
Abstract
The genes of the major histocompatibility complex (MHC) are attractive candidates for investigating the link between adaptive variation and individual fitness. High levels of diversity at the MHC are thought to be the result of parasite-mediated selection and there is growing evidence to support this theory. Most studies, however, target just a single gene within the MHC and infer any evidence of selection to be representative of the entire gene region. Here we present data from three MHC class II beta genes (DPB, DQB, and DRB) for brown hares in two geographic regions and compare them against previous results from a class II alpha-chain gene (DQA). We report moderate levels of diversity and high levels of population differentiation in the DQB and DRB genes (Na = 11, D est = 0.071 and Na = 15, D est = 0.409, respectively), but not for the DPB gene (Na = 4, D est = 0.00). We also detected evidence of positive selection within the peptide binding region of the DQB and DRB genes (95% CI, ω > 1.0) but found no signature of selection for DPB. Mutation and recombination were both found to be important processes shaping the evolution of the class II genes. Our findings suggest that while diversifying selection is a significant contributor to the generally high levels of MHC diversity, it does not act in a uniform manner across the entire MHC class II region. The beta-chain genes that we have characterized provide a valuable set of MHC class II markers for future studies of the evolution of adaptive variation in Leporids.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
How variation at functional gene regions arises and is maintained in natural populations is a key remaining question of evolutionary genetics (reviewed in Piertney and Oliver 2006). The prime candidate gene region to investigate this question is the major histocompatibility complex (MHC). It is hypothesized that positive selection, acting in concert with a “Red Queen” arms race between pathogens and the immune system (Ladle 1992), drives the maintenance of diversity at the MHC. However, empirical evidence for high levels of selection within the MHC has been mixed, depending on which species, MHC region, or method of analysis is employed. Numerous studies have reported evidence of selection on MHC genes in mammals (Aguilar et al. 2004; Amills et al. 2008; Čížková et al. 2011; Goüy de Bellocq et al. 2009), fish (Evans et al. 2010; Gomez et al. 2010; Schaschl et al. 2008), and birds (Alcaide et al. 2008; Anmarkrud et al. 2010; Loiseau et al. 2009). Few researchers have considered, however, whether selection acts in a constant and consistent way across all MHC genes and if there are differences in selection signatures between alpha and beta class genes of the MHC (Averdam et al. 1992).
Much of the work to date on MHC selection processes has been based on model species including humans, rodents, and fish. More recently, attention has turned to natural populations in an attempt to understand how ecological factors interact to influence the dynamics of MHC evolution and how MHC diversity affects individual fitness and population level processes (Piertney and Oliver 2006). While many of the initial hurdles in the isolation and characterization of individual, functional, highly polymorphic MHC loci have been overcome (see review in Bernatchez and Landry 2003), most studies have restricted themselves to investigations of single MHC loci or single populations (Acevedo-Whitehouse and Cunningham 2006). To better understand how the full range of MHC variation contributes to individual fitness and is maintained in natural populations, it is important to extend the breadth of MHC studies to encompass multiple loci and a greater spatial range of populations (Čížková et al. 2011; Garrigan and Hedrick 2003). While, for some studies, it makes sense to only consider single locus interactions (e.g., MHC-related matechoice or pathogen driven selection) and, for some species, there are difficulties in amplifying just single interpretable loci, there are clear advantages to a multiple gene approach to MHC. By contrasting patterns of selection at the different MHC gene regions, it has been shown that in many species (e.g., human and nonhuman primates, rats), variability at class II alpha-chain genes is lower than for beta-chain genes (Averdam et al. 2010; Bondinas et al. 2007). On the other hand, in salmonids, the difference is less pronounced (Gomez et al. 2010), reflecting perhaps a different evolutionary trajectory for the MHC of teleost fishes. Comparisons of the levels of diversity and the strengths of selection acting on the different gene regions of the MHC are essential first steps to understanding how adaptive genetic variation arises and is maintained in an ecological framework.
The brown hare (Lepus europaeus) represents an excellent species to examine MHC gene interactions. It belongs to a Pleistocene species flock that occurs in a wide range of ecologically different habitats from arctic/alpine to desert environments. Hares are virtually the only small- or medium-sized mammals in Europe that raise their young above ground right from the day of birth onwards. Their development is incredibly rapid with an increase in body weight by eight to tenfold of birth weight within the first month of life. Living above ground, they receive little protection against harsh environmental conditions and are usually left alone and only visited by their mother once a day for a very short period of time to suckle (Göritz et al. 2001; Hartl et al. 1995). The sudden exposure to aboveground environmental challenges after an intrauterine life, with virtually no protection from their mother, together with their rapid development suggests a very efficient immune system to guarantee sufficient energy allocation to growth. A diverse set of infectious and parasitic diseases such as European brown hare syndrome (a Lepus-specific calicivirus), tularemia and pseudotuberculosis (bacterial diseases), and coccidiosis (sporozoan infection) are known to strongly affect regional survival (reviewed in Wibbelt and Frölich 2005). Previously, we have examined diversity and selection acting on an alpha-chain gene (DQA; Campos et al. 2011; Goüy de Bellocq et al. 2009; Smith et al. 2010), but no work has been carried out, to date, on other genes of the MHC. Here, we compare those results to additional brown hare data from the three MHC class II genes DQB, DPB, and DRB. Furthermore, we examine the scale of the population recombination rate in this species as compared to point mutations, in order to assess the extent to which recombination and mutation contributes to the diversity at the MHC class II beta loci. We expect the levels of diversity at the beta-chain genes to be higher than the moderate level reported for the DQA locus (Goüy de Bellocq et al. 2009), but in line with other studies of mammalian class II genes (Aguilar et al. 2004; Amills et al. 2008; Averdam et al. 2010; Bryja et al. 2006; Čížková et al. 2011; Oliver and Piertney 2006).
Materials and methods
Animal sampling and sample preparation
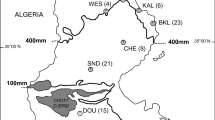
All samples were derived during autumnal hunts in 2006 and 2007 from three localities in eastern Austria and three in northern Belgium (Fig. 1). In total, 518 individuals were sampled, 291 from Austria and 227 from Belgium. For each individual, a small piece of liver was collected and preserved at −20°C. Total DNA was extracted using the GeneElute Mammalian Genomic DNA Miniprep Kit (Sigma) and eluted in 200 μl of water. Additionally, RNA was obtained from four individuals from our breeding station which were to be euthanized for the purpose of an unrelated experiment. A further piece of liver was preserved in RNAlater at −20°C. Total RNA was isolated using RNAeasy Mini Kit with on-column RNase-free DNase set (Qiagen) according to the manufacturer’s instructions. Total RNA was eluted in 30 μl RNase-free water and stored at −70°C.

Sampling localities of brown hares from Belgium (hatched circles) and Austria (stars). BK Bulskamp, SL Sint Laureins, MB Moerbeke, OW/STR Oberweiden/Stripfing, ZW Zwerndorf, BG/LA Baumgarten/Lassee
Design of beta gene exon 2-specific primers for the hare
To ensure specificity of the primer design process for brown hares, we performed a RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) using a GeneRacer kit (Invitrogen) following the specifications of the manufacturer. Specific conditions for the method are described in detail in Goüy de Bellocq et al. (2009). In brief, RNA from four individuals was pooled two by two into two samples and used as a template for cDNA synthesis. RACE-polymerase chain reaction (PCR) amplification of the 5′ end of the cDNA was performed using the GeneRacer 5′ primer provided with the kit and the universal JS2 primer (Schad et al. 2004) which has been designed in a part of exon 2 relatively conserved in beta genes. PCR was performed as described in Goüy de Bellocq et al. (2009). Bands representing PCR products within the expected size range (~400 bp) were excised from 1.5% agarose gel, purified, cloned, and sequenced as described in Goüy de Bellocq et al. (2009). We obtained 11 sequences of the 5′ end of the cDNA, one corresponding to the DPB gene (BLAST, Max ident >94% with RLA-DPB of Oryctolagus cuniculus), eight corresponding to DQB gene (BLAST, Max ident >92% with RLA-DQB of O. cuniculus), and two corresponding to DRB gene (BLAST, Max ident 86% with RLA-DRB of O. cuniculus; GenBank AN: NC_013680.1). Based on the cDNA sequences obtained with the RACE and rabbit RLA sequences from GenBank, we designed forward and reverse primers embedded within exon 2 to amplify portions of the exon in each of the three beta genes (see Table 1 for primer sequences). The DRB primers within exon 2 amplified an additional amplicon from genomic DNA that was not detected when RNA was used as a template. The sequences generated from gDNA contained a mutation in the exon 2 portion which altered the reading frame such that a stop codon was introduced midsequence. To avoid amplification of this nonfunctional gene from gDNA or the time-consuming process of genotyping from RNA, it was necessary to design a new forward primer within intron 1 at the 5′ end of exon 2. We used a primer designed in exon 1 of the DRB gene (Lepus_DRBex1_F2, identified from the original 5′ RACE-PCR) in combination with primer JS2 to amplify across intron 1 and provide sequences for alignment and primer design. The sequence divergence in this intron was sufficient to design a forward primer (DRB_Int_1f) that excluded the more variable nonfunctional copies and only amplified the functional alleles.
PCR amplification and CE-SSCP
Fragments of exon 2 for each gene (DPB 188 bp, DQB 210 bp, DRB 225 bp excluding primers) were amplified with 6′ FAM-labeled forward and NED-labeled reverse primers using the Multiplex PCR kit (Qiagen) following the manufacturer instructions in a final reaction volume of 10 μl. The thermal profile started with an initial HotStarTaq DNA polymerase activation at 95°C for 15 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s and extension at 72°C for 1 min, and final extension at 72°C for 10 min. One microliter of 25× diluted PCR product was mixed with 13.75 μl of Hi-DI formamide (ABI) and 0.25 μl of GeneScan 350 ROX size standard and denaturated at 96°C for 3 min and immediately placed on ice. Capillary electrophoresis single-strand conformation polymorphism (CE-SSCP) analysis was carried out using 5% Genescan polymer (ABI) with the addition of 10% glycerol in an ABI 3130xl genetic analyzer. TBE (1×) with 10% glycerol was used as running buffer. Electrophoresis was conducted at 18 and 22°C, samples were injected for 18 s and electrophoresis voltage was set to 12 kV. Samples were run for 36 min. Chromatograms were analyzed using the software program GeneMapper 3.7 (ABI).
Cloning and sequencing
We selected individuals with diverse SSCP patterns, representing all identified alleles, to investigate sequence variation at the second exon of the three beta-chain loci, respectively. The genes were amplified as described above, but using nonlabeled primers. Where homozygous individuals were detected by SSCP analysis, we directly sequenced the PCR product. The PCR products from heterozygote individuals were ligated into pCR2.1 vectors, and chemically competent TOP 10 cells were transformed with the ligation products using a TOPO TA cloning kit (Invitrogen). Transformants containing inserts were identified via PCR screening and plasmid DNA was extracted by QIAprep Spin Miniprep Kit (Qiagen), and inserts sequenced (Macrogen). Each potential allele (unique conformation pattern) was sequenced at least twice from two different individuals that were taken, if possible, across the Austrian and Belgium regions. The sequences were edited in Sequencing Analysis 5.1 and aligned in BioEdit (Hall 1999) using ClustalW multiple alignment.
Population genetic parameters and analysis of selection
Population allele frequencies, observed and expected heterozygosities (Ho and He), and tests of deviation from Hardy–Weinberg equilibrium were calculated using GENEPOP 4.0 (Rousset 2008). The allelic numbers for the brown hare sequences were assigned according to the guidelines of Klein et al. (1990). Population differentiation was determined by calculation of Jost’s D (Jost 2008) as a more robust measure than Fst. Calculations were performed using the DEMEtix package within the R statistical framework (R Development Core Team), and p-values were calculated by performing 10,000 bootstrap iterations. In addition, a hierarchical analysis of molecular variance from the MHC data was calculated for populations within the two regions, Belgium and Austria, using the Arlequin 3.5.1.2 program (Excoffier and Lischer 2010). In order to address the evidence for positive selection in the MHC genes, we used the OmegaMap program (Wilson and McVean 2006). This program detects positively selected sites in sequences in the presence of recombination. It employs a Bayesian population genetics approximation to the coalescent theory and generates means and credible intervals for the selection parameter (dN/dS = ω) and recombination rate (ρ = 4Nr) for each codon (N and r represent the effective population size and the per codon rate of recombination). The probable values of the mutation rate (μ) and the transition/transversion rate ratio (κ) were adjusted to follow improper inverse distributions: starting values for μ and κ were set at 0.1 and 3.0, respectively, and the selection parameter (ω) and the recombination rate (ρ) were adjusted to follow inverse distributions in the range between 0.01 and 20 for ω and 0.01 and 100 for ρ, respectively. We chose to estimate ω for each codon independently and for ρ a block-like structure of 10 codons. Means for ω and ρ per codon were calculated using the posterior distributions generated with the objective prior set. Two Markov chain Monte Carlo tests were run for 250,000 iterations on population allele frequencies at each MHC locus, with a 20,000 iteration burn-in. The data was visualized in R with the included R-script provided by the authors. The program MEGA 4 (Tamura et al. 2007) was used to compute average pairwise nucleotide distances (Kimura 2-parameter model or K2P) and average pairwise Poisson-corrected amino acid distances. Standard errors were obtained with 1,000 bootstrap replicates.
Analysis of population recombination rate
The LDhat program was used to estimate rates of population mutation (from Waterson’s θ, where θ = 4N e μ) and population recombination ρ (where ρ = 4N e r; McVean et al. 2002). This program estimates population recombination from a set of aligned sequences using the composite likelihood method (Hudson 2001) within a coalescent framework. This analysis is reliable and valid even for low variability loci. The program considers the population frequency of each allele and each codon independently. Specifically, it uses a population genetics approximation to the coalescent with recombination (ρ) and then applies the reversible-jump MCMC method to perform Bayesian inference on the selection parameter omega. We used the likelihood permutation-based approach to test significance against the null hypothesis of no recombination (i.e. 4N e r = 0). We also calculated the ratio of ρ/θ as an estimate of the relative amount of recombination compared to point mutations (Fearnhead and Donnelly 2001). Additionally, D′ and r 2 statistics (Awadalla et al. 1999), which assess the correlation of linkage disequilibrium among pairs of polymorphic sites with the distance between them, were calculated as alternate indicators of recombination. In these cases, recombination is inferred if there is a significant decay of linkage disequilibrium with distance.
Results
Population genetic parameters
From the entire sample, we identified 15 DRB alleles, 11 DQB alleles, and four DPB alleles. All are previously undescribed. The same individuals were genotyped for all loci except DPB which showed very low variability after the initial screen of a subset of 71 individuals (51 from Austria and 20 from Belgium), so it was decided to discontinue the population level analysis for this locus. Allele frequencies and basic diversity parameters for the three MHC class II beta-chain exon 2 loci in the different localities as well as the overall values for the Austrian and Belgian regions are displayed in Table 2. For DRB, there were eight alleles shared between the two regions. Alleles Leeu-DRB*06, Leeu-DRB*10, Leeu-DRB*11, Leeu-DRB*12, Leeu-DRB*14, and Leeu-DRB*15 only occurred in Austria; whereas allele Leeu-DRB*07 only occurred in Belgium. For DQB, nine alleles were shared with alleles Leeu-DQB*05 and Leeu-DQB*06 only occurring in Austria. Of the four DPB alleles, Leeu-DPB*03 and Leeu-DPB*04 were not detected in Belgium, but it is important to note the small sample sizes for the Belgium subpopulations. HWE analyses highlighted that the DRB locus contained significantly fewer heterozygotes than expected under equilibrium in every subpopulation except OW/STR and also for each region overall. DRB and DQB showed significant linkage disequilibrium, however, DPB showed no linkage with the other two loci, perhaps reflecting its physical distance from them (as shown for humans and rabbits: Chouchane and Kindt 1992; Trowsdale 1995) and/or as a consequence of the low number of individuals included in analyses involving DPB.
Population differentiation, as determined by mean D est, across all three loci was high and significant (mean D est = 0.16, p < 0.001). For the individual loci, significant differentiation was detected for DRB (D est = 0.409, p < 0.001) and DQB (D est = 0.071, p < 0.001), but not for DPB (D est = 0.00, p = 0.43). When data from the previous study for the DQA locus was included (D est = 0.594, p < 0.001), the mean D est value increased sharply (D est = 0.268, p < 0.001). A hierarchical population structure was evident in the genetic differentiation across all three loci, with 12.4% of the variance attributable to between region differences, 1.9% between localities within regions, and 85.7% to within sampling localities (P < 0.001, P ≪ 0.0001, and P ≪ 0.0001, respectively).
Selection and recombination
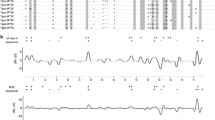
The average pairwise nucleotide distance (K2P) for the DQB gene was 0.096 (±0.014) and for the DRB gene 0.101 (±0.014). The average pairwise Poisson-corrected amino acid distances for DQB and DRB were 0.195 (±0.039) and 0.180 (±0.034), respectively. We identified, via OmegaMap, two codons (26S, 60 N) in the DQB gene and six codons in the MHC DRB gene (11 V, 57I, 67 L, 71R, 74S, and 78Y) that showed strong and significant effects of positive selection. In both genes, the positively selected amino acid sites had posterior probabilities greater than 95% (Fig. 2). In the DPB gene, no sites were indentified to be under strong positive selection. All of the positively selected sites identified for the DQB and DRB genes correspond to proposed peptide-binding sites of human HLA class II molecules (Bondinas et al. 2007; Brown et al. 1993; Stern et al. 1994). OmegaMap identified no particular recombination hot spots for any of the genes. The estimates of population mutation (θ) and recombination (ρ) obtained with the LDhat program show that each of these processes has played a significant role in generating the diversity seen among the hare MHC beta genes DRB, DQB, and DQA (Table 3). The likelihood permutation test (LPT) indicated that the estimates of ρ were significantly different from zero for the DRB, DQB, and DQA in the study (Table 3), but not for DPB. The two alternative statistical tests for recombination, r 2 and D′, also detected recombination rates significantly different from zero in most cases. For r 2, however, this was only the case for DRB, and all but DPB were significant for D′ (Table 3). The ratios of ρ/θ for the MHC loci DRB, DQB, and DQA (0.6, 1.4, and 0.2, respectively) indicate that the relative contribution of recombination and point mutations has been close to even in the evolution of these genes in brown hares (ρ/θ < 1 indicates a greater role for point mutations, whereas ρ/θ > 1 indicates more influence from recombination).

Omega Plot output for a the DQB gene and b the DRB gene. The dotted line indicates omega (ω) = 1. The unbroken line shows the mean value for ω from posterior distribution, and the shadowed area displays the 95% confidence interval. Amino acid codes are shown above each plot, and those presumed to be involved in peptide binding based on the human sequence are in bold. Codon positions are given with respect to the standard human HLA numbering and are considered to be significantly under the influence of positive selection if the mean and 95% confidence interval for ω > 1. Positively selected sites for DQB are 26S and 60 N and for DRB are 11 V, 57I, 67 L, 71R, 74S, and 78Y. All positively selected sites correspond to proposed peptide-binding regions
Discussion
We have previously reported moderate diversity and evidence of selection (10 alleles, ω = 2.69) acting on the MHC class II DQA locus in European brown hares (Goüy de Bellocq et al. 2009). In this study, we extend the coverage of the brown hare MHC to include three other class II genes (DPB, DQB, and DRB). Heterozygosities for the three genes varied substantially from 0.18 for DPB in Belgium to 0.784 for DRB in Austria. Austria tended to exhibit higher He values than Belgium for the three genes, but only significantly so for DQB (t = 4.10, p = 0.015). The lower diversity levels detected for Belgium populations fits with a pattern of recent founder events as the species has expanded northwards from refugia in the Balkans since the last glacial maximum (Kasapidis et al. 2005; Stamatis et al. 2009). Spatial differences were clearly evident in the distribution of genetic variation with significant structure reported between the regions as well as between populations within each region. Although it is possible that demographic processes may explain a portion of this structuring, the lack of population differentiation at one locus (DPB) and the departure from HWE for the DRB locus suggests that differential selection pressures acting on these immune system genes are likely to also play a role. Some caution is needed in interpreting these data, however, as only a subset of individuals (n = 71) were genotyped for the DPB locus and the departure from HWE at DRB may also be the result of some alleles being missed due to the specificity of the forward primer placed in the intron.
The DQB and DRB genes showed evidence of positive selection at putative peptide binding sites similar to that revealed for the DQA gene. For DPB, however, we found no statistical evidence for positive selection and very low levels of diversity, indicating that this locus may be operating under a different selection regime to the others. Our data corresponds with human HLA allele frequency data for class II beta genes (Begovich et al. 1992), which suggests that DRB, DQA, and DQB loci have been subjected to balancing selection, whereas DPB has shown a different evolutionary past. This difference in the evidence for selection and the reduced diversity also reported for the human DPB (Bodmer et al. 1999) is presumably, at least in part, a result of the lower levels of linkage disequilibrium between DPB and the other class II genes (Begovich et al. 1992; Petersdorf et al. 2001). We also found no evidence of recombination (Table 3) or linkage disequilibrium at the DPB locus which supports the notion of a different evolutionary history for this locus compared to the other class II genes in the study.
The evidence for positive selection at DRB and DQB is consistent with findings for class II genes of other mammalian species (Amills et al. 2008; Averdam et al. 2010; Čížková et al. 2011; Schaschl et al. 2006). However, the rather surprising finding is how few sites were identified as being under positive selection compared to other studies. Much of this is probably due to the conservative approach adopted in our analysis to ensure robust extrapolation of positively selected sites. The task of disentangling the cooccurring signals of selection and recombination is a complicated process, but an important one in a system where both are known to play significant roles (Begovich et al. 1992). The evidence for recombination in brown hare MHC genes, while not as strong as for ungulate DRB genes (average ρ/θ = 8.03, Schaschl et al. 2006), suggests that its effects need to be controlled for in tests of selection. We chose to analyze our data via the OmegaMap program which does take the potential influence of recombination into account in the detection of selection (Wilson and McVean 2006). Other studies which have not acknowledged the likely presence of recombination within their datasets must potentially be treated with caution and are perhaps prone to type 1 error.
To better understand how immune genes are interacting with parasites or pathogens, a detailed analysis of MHC genes is needed, particularly for nonmodel species. MHC molecules play a key role in directing and shaping the T-cell repertoire during T-cell maturation. Heterozygosity or specific alleles at MHC loci may therefore enhance resistance to infectious diseases by generating a more diverse or specific T-cell repertoire. Combined with the previously characterized DQA gene, there is now a useful panel of MHC class II genes available to investigate how adaptive variation is maintained at a broader level in this wild species as well as allowing researches more choice in which gene or genes to select for studies of important associations between fitness and immunogenetic diversity.
References
Acevedo-Whitehouse K, Cunningham AA (2006) Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol 21:433–438
Aguilar A, Roemer G, Debenham S et al (2004) High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc Natl Acad Sci USA 101:3490–3494
Alcaide M, Edwards SV, Negro JJ, Serrano D, Tella JL (2008) Extensive polymorphism and geographical variation at a positively selected MHC class II B gene of the lesser kestrel (Falco naumanni). Mol Ecol 17:2652–2665
Amills M, Ramirez O, Tomas A, Obexer-Ruff G, Vidal O (2008) Positive selection on mammalian MHC-DQ genes revisited from a multispecies perspective. Genes Immunol 9:651
Anmarkrud JA, Johnsen A, Bachmann L, Lifjeld JT (2010) Ancestral polymorphism in exon 2 of bluethroat (Luscinia svecica) MHC class II B genes. J Evol Biol 23:1206–1217
Averdam A, McClure G, Suraj V et al (1992) Polymorphism, recombination, and linkage disequilibrium within the HLA class II region. J Immunol 148:249–258
Averdam A, Kuschal C, Otto N et al (2010) Sequence analysis of the grey mouse lemur (Microcebus murinus) MHC class II DQ and DR region. Immunogenetics 63:1–9
Awadalla P, Eyre-Walker A, Smith JM (1999) Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286:2524–2525
Begovich A, McClure G, Suraj V et al (1992) Polymorphism, recombination, and linkage disequilibrium within the HLA class II region. J Immunol 148:249–258
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bodmer JG, Marsh SGE, Albert ED et al (1999) Nomenclature for factors of the HLA system, 1998. Tissue Antigens 53:407–446
Bondinas G, Moustakas A, Papadopoulos G (2007) The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics 59:539–553
Brown JH, Jardetzky TS, Gorga JC et al (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Bryja J, Galan M, Charbonnel N, Cosson JF (2006) Duplication, balancing selection and trans-species evolution explain the high levels of polymorphism of the DQA MHC class II gene in voles (Arvicolinae). Immunogenetics 58:191–202
Campos JL, Bellocq JGd, Schaschl H, Suchentrunk F (2011) MHC class II DQA gene variation across cohorts of brown hares (Lepus europaeus) from eastern Austria: testing for different selection hypotheses. Mamm Biol Z Saugetierkunde 76:251–257
Chouchane L, Kindt T (1992) Mapping of the rabbit MHC reveals that class I genes are adjacent to the DR subregion and defines an insertion/deletion-related polymorphism in the class II region. J Immunol 149:1216–1222
Čížková D, Goüy de Bellocq J, Baird SJE, Piálek J, Bryja J (2011) Genetic structure and contrasting selection pattern at two major histocompatibility complex genes in wild house mouse populations. Heredity 106(5):727–740
Evans ML, Neff BD, Heath DD (2010) MHC genetic structure and divergence across populations of Chinook salmon (Oncorhynchus tshawytscha). Heredity 104:449–459
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resourc 10:564–567
Fearnhead P, Donnelly P (2001) Estimating recombination rates from population genetic data. Genetics 159:1299–1318
Garrigan D, Hedrick PW (2003) Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57:1707–1722
Gomez D, Conejeros P, Marshall SH, Consuegra S (2010) MHC evolution in three salmonid species: a comparison between class II alpha and beta genes. Immunogenetics 62:531–542
Göritz F, Faßbender M, Broich A et al (2001) Untersuchungen zur reproduktiven Fitness lebender weiblicher Feldhasen aus unterschiedlichen Habitaten. Z Jagdwissenschft 47:92
Goüy de Bellocq J, Suchentrunk F, Baird S, Schaschl H (2009) Evolutionary history of an MHC gene in two leporid species: characterisation of Mhc-DQA in the European brown hare and comparison with the European rabbit. Immunogenetics 61:131–144
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis rogram for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 41:95–98
Hartl GB, Suchentrunk F, Willing R, Petznek R (1995) Allozyme heterozygosity and fluctuating asymmetry in the brown hare (Lepus europaeus): a test of the developmental homeostasis hypothesis. Philos Trans R Soc Lond B Biol Sci 350:313–323
Hudson RR (2001) Two-locus sampling distributions and their application. Genetics 159:1805–1817
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kasapidis P, Suchentrunk F, Magoulas A, Kotoulas G (2005) The shaping of mitochondrial DNA phylogeographic patterns of the brown hare (Lepus europaeus) under the combined influence of Late Pleistocene climatic fluctuations and anthropogenic translocations. Mol Phylogenet Evol 34:55–66
Klein J, Kasahara M, Gutknecht J, Figueroa F (1990) Origin and function of mhc polymorphism. Chem Immunol 49:35–50
Ladle RJ (1992) Parasites and sex: catching the red queen. Trends Ecol Evol 7:405–408
Loiseau C, Richard M, Garnier S et al (2009) Diversifying selection on MHC class I in the house sparrow (Passer domesticus). Mol Ecol 18:1331–1340
McVean G, Awadalla P, Fearnhead P (2002) A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160:1231–1241
Oliver MK, Piertney SB (2006) Isolation and characterization of a MHC class II DRB locus in the European water vole (Arvicola terrestris). Immunogenetics 58:390–395
Petersdorf EW, Gooley T, Malkki M et al (2001) The biological significance of HLA-DP gene variation in haematopoietic cell transplantation. Br J Haematol 112:988–994
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Rousset F (2008) GENEPOP'007: A complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources 8:103–106
Schad J, Sommer S, Ganzhorn JU (2004) MHC variability of a small lemur in the littoral forest fragments of southeastern Madagascar. Conservation Genetics 5:299–309
Schaschl H, Wandeler P, Suchentrunk F, Obexer-Ruff G, Goodman SJ (2006) Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 97:427–437
Schaschl H, Tobler M, Plath M, Penn DJ, Schlupp I (2008) Polymorphic MHC loci in an asexual fish, the amazon molly (Poecilia formosa; Poeciliidae). Mol Ecol 17:5220–5230
Smith S, Mang T, Goüy de Bellocq J et al (2010) Homozygosity at a class II MHC locus depresses female reproductive ability in European brown hares. Mol Ecol 19:4131–4143
Stamatis C, Suchentrunk F, Moutou KA et al (2009) Phylogeography of the brown hare (Lepus europaeus) in Europe: a legacy of south-eastern Mediterranean refugia? J Biogeogr 36:515–528
Stern LJ, Brown JH, Jardetzky TS et al (1994) Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 368:215–221
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24:1596–1599
Trowsdale J (1995) ‘Both man and bird and beast’: comparative organization of MHC genes. Immunogenetics 41:1–17
Wibbelt G, Frölich K (2005) Infectious diseases in European brown hare (Lepus europaeus). Wildl Biol Pract 1:86–93
Wilson DJ, McVean G (2006) Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172:1411–1425
Acknowledgments
We thank H. Ben Slimen (Vienna, Beja) and Th. Ceulemans (Borsbeek) for assisting in the field collection of samples, A. Haiden for supporting with laboratory work, and D. Penn for providing access to the DNA sequencer. This study was funded by the Austrian Science Fund (FWF): #P18534-B03 and the government of Lower Austria. JGB is presently a postdoctoral fellow with FWO.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Smith, S., Goüy de Bellocq, J., Suchentrunk, F. et al. Evolutionary genetics of MHC class II beta genes in the brown hare, Lepus europaeus . Immunogenetics 63, 743–751 (2011). https://doi.org/10.1007/s00251-011-0539-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-011-0539-3