, and the gene sequence revealed that this enzyme belongs to the molybdopterin binding oxidoreductase family containing molybdopterins, FAD, and iron-sulfur clusters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Simple alicyclic compounds like cyclohexanol and cyclohexanone can be degraded by some microbes via both aerobic and anaerobic pathways (Trudgill 1984; Dangel et al. 1988, 1989). The aerobic degradation of acyclic compounds has been extensively studied (Norris and Trudgill 1971; Donoghue et al. 1976). In this pathway, monooxygenases are required to cleave the ring by Baeyer–Villiger oxidation where an oxygen atom originating from molecular oxygen is inserted. On the other hand, the anaerobic degradation of alicyclic compounds is less well studied, and only limited information is available (Bakker 1977; Williams and Evans 1975; Balba and Evans 1980). So far, three hypotheses for the anaerobic metabolism of cyclohexanone have been proposed. The first hypothesis is the direct ring fission of cyclohexanone yielding caproate (Bakker 1977). The second hypothesis is the hydroxylation of cyclohexanone forming 1,2-cyclohexanediol followed by subsequent oxidation to 2-hydroxycyclohexanone. The oxidation product is further cleaved and oxidized to adipate (Williams and Evans 1975; Balba and Evans 1980). In the third hypothesis, cyclohexanone is oxidized to 2-cyclohexenone which is further hydrated to 3-hydroxycyclohexanone. The hydration product is then oxidized to 1,3-cyclohexanedione which is subsequently cleaved to 5-oxohexanoic acid (Dangel et al. 1988, 1989). Direct experimental evidence for the formation of 2-cyclohexenone and 1,3-cyclohexanedione was obtained in the anaerobic growth of the denitrifying bacterium Alicycliphilus denitrificans DSMZ 14773 with cyclohexanol (Dangel et al. 1988, 1989; Mechichi et al. 2003). This strain was isolated from a municipal sewage plant and is able to metabolize aromatic and alicyclic compounds under aerobic or anaerobic conditions. Phylogenetic analysis showed that the strain is a member of the family Comamonadaceae in the β-subclass of Proteobacteria (Mechichi et al. 2003). In the proposed degradation pathway of cyclohexanol (Fig. 1), the alcohol 1 is first oxidized to the corresponding ketone 2, followed by the introduction of a C=C double bond at the α–β position forming the vinylketone 3. Michael addition of water to 3 leads to the β-hydroxy ketone 4. The degradation proceeds with oxidation of 4 yielding 1,3-dione 5 and subsequent hydrolytic cleavage of 5. In this proposed pathway, a putative hydro-lyase, 2-cyclohexenone hydratase, and a putative alcohol dehydrogenase, 3-hydroxycyclohexanone dehydrogenase (Hdh), are involved in the hydration of 3 and oxidation of 4. However, the hydration product 4 was not directly observed; only indirect evidence for this highly unusual Michael addition of water was provided. Instead, the conversion of 3 to 5 could occur via an oxidative addition of water. Indeed, some hydroxylases can introduce a hydroxyl group using water as the ultimate source of oxygen, for example, xanthine dehydrogenase (Xdh), quinoline 2-oxidoreductase (Qor), and nicotinic acid dehydrogenase (NDH; Hille 2005; Schräder et al. 2002; Bonin et al. 2004). Therefore, it is of interest to establish whether the conversion of 3 to 5 is catalyzed by two enzymes (a hydro-lyase and a dehydrogenase) or one enzyme (a hydroxylase).

Anaerobic degradation of cyclohexanol by A. denitrificans DSMZ 14773. Compounds: 1 cyclohexanol, 2 cyclohexanone, 3 2-cyclohexenone, 4 3-hydroxycyclohexanone, 5 1,3-cyclohexanedione, 6 5-oxohexanoic acid. Enzymes: A cyclohexanol dehydrogenase; B cyclohexanone dehydrogenase; C 2-cyclohexenone hydratase; D 3-hydroxycyclohexanone dehydrogenase, methylene blue is used as artificial acceptor; E 1,3-cyclohexanedione hydrolase. The figure is based on the scheme from Dangel et al. (1989)
Here, we report on the isolation, characterization, and cloning of a bifunctional hydratase/alcohol dehydrogenase from A. denitrificans DSMZ 14773. The enzyme is able to catalyze both the addition of water to the C=C bond of α,β-unsaturated carbonyl compounds and the oxidation of the resulting hydration product.
Materials and methods
Chemicals
Methylene blue, 2,6-dichlorophenol indophenol (DCPIP), and nitro blue tetrazolium chloride (NBT) were purchased from AcrosOrganics. Acrylic acid, 2-cyclohexenone, 3-buten-2-one, 3-penten-2-one, trans-2-pentenal, trans-2-hexenal, trans-2-octenal, 3-hexen-2-one, 3-octen-2-one, cinnamaldehyde, benzalacetone, oxine, 3-methoxy-4-hydroxymandelic acid, sodium molybdate, and sodium chlorate were obtained from Sigma-Aldrich. Mercury (AR grade), perchloric acid, and sulfuric acid were obtained from Merck.
Microorganism and culture conditions
A. denitrificans DSMZ 14773 was purchased from DSMZ (Germany). The medium used for cultivation contained (per liter distilled water) Na2HPO4·2H2O 3.5 g, KH2PO4 1.0 g, (NH4)2SO4 0.5 g, MgCl2·6H2O 0.1 g, Ca(NO3)2·4H2O 0.05 g, and 1.0 mL trace element solution SL-4. The trace element solution SL-4 contained (per liter distilled water) EDTA 0.5 g, FeSO4·7H2O 0.2 g, and trace element solution SL-6 100 mL. The trace element solution SL-6 contained (per liter distilled water) ZnSO4·7H2O 0.10 g, MnCl2·4H2O 0.03 g, H3BO3 0.30 g, CoCl2·6H2O 0.20 g, CuCl2·2H2O 0.01 g, NiCl2·6H2O 0.02 g, and Na2MoO4·2H2O 0.03 g. The final pH was 7.2–7.4. The culture was shaken reciprocally in a 50-mL flask containing 5 mL medium at 28°C.
Analytical methods
The coupled activity assay of 2-cyclohexenone hydratase and 3-hydroxycyclohexanone dehydrogenase was performed at room temperature in a quartz cuvette containing 0.9 mL reaction mixture containing 1 mM substrate and 60 μM methylene blue or DCPIP (accept two electrons) in 100 mM Tris–HCl (pH 7.8). The reaction was initiated by adding 0.1 mL cell extract or enzyme solution. The spectra of the reaction mixture were recorded on a Hewlett-Packard 8452A diode array spectrophotometer at an interval of 2 s and absorbance change at 578 nm (methylene blue, ε = 20.7 cm−1 mmol−1; DCPIP, ε = 16.8 cm−1 mmol−1) was determined (Dangel et al. 1988). One unit is defined as the amount of enzyme that reduces 1 μmol of methylene blue or DCPIP at pH 7.8 and 25°C in 1 min.
The hydration reaction was performed in 10 mL of Tris–HCl buffer (100 mM, pH 7.8) containing 0.1 mmol 2-cyclohexenone (3) and 3.1 U Michael hydratase/alcohol dehydrogenase (MhyADH) activity; 0.5-mL samples were taken from the reaction mixture every 30 min and extracted with 1.0 mL of ethyl acetate for GC and GC–MS analysis.
1H and 13C NMR spectra were recorded with a Bruker Avance 400 (400 and 100 MHz, respectively) or a Varian Unity Inova 300 (300 and 75 MHz, respectively) instrument. Chemical shifts are expressed in parts per million (δ) relative to tetramethylsilane. Coupling constants J are expressed in Hertz. GC analysis of the reactions were performed with a Shimadzu GC 2014 equipped with a CP-Wax 52 CB column (2.0 μm × 50 m × 0.5 mm) using N2 as the carrier gas. Mass spectra were determined with a Shimadzu GC-2010 Gas Chromatograph coupled to a Shimadzu GCMS-QP2010S Gas Chromatographic Mass Spectrometer.
UV–Vis analysis
The UV–visible absorption spectra of MhyADH were recorded using a Hewlett-Packard 8452A diode array spectrophotometer. Spectral analyses were performed for purified enzyme in 20 mM Tris–HCl (pH 7.8). The spectrum of reduced enzyme was obtained after addition of 1 mM 3-hydroxycyclohexanone.
Synthesis of 3-hydroxycyclohexanone
N-hydroxyphthalimide (NHPI) and Co(acac)3-mediated synthesis of 3-hydroxycyclohexanone: A solution of 1,3-cyclohexanediol (500 mg, 4.3 mmol), N-hydroxyphthalimide (49 mg, 10 mol%), and Co(acac)3 (11 mg, 1 mol%) in 5 mL acetonitrile was stirred at 65°C under O2 (balloon; Iwahama et al. 2000). After 24 h, the reaction mixture was loaded with the aid of a little ethanol onto a silica gel column and eluted with ethyl acetate/n-hexane (3/1, v/v). 3-Hydroxycyclohexanone was obtained in 56% yield as a light yellow oil. The spectroscopic data are in accordance with the literature: 1H NMR (400 MHz, CDCl3) δ 1.65–1.83 (m, 2H), 1.96–2.13 (m, 2H), 2.32 (t, 2H, J = 6.6), 2.39 (dd, 1H, J = 7.5, 14.1), 2.64 (dd, 1H, J = 4.0, 14.1), 2.85 (bs, 1H, OH), 4.19 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 20.69, 32.67, 40.95, 50.37, 69.63, 210.61; MS m/z = 114 (M+), 96 (M+–H2O) (Arai et al. 2005).
Native PAGE and activity staining of cyclohexanol dehydrogenase, cyclohexanone dehydrogenase, and 3-hydroxycyclohexanone dehydrogenase (MhyADH)
Native polyacrylamide gel electrophoresis (PAGE) was performed in 8–25% gradient gels (GE Healthcare Life Sciences). Extracts were applied of cells anaerobically grown on cyclohexanol, and a standard protein calibration kit (GE Healthcare Life Sciences) containing albumin (66,000), lactate dehydrogenase (140,000), catalase (232,000), ferritin (440,000), and thyroglobulin (669,000) was used. For activity staining, the gel was cut into slips and soaked anaerobically in 20 mL of 100 mM Tris–HCl (pH 7.8) containing 0.6 mM NAD+ (for cyclohexanol dehydrogenase and cyclohexanone dehydrogenase), 60 μM methylene blue (for 3-hydroxycyclohexanone dehydrogenase), and 0.3 mM nitro blue tetrazolium chloride. The staining reaction was started with cyclohexanol (1 mM; for cyclohexanol dehydrogenase), cyclohexanone (1 mM; for cyclohexanone dehydrogenase), or 2-cyclohexenone (1 mM) or 3-hydroxycyclohexanone (1 mM; for 3-hydroxycyclohexanone dehydrogenase, MhyADH). When bands appeared, the staining mixture was replaced by water. Standard proteins and the cell extract were stained with 0.1% Coomassie brilliant blue R-250, 50% methanol, and 10% glacial acetic acid. After staining, the gel was unstained with 40% methanol and 10% glacial acetic acid to visualize the protein bands.
SDS-PAGE
SDS-PAGE was performed in 4–12% NuPAGE®Novex Bis-Tris gels (Invitrogen). Precision Plus Protein™ standards (10–250 kDa; Bio-Rad) were applied for the estimation of the molecular weight of the proteins. The gel was stained in SimplyBlue™ SafeStain (Invitrogen).
Purification of 3-hydroxycyclohexanone dehydrogenase (MhyADH)
For the preparation of cell extracts, 1.5-g cells (wet weight) were suspended in 10 mL Tris–HCl buffer (100 mM, pH 7.8). Cells were disrupted by passage through a cooled French pressure cell with a pressure difference of 139 MPa. Unbroken cells and debris were removed by centrifugation at 100,000×g for 1 h. The cell-free extract of A. denitrificans was applied to a DEAE Sepharose column (28 mL) previously equilibrated with buffer A (20 mM Tris–HCl, pH 7.8). The elution was performed with buffer B (20 mM Tris–HCl, 1 M NaCl, pH 7.8) with a gradient from 0% to 50% at a flow rate of 5 mL/min. Fractions showing activity were pooled and concentrated using Centricon YM30 centrifugal filter devices (Millipore) in a centrifuge at 3,000×g. After desalting with a PD-10 column, pooled fractions containing 3-hydroxycyclohexanone dehydrogenase (MhyADH) activity were applied to a MonoQ 5/50 GL column (1 mL; GE Healthcare) equilibrated with buffer A. The bound 3-hydroxycyclohexanone dehydrogenase (MhyADH) activity was eluted by a linear gradient from 0% to 50% of buffer B at a flow rate of 0.4 mL/min. Active fractions were pooled and concentrated using Centrion YM30 filter devices. The purified protein was stored at −20°C. The protein content was determined using a Pierce® BCA protein assay kit (Pierce Biotechnology).
Determination of cofactor content
Electrochemical measurements were performed with a digitally controlled PSTAT10 potentiostat using GPES 4.8 software (Eco-chemie, The Netherlands) equipped with a hanging mercury drop working electrode (HMDE 663 VA stand, Metrohm Switzerland), a glassy carbon counter electrode, and a double-junction Ag/AgCl (3 M KCl) reference electrode (Metrohm).
Molybdenum was determined by catalytic-adsorptive stripping voltammetry (AdSV) according to a previously described method (Hagedoorn et al. 2001). A sample containing 4.7 μM 3-hydroxycyclohexanone dehydrogenase was deproteinized by the addition of 10% (w/v) perchloric acid with subsequent centrifugation for 5 min at 9,000 rpm. Aliquots of 5 μL supernatant or 10 μM sodium molybdate standard solution were added to a 20-mL blank solution containing 10 mM H2SO4, 0.5 mM oxine, 0.05 mM 3-methoxy-4-hydroxymandelic acid, and 50 mM NaClO3. The potentials are reported with reference to the Ag/AgCl electrode. For the AdSV, the following conditions were used: pulse height 0.025 V, step potential 0.005 V, deposition potential 0.10 V, deposition time 120 s, stirring speed during deposition 1,000 rpm, and scan range 0.10 to −1.0 V. The mercury drop surface was approximately 0.52 mm2. Prior to measurement, the solution was purged with high-purity argon for 300 s. The molybdenum peak was observed at −0.203 V. Three replicates of the protein sample and three molybdate standard additions were measured.
Tandem MS analysis and protein identification
After SDS-PAGE of the purified 3-hydroxycyclohexanone dehydrogenase (MhyADH) protein sample, protein bands of interest were excised from the gel and subjected to in-gel proteolytic digestion with trypsin (10 mg/L in 25 mM ammonium bicarbonate, pH 8.1) overnight at 37°C. Prior to digestion, cysteines were reduced with 5 mM DTT in 25 mM ammonium bicarbonate, pH 8.0 for 30 min, and subsequently alkylated with 10 mM iodoacetamide in 25 mM ammonium bicarbonate, pH 8.0, in the dark for 45 min. In-gel digests were acidified with 5% formic acid (final concentration) and were analyzed using nano-LC-ESI-QTOF tandem mass spectrometry. Nano-LC was performed on a nanoAcquity UPLC (Waters, Manchester, UK) directly coupled to a Q-Tof Premier (Waters, Manchester, UK). Peptides were delivered to a trap column (100 μm ID × 20 mm Length) packed in-house with ReproSil-Pur C18-AQ, 5 μm particles, 12 nm (Dr. Maisch, GmbH, Ammerbuch, Germany) at a flow rate of 5 μL/min in 100% Solvent A (0.1 M acetic acid in water) for 10 min. After trapping, the trap column was switched in-line with the analytical column (Reprosil C18-iso_AQ, 50 μm ID × 250 mm length, 3 μm particles, pore size 12 nm, packed in-house), and the flow rate was reduced to 150 nL/min. A linear gradient from 0% to 40% solvent B (0.1 M acetic acid in 8/2 v/v acetonitrile/water) at 1% min−1 was used to analytically separate the peptides eluting from the trap column. The column effluent was directly electrosprayed in the ESI source of the mass spectrometer using a nano-ESI emitter (New Objective, Woburn, MA, USA). The mass spectrometer was programmed in a data-dependent analysis mode. Survey scans were acquired in positive ion centroid mode from m/z 400–1,500 at a scan time of 1 s using an interscan time of 0.02 s. Up to three multiply charged precursors were allowed for low-energy collision-induced dissociation at a selection threshold 1,000 counts/s. MS/MS spectra were acquired in continuous mode for 1 s. Fragmentation spectra were interpreted manually using MassLynx 4.1 software (Waters, Manchester, UK). BLAST searches were performed to find sequence similarity with other proteins in the NCBI non-redundant database.
DNA manipulation
The chromosome DNA from A. denitrificans was isolated from cells harvested at early stationary growth phase using a modified protocol of Marmur (1961). The DNA fragments of 3-hydroxycyclohexanone dehydrogenase (MhyADH) were amplified by Taq DNA polymerase (Invitrogen) using the chromosomal DNA of A. denitrificans as the template. Two pairs of degenerate primers with the following sequence were designed: p_forward1, 5′ttyggnccngtngaygaygaygay3′; p_forward2, 5′gtnaayacngtnccngtnaar3′; p_reverse, 5′ccanccytcraartcraacat3′ where N = T, C, A or G; R = A or G; and Y = C or T. For the first pair of primers, p_forward1 was derived from the peptide FGPVDDD, and for the second pair of primers, p_forward2 was derived from the peptide VNTVPVK. The reverse primer for both amplification reactions was derived from the same peptide MFDFEGW. PCRs were performed under denaturing conditions at 94°C for 3 min and then 30 cycles consisting of 94°C for 60 s, 45°C to 55°C for 45 s, and 72°C for 60 s at primer concentrations from 0.4 to 12 μM.
For the amplification of the whole gene cluster of 3-hydroxycyclohexanone dehydrogenase (MhyADH), walking primers p_fw1 5′aatgaacttggctggccgccctg3′, p_fw4 5′gcggatgaaatcggacaacag3′, p_rev1 5′tggcgctgcgttataccgttg3′, and p_rev4 5'cccgaagattctgaaggagtg3′ were used to amplify the gene, and sequencing primers p_fw2 5′cgaaagcgatgccgtcagaac3′, p_fw3 5′gtgtcgaaggcgggattgtcacttc3′, p_fw5 5′acacttcgcgctcgatcttcc3′, p_fw6 5′agttcatcatcggcaccaagc3′, p_rev2 5′gtacctgcaatacggctatgg3′,p_rev3 5′gtgtcgaaggcgggattgtcacgtc3′, p_rev5 5′gtaaatcggacgggcgatgag3′, and p_rev6 5′gccgaacaatgtcccgatgag3′ were used to sequence the amplified DNA. The amplified PCR products were cloned to pCR2.1-TOPO cloning vector (Invitrogen). PCRs were performed by Pfx DNA polymerase (Invitrogen) under denaturing conditions at 94°C for 3 min, two rounds of 30 cycles consisting of 94°C for 30 s, 50°C to 60°C for 30 s, and 68°C for 3 min, and between the two 30-cycle rounds, there was one cycle with annealing at 40°C.
Results
Staining of 3-hydroxycyclohexanone dehydrogenase activity in native gel
According to Dangel et al. (1989), 2-cyclohexanone hydratase and 3-cyclohexanone dehydrogenase are involved in the conversion of 2-cyclohexenone 3 to 1,3-cyclohexanedione 5 (Fig. 1). To isolate 2-cyclohexenone hydratase from the cell extract of A. denitrificans, a fast and easy activity assay that can be measured spectrophotometrically is preferred. However, no direct measurement of the activity of 2-cyclohexenone hydratase has been reported so far, but a coupled activity assay has been described (Dangel et al. 1989). In this coupled assay, the reaction started with the hydration of 2-cyclohexenone 3 followed by the oxidation of the resulting hydroxyl group in 3-hydroxycyclohexanone 4 into an oxo group in 1,3-cyclohexanedione in the presence of electron acceptors such as methylene blue or DCPIP. Because methylene blue and DCPIP accept two electrons in the oxidation step resulting in the color change of the dye, the reaction can be followed spectrophotometrically by recording the decrease of the absorbance. Since we synthesized 4 via selective oxidation of 1,3-cyclohexanediol as described (Iwahama et al. 2000), the activity of 3-hydroxycyclohexanone dehydrogenase could be measured separately, and it was possible to purify this dehydrogenase accordingly. Therefore, it was decided to begin with the purification of 3-hydroxycyclohexanone dehydrogenase and then purify 2-cyclohexenone hydratase using the purified dehydrogenase in the coupled activity assay.
The existence of 3-hydroxycyclohexanone dehydrogenase could be determined by activity staining of the native protein gel. After native gel electrophoresis of the cell extract of A. denitrificans, the gel was soaked in the staining mixture containing methylene blue, nitro blue tetrazolium chloride, 3-hydroxycyclohexanone 4, or 2-cyclohexenone 3. The activity staining surprisingly showed that the same band was stained by 3 and 4 (Fig. 2). This result suggests that the hydration of 3 and oxidation of 4 could be catalyzed by one enzyme.

Activity staining of the native gel. Lanes 1–4, cell extract stained by different substrates, methylene blue and NBT (lane 1: 2-cyclohexenone, lane 2: 3-hydroxycyclohexanone, lane 3: cyclohexanol, lane 4: cyclohexanone); lanes 5 and 6, cell extract stained by Coomassie brilliant blue R-250; lane 7, protein marker (albumin (66,000), lactate dehydrogenase (140,000), catalase (232,000), ferritin (440,000), thyroglobulin (669,000))
Purification of 3-hydroxycyclohexanone dehydrogenase
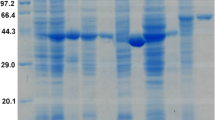
To prove the hypothesis that the hydration of 2-cyclohexenone and dehydrogenation of 3-hydroxycyclohexanone were catalyzed by the same enzyme, 3-hydroxycyclohexanone dehydrogenase was purified from cell-free extract of A. denitrificans. The cell-free extract was applied onto a DEAE Sepharose anion exchange column, and 3-hydroxycyclohexanone dehydrogenase started to elute at ~0.23 M NaCl. The active fractions were pooled, desalted, and loaded onto a MonoQ anion exchange column. After two steps of purification, the enzyme was purified 43-fold (Table 1). Three dominant protein bands could be clearly observed in the SDS denaturing gel (Fig. 3). The protein band showing 3-hydroxycyclohexanone dehydrogenase activity in the native gel was excised and loaded onto the denaturing gel, and also three protein bands were visualized albeit a lag of ~20 kDa of the bands compared to those of the purified protein, which might be caused by the insufficient denaturing of the protein in the native gel pieces. The active fractions showed activity toward both 2-cyclohexenone and 3-hydroxycyclohexanone indicating that this enzyme has both hydratase and dehydrogenase activity, and it is thus a bifunctional hydratase/alcohol dehydrogenase. Since the hydratase acts according to the Michael regiochemistry, we abbreviate the enzyme MhyADH for Michael hydratase alcohol dehydrogenase.

Partial purification of 3-hydroxycyclohexanone dehydrogenase from A. denitrificans DSMZ 14773. Lane 1, protein marker; lane 2, active fractions from the MonoQ column; lane 3, active fractions from the DEAE Sepharose column; lane 4, cell free extract; lane 5, active protein band from cell free extract showing 3-hydroxycyclohexanone dehydrogenase activity in the native gel
Properties of MhyADH
MhyADH was partially purified by two steps of chromatographic purification. The purified enzyme showed three protein bands in the SDS-PAGE gel (Fig. 3). The molecular weight of the three subunits was estimated to be 20, 30, and 90 kDa, respectively. This observation is consistent with that of Xdh, Qor, NDH, and ketone dehydrogenase (KDH; Hille 2005; Schräder et al. 2002; Bonin et al. 2004; Schenk et al. 1998). These three subunits were assigned to MhyADHS, MhyADHM, and MhyADHL, respectively. For nicotinic acid dehydrogenase, it was reported to be a dimer of heterotrimers, but some subunits could be lost during purification (Bonin et al. 2004). We observed a similar phenomenon for MhyADH in native protein gel electrophoresis (Fig. 2) and size-exclusion chromatography (data not shown). The apparent molecular weight is estimated to be ~170 kDa suggesting a heterotrimer (MhyADHS, MhyADHM, MhyADHL) plus an additional small subunit or medium subunit.
The purified MhyADH showed absorption maxima in the UV region at 271 nm, in the visible region at 440 nm, and broad shoulders around 350 and 550 nm (Fig. 4), corresponding to the spectra of other molybdoflavin proteins including ketone dehydrogenase and nicotinic acid dehydrogenase (Schräder et al. 2002; Baitsch et al. 2001). Cofactor content was determined by coupled plasma-mass spectrometry (ICP-MS) and catalytic AdSV. Metal analysis by ICP-MS showed that the purified enzyme contained molybdenum, iron, and zinc in a ratio of 1:4.05:0.8. A similar ratio of 1: 4.6: 1.1 was reported in the study of nicotinic acid dehydrogenase (Schräder et al. 2002). Molybdenum was determined by AdSV, and the result showed that one monomer contains 1.19 ± 0.15 Mo/protein molecule. The optimum pH and thermal stability of MhyADH were determined. MhyADH has a bell-shaped pH profile with 80% activity between pH 6.5 and 8.0. The enzyme showed the highest activity at pH 7.5. In the potassium phosphate buffer at pH 7.5, MhyADH showed a half-life of ~120 min at 50°C but is quickly deactivated after 15 min at 70°C.

UV–visible spectrum of purified 3-hydroxycyclohexanone dehydrogenase (MhyADH). The spectrum of the oxidized enzyme (2.4 μM) was recorded in 20 mM Tris–HCl, pH 7.8. The inset shows the spectra of oxidized (solid line) and reduced enzyme (dotted line). The reduction of the enzyme was performed by addition of 1 mM 3-hydroxycyclohexanone
A substrate screening was performed to explore the substrate specificity of MhyADH As shown in Table 2, in addition to the cyclic substrate 2-cyclohexenone, the enzyme also accepts unsaturated aliphatic ketones and aldehydes. For the substrates of the same carbon chain length, MhyADH has higher activity toward aldehydes than ketones. The activity of MhyADH toward trans-2-pentenal was 1.6-fold of that toward 3-penten-2-one. The enzyme readily accepted trans-2-hexenal, trans-2-octenal, and cinnamaldehyde but had no activity toward 3-hexen-2-one, 3-octen-2-one, and benzalacetone. In addition, no activity toward unsaturated acids such as acrylic acid and 2-furoic acid was observed.
To probe whether the enzyme catalyzes the two steps really independently and whether the intermediate 3-hydroxycyclohexanone could be isolated, the MhyADH catalyzed hydration of 2-cyclohexenone was performed in the absence of the reducing equivalent, to prevent consecutive oxidation. After 1.5 h, 15% of 2-cyclohexenone was converted to 3-hydroxycyclohexanone as calculated based on the consumption of the substrate and appearance of the product. No further conversion was observed (Fig. S1). Follow-up work should reveal if this limited yield is due to equilibrium restrictions. The V max of oxidation of racemic 3-hydroxycyclohexanone is 36.6 ± 2.2 nmol/min/mg protein. The V max of 2-cyclohexenone is 32.9 ± 0.8 nmol/min/mg protein as determined by the coupled activity assay with methylene blue suggesting that the hydration reaction is the rate-limiting step. The concentration of substrate also affected the activity. For both 2-cyclohexenone and 3-hydroxycyclohexanone, high substrate concentrations led to a decrease of the reaction rate (Fig. S2). The formation of 3-hydroxycyclohexanone indicates that MhyADH first catalyzes the addition of water to the conjugated C=C bond and then catalyzes the oxidation of the hydration product.
Primary structure of putative 3-hydroxycyclohexanone dehydrogenase
To obtain information about the primary amino acid sequence of the three subunits of MhyADH, the three dominant protein bands seen on the SDS denaturing gel (Fig. 3) were excised from the gel and subjected to in-gel tryptic digestion. The peptides were extracted out of the gel pieces and analyzed using reversed-phase nano-HPLC and tandem mass spectrometry. After manual interpretation of the acquired peptide fragmentation spectra, several peptides were sequenced with high coverage. Among these fragments, nine peptides were found for the MhyADHL, five for MhyADHM, and three for MhyADHS (Table S1). Using BLAST searches, the peptides of MhyADHL showed high similarity with the sequence of the large subunit of molybdopterin binding oxidoreductase from Roseobacter litoralis Och 149 (α Proteobaceria; gi:163733669). The MhyADHM-derived peptides showed high similarity with the sequence of carbon monoxide dehydrogenase from R. litoralis Och 149 (gi:163733671) and to a lesser extent to the medium subunit of ketone dehydrogenase from Arthrobacter nicotinovorans (Actinobacteria; gi: 14495295). The MhyADHS-derived peptides showed a high similarity with the sequence of the [2Fe-2S]-binding protein from R. litoralis Och 149 (gi:163733670). The KDH from A. nicotinovorans is involved in nicotine catabolism and catalyzes the hydroxylation at the pyridine ring of (3-(6-hydroxypyridyl)-γ-N-methyl-aminopropyl-) ketone (Schenk et al. 1998; Baitsch et al. 2001). It has been proven that ketone dehydrogenase has three subunits. The small subunit (KDH-B) contains two iron-sulfur clusters, the medium subunit (KDH-A) contains a FAD cofactor, and the large subunit (KDH-C) has a molybdopterin cofactor (MoCo; Baitsch et al. 2001). The MS result of MhyADH indicated that this enzyme might be a KDH-like enzyme with three functional domains.
Amplification of the gene fragment of MhyADH
Based on the peptide sequences deduced from LC–MS, two pairs of degenerate primers were designed. A fragment of 700-bp DNA fragment was amplified in the reaction using the first pair of primers. Sequence analysis revealed that this 700-bp DNA fragment had 59% identity with the large subunit of molybdopterin binding oxidoreductase from R. litoralis Och 149 (gi:16373371). The walking primers were designed based the 700-bp DNA to amplify the rest part of the gene of MhyADH. After two rounds of primer walking in both upstream and downstream of the 700-bp DNA fragment, the amplified DNA was sequenced and assembled to a 7-kb DNA fragment containing the full structural genes of MhyADH.
The assembled DNA includes the complete ORFs of three MhyADH subunits, a possible promoter region, and part of the gene of a flavin-containing dehydrogenase (Fig. 5). The first ORF of MhyADH contains a 879-bp fragment that codes for a polypeptide of 292 aa corresponding to the medium subunit (MhyADHM; GenBank accession number: BankIt1385919 HQ201715). The second ORF is located 33 bp downstream of MhyADHM and contains 498 bp coding for the small subunit (MhyADHS) of 165 aa (GenBank accession number: BankIt1385935 HQ201716). The third ORF uses the first two bases of the stop codon TGA of MhyADHS to form the start codon ATG. The shifted frame contains 2475 bp and translates into a polypeptide of 824 aa that corresponds to the large subunit (MhyADHL; GenBank accession number: BankIt1385935 HQ201716). An incomplete ORF was found 750 bp downstream of MhyADHL. The 1,200-bp DNA sequence shares 41% identity with the flavin-containing domain of the formate reductase/succinate dehydrogenase from Frankia sp. EAN1pec (Actinobacteria; gi:58616423) and 3-ketosteroid dehydrogenase (KSTD) from Stigmatella aurantiaca DW4/3-1 (δ Proteobacteria; gi:115375375; Rabus et al. 2005). By comparing the reactions catalyzed by KSTD, we assume that this ORF probably codes for cyclohexanone dehydrogenase.

The gene structure of MhyADH. The medium subunit, small subunit, and large subunit are indicated as M, S, and L. The putative binding sites for [2Fe-2S] clusters and molybdopterins are indicated as blocks
Discussion
In the previous studies of the anaerobic metabolism of cyclohexanol by A. denitrificans, it was proposed that a putative hydratase and a dehydrogenase were involved in the hydration of 2-cyclohexenone and subsequent oxidation of the resulting hydration product 3-hydroxycyclohexanone to 1,3-cyclohexanedione (Dangel et al. 1988, 1989). In this study, we discovered that these two steps of reactions were catalyzed by one enzyme. In the absence of an electron acceptor, the hydration product could be identified. In the presence of electron acceptors, the hydration product was further oxidized to the corresponding ketone. MhyADH showed this catalytic activity for a range of C=C conjugated aldehydes and ketones.
Metal analysis showed that the purified enzyme contains stoichiometric amounts of molybdenum and zinc and four iron atoms per protein molecule. Similar results were reported in the study of molybdenum-containing oxidoreductases (Schräder et al. 2002; Lehmann et al. 1994; Gremer and Meyer 1996; Bonam and Ludden 1987). In this group of enzymes, the molybdenum center is the catalytic site, and irons are bound in iron-sulfur clusters which transfer electrons (Hille 1996). In addition, nickel and copper are also found in these enzymes (Lehmann et al. 1994; Gremer and Meyer 1996; Bonam and Ludden 1987). However, the function of these metals is not yet clearly known.
A BLAST search revealed that the sequence of MhyADH is homologous to carbon monoxide dehydrogenase (CO-DH), Xdh, and other molybdenum-containing oxidoreductases. These enzymes are heterodimers or heterotrimers containing iron-sulfur clusters, FAD and MoCo (Hille 2005). The sequence of the small subunit of MhyADH shows a remarkable sequence identity with the (2Fe-2S)-binding protein from R. litoralis Och 149 (gi:163733670; 66%), with the small subunit of aldehyde oxidase from Thermomicrobium roseum DSM 5159 (Chloroflexi; gi: 221632602), and the small subunit of CO-DH from T. roseum DSM 5159 (gi: 221636182), which all contain [2Fe-2S] clusters (Wu et al. 2009). The motifs for binding of [2Fe-2S] clusters C-X4-C-X2-C-X n -C and C-X2-C-X n -C-X-C were found in prokaryotic and eukaryotic ferredoxins, xanthine dehydrogenase, and succinic dehydrogenase (Harayama et al. 1991; Glatigny and Scazzocchio 1995; Hughes et al. 1992; Wootton et al. 1991; Bläse et al. 1996; Schübel et al. 1995; Kang and Kim 1999). In the small subunit of MhyADH, the first motif contains the conserved C44, C49, C52, and C64 with n = 11. In the second motif, C103, C106, C138, and C140 are found with n = 31.
The sequence of the medium subunit exhibits significant identity with the medium subunit of CO-DH from R. litoralis Och 149 (163733671; 53%) and the FAD-binding domain of a molybdopterin dehydrogenase from Sphaerobacter thermophilus DSM 20745 (Chloroflexi; gi: 269928727; 50%). Although there are no evident sequence data on FAD-binding in prokaryotic molybdo-iron-flavoproteins, the medium subunit of MhyADH should contain an FAD cofactor as other molybdenum-containing oxidoreductases. This is also supported by the UV data (Fig. 4).
The sequence of the large subunit shows a remarkable degree of identity with the large subunit of molybdopterin binding oxidoreductase from R. litoralis Och 149 (163733669) (60%) and aldehyde oxidase and xanthine dehydrogenase from S. thermophilus DSM 20745 (269838325; 57%). The G-X-G-X-X-G/A motif is known as the dinucleotide-binding motif (Thoenes et al. 1994; Romão et al. 1995; Huber et al. 1996). In MhyADHL, three conserved regions GYGTHG (343–348), GTGPGG (559–564), GGGEGG (826–831), and another molybdopterin binding motif GGSFGSK (314–321) are found to be putative molybdopterin-contacting regions (Wootton et al. 1991).
Based on the above sequence information and the reaction type, MhyADH can be grouped into the xanthine oxidase family, which introduce a hydroxyl group to the substrate and use water as source of the oxygen atom (Bonam and Ludden 1987; Howes et al. 1996; Leimkühler et al. 2004; Okamoto et al. 2004). The resulting products are usually in the enol rather than oxo form in aqueous solution. It can also be concluded that such enzymes are present in bacteria that are not phylogenetically related, indicating that such type of hydroxylation is very common in bacteria and more similar reactions can be discovered. So far, the known molybdenum-containing oxidoreductases catalyze the addition of water to imine (xanthine dehydrogenase, quinoline 2-oxidoreductase, nicotinic acid dehydrogenase) and carbonyl (aldehyde oxidase) followed by oxidation (Thoenes et al. 1994; Romão et al. 1995; Huber 1996). The only example of molybdopterin-binding protein that can catalyze the addition of water to unsaturated C–C bond is acetylene hydratase, which contains one [4Fe-4S] cluster and a tungsten center coordinated to molybdopterins (Meckenstock et al. 1999). However, the addition of water to acetylene catalyzed by acetylene hydratase does not involve electron transfer and the oxidation states of W and the [4Fe:4S] cluster does not change during catalysis (Seiffert et al. 2007). MhyADH represents a new member of molybdenum-containing oxidoreductase which is able to catalyze Michael addition of water to α,β-unsaturated carbonyl compounds and in situ oxidation. Our preliminary result indicates a new route to the synthesis of β-hydroxy carbonyl compounds by using such type of enzymes.
References
Arai MA, Tsutusmi K, Hara H, Chen TC, Sakaki T, Urushino N, Inouye K, Kittaka A (2005) Synthesis of 25-hydroxy-19-norvitamin D3 analogs and their antiproliferative activities on prostate cells. Heterocycles 66:469–479
Baitsch D, Sandu C, Brandsch R, Igloi GL (2001) Gene cluster on pAO1 of Arthrobacter nicotinovorans involved in degradation of the plant alkaloid nicotine: cloning, purification, and characterization of 2,6-dihydroxypyridine 3-hydroxylase. J Bacteriol 183:5262–5267
Bakker G (1977) Anaerobic degradation of aromatic compounds in the presence of nitrate. FEMS Microbiol Lett 1:103–108
Balba MT, Evans WC (1980) The methanogenic biodegradation of catechol by a microbial consortium: evidence for the production of phenol through cis-benzenediol. Biochem Soc Trans 8:452–453
Bläse M, Bruntner C, Tshisuaka B, Fetzner S, Lingens F (1996) Cloning, expression, and sequence analysis of three genes encoding quinoline 2-oxidoreductase, a molybdenum-containing hydroxylase from Pseudomonas putida 86. J Biol Chem 271:23068–23079
Bonam D, Ludden PW (1987) Purification and characterization of carbon monoxide dehydrogenase, a nickel, zinc, iron-sulfur protein, from Rhodospirillum rubrum. J Biol Chem 262:2980–2987
Bonin I, Martins BM, Purvanov V, Fetzner S, Huber R, Dobbek H (2004) Active site geometry and substrate recognition of the molybdenum hydroxylase quinoline 2-oxidoreductase. Structure 12:1425–1435
Dangel W, Tschech A, Fuchs G (1988) Anaerobic metabolism of cyclohexanol by denitrifying bacteria. Arch Microbiol 150:358–362
Dangel W, Tschech A, Fuchs G (1989) Enzyme reactions involved in anaerobic cyclohexanol metabolism by a denitrifying Pseudomonas species. Arch Microbiol 152:273–279
Donoghue NA, Norris DB, Trudgill PW (1976) The purification and properties of cyclohexanone monooxygenase from Nocardia globerula CLl and Acinetobacter NCIB 9871. Eur J Biochem 63:175–192
Glatigny A, Scazzocchio C (1995) Cloning and molecular characterization of hxA, the gene coding for the xanthine dehydrogenase (purine hydroxylase I) of Aspergillus nidulans. J Biol Chem 270:3534–3550
Gremer L, Meyer O (1996) Characterization of xanthine dehydrogenase from the anaerobic bacterium Veillonella atypical and identification of a molybdenum-cytosine-dinucleotide-containing molybdenum cofactor. Eur J Biochem 238:862–866
Hagedoorn PL, van't Slot P, van Leeuwen HP, Hagen WR (2001) Electroanalytical determination of tungsten and molybdenum in proteins. Anal Biochem 297:71–78
Harayama S, Polissi A, Rekik M (1991) Divergent evolution of chloroplast-type ferredoxins. FEBS Lett 285:85–88
Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96:2757–2816
Hille R (2005) Molybdenum-containing hydroxylases. Arch Biochem Biophys 433:107–116
Howes BD, Bray RC, Richards RL, Turner NA, Bennett B, Lowe DJ (1996) Evidence favoring molybdenum–carbon bond formation in xanthine oxidase action: 17O- and 13C-ENDOR and kinetic studies. Biochemistry 35:1432–1443
Huber R, Hof P, Duarte RO, Moura JJ, Moura I, Liu MY, LeGall J, Hille R, Archer M, Romão MJ (1996) A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc Natl Acad Sci USA 93:8846–8851
Hughes RK, Doyle WA, Chovnick A, Whittle JRS, Burke JF, Bray RC (1992) Use of rosy mutant strains of Drosophila melanogaster to probe the structure and function of xanthine dehydrogenase. Biochemistry 31:3073–3083
Iwahama T, Yoshino Y, Keitoku T, Sakaguchi S, Ishii Y (2000) Efficient oxidation of alcohols to carbonyl compounds with molecular oxygen catalyzed by N-hydroxyphthalimide combined with a Co species. J Org Chem 65:6502–6507
Kang BS, Kim YM (1999) Cloning and molecular characterization of the genes for carbon monoxide dehydrogenase and localization of molybdopterin, flavin adenine dinucleotide, and iron-sulfur centers in the enzyme of Hydrogenophaga pseudoflava. J Bacteriol 181:5581–5590
Lehmann M, Tshisuaka B, Fetzner S, Röger P, Lingens F (1994) Purification and characterization of isoquinoline 1-oxidoreductase from Pseudomonas diminuta 7, a novel molybdenum-containing hydroxylase. J Biol Chem 269:11254–11260
Leimkühler S, Stocker AL, Igarashi K, Nishino T, Hille R (2004) The role of active site glutamate residues in catalysis of Rhodobacter capsulotus xanthine dehydrogenase. J Biol Chem 279:40437–40444
Marmur J (1961) A procedure for the isolation of deoxyribonucleic acid from microorganisms. J Mol Biol 3:208–218
Mechichi T, Stackebrandt E, Fuchs G (2003) Alicycliphilus denitrificans gen. nov., sp. nov., a cyclohexanol-degrading, nitrate-reducing β-proteobacterium. Int J Syst Evol Microbiol 53:147–152
Meckenstock RU, Krieger R, Ensign S, Kroneck PMH, Schink B (1999) Acetylene hydratase of Pelobacter acetylenicus: molecular and spectroscopic properties of the tungsten iron-sulfur enzyme. Eur J Biochem 264:176–182
Norris DB, Trudgill PW (1971) The metabolism of cyclohexanol by Nocardia globerula CL1. Biochem J 121:363–370
Okamoto K, Matsumoto K, Hille R, Eger BT, Pai EF, Nishino T (2004) The crystal structure of xanthine oxidoreductase during catalysis: implications for reaction mechanism and enzyme inhibition. Proc Natl Acad Sci USA 101:7931–7936
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Romão MJ, Archer M, Moura I, Moura JJ, LeGall J, Engh R, Schneider M, Hof P, Huber R (1995) Crystal structure of the xanthine oxidase-related aldehyde oxido-reductase from D. gigas. Science 270:1170–1176
Schenk S, Hoelz A, Krau B, Decker K (1998) Gene structures and properties of enzymes of the plasmid-encoded nicotine catabolism of Arthrobacter nicotinovorans. J Mol Biol 284:1323–1339
Schräder T, Thiemer B, Andreesen JR (2002) A molybdenum-containing dehydrogenase catalyzing unusual 2-hydroxylation of nicotinic acid. Appl Microbiol Biotechnol 58:612–617
Schübel U, Kraut M, Morsdorf G, Meyer O (1995) Molecular characterization of the gene cluster coxMSL encoding the molybdenum-containing carbon monoxide dehydrogenase of Oligotropha carboxidovorans. J Bacteriol 177:2197–2203
Seiffert GB, Ullmann GM, Messerschmidt A, Schink B, Kroneck PMH, Einsle O (2007) Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc Natl Acad Sci USA 104:3073–3077
Thoenes U, Flores OL, Neves A, Devreese B, van Beeumen JJ, Huber R, Romão MJ, LeGall J, Moura JJ, Rodrigues-Pousada C (1994) Molecular cloning and sequence analysis of the gene of the molybdenum-containing aldehyde oxido-reductase of Desulfovibrio gigas. Eur J Biochem 220:901–910
Trudgill PW (1984) Microbial degradation of organic compounds. Marcel Dekker, New York
Williams RJ, Evans WC (1975) The metabolism of benzoate by Moraxella species through anaerobic nitrate respiration. Evidence for a reductive pathway. Biochem J 148:1–10
Wootton JC, Nicolson RE, Cock JM, Walters DE, Burke JF, Doyle WA, Bray RC (1991) Enzymes depending on the pterin molybdenum cofactor: sequence families, spectroscopic properties of molybdenum and possible cofactor-binding domains. Biochim Biophys Acta 1057:157–185
Wu D, Raymond J, Wu M, Chatterji S, Ren Q, Graham JE, Bryant DA, Robb F, Colman A, Tallon LJ, Badger JH, Madupu R, Ward NL, Eisen JA (2009) Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS ONE 4:E4207
Acknowledgments
The authors thank Dr. Peter-Leon Hagedoorn and Ana M. Sevcenco (Enzymology group, Department of Biotechnology, Delft University of Technology) for induced coupled plasma-mass spectrometric analysis and catalytic-adsorptive stripping voltammetric analysis. This study has been financially supported by the Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (www.b-basic.nl) through B-Basic, a public-private NWO-Advanced Chemical Technologies for Sustainability (ACTS) program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Peptide sequences of MhyADH obtained by de novo peptide sequencing (DOC 37.5 kb)
Figure S1
The conversion of 2-cyclohexenone to 3-hydroxy cyclohexanone by MhyADH. No oxidation product was detected (DOC 114 kb)
Figure S2
The specific rates of MhyADH for the substrates 3-hydroxy cyclohexanone (racemic) (A) and 2-cyclohexenone (B) as determined by the coupled activity assay using methylene blue as the electron acceptor (DOC 32.5 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jin, J., Straathof, A.J.J., Pinkse, M.W.H. et al. Purification, characterization, and cloning of a bifunctional molybdoenzyme with hydratase and alcohol dehydrogenase activity. Appl Microbiol Biotechnol 89, 1831–1840 (2011). https://doi.org/10.1007/s00253-010-2996-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2996-2