
Abstract
Short cationic antimicrobial peptides (AMPs) are believed to act either by inducing transmembrane pores or disrupting membranes in a detergent-like manner. For example, the antimicrobial peptides aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1, despite being closely related, appear to act by fundamentally different mechanisms depending on their length. Using molecular dynamics simulations, the structural properties of these four peptides have been examined in solution as well as in a variety of membrane environments. It is shown that each of the peptides has a strong preference for binding to regions of high membrane curvature and that the structure of the peptides is dependent on the degree of local curvature. This suggests that the shorter peptides aurein 1.2 and citropin 1.1 act via a detergent-like mechanism because they can induce high local, but not long-range curvature, whereas the longer peptides maculatin 1.1 and caerin 1.1 require longer range curvature to fold and thus bind to and stabilize transmembrane pores.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antimicrobial peptides (AMPs) occur widely in both animals and plants where they act as innate modulators of the immune response and/or direct anti-infection agents (Castro and Fontes 2005; Hancock and Sahl 2006). AMPs from various sources have attracted much interest over recent years because of their potential to form the basis of novel therapeutic agents. While there is a wide range of AMPs that act via a variety of mechanisms, those of primary therapeutic interest are short (~10–50 residues), cationic and amphiphilic, and directly disrupt the integrity of cell membranes. In particular, the skin secretions of Australian frogs (Litoria genus) and toadlets (Uperoleia genus) have been shown to be a rich resource of short cationic AMPs with more than 80 peptides with distinct structural properties (amino acid sequence, length and overall charge) having been isolated and characterized (Apponyi et al. 2004). Of these, four peptides, namely aurein 1.2 (GLFDI IKKIA ESF-NH2), citropin 1.1 (GLFDV IKKVA SVIGG L-NH2), maculatin 1.1 (GLFGV LAKVA AHVVP AIAEH F-NH2) and caerin 1.1 (GLLSV LGSVA KHVLP HVVPV IAEHL-NH2), have been the most extensively studied (Apponyi et al. 2004). Aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1 contain 13, 16, 21 and 25 amino acids, respectively, and have net charges of +1, +2, +3 and +4 at neutral pH. All show high activity against gram-positive bacteria, and each is believed to act by directly disrupting the integrity of cell membranes (Boland and Separovic 2006). For example, exposure of POPC (2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine) giant unilamellar vesicles (GUV) containing carboxyfluroscein to 5 μM aurein 1.2 or 5 μM maculatin 1.1 leads to leakage of the dye within 10–20 min (Ambroggio et al. 2005). However, while it is clear that the peptides do compromise the integrity of model membranes, the precise mechanism by which this occurs is uncertain (Ambroggio et al. 2005; Gehman et al. 2008).
Short cationic AMPs are believed to act either by inducing transmembrane pores or disrupting membranes in a detergent-like manner (Almeida and Pokorny 2009). Various models for how such peptides may stabilize a transmembrane pore or disrupt a membrane have been proposed. In one model, the barrel-stave model, the peptides are assumed to adopt a helical conformation and to aggregate into a ring-like structure that spans the membrane with the peptides lying perpendicular to the plane of the membrane. A notable feature of this model is that there is little disruption of the membrane itself. In an alternative model, the toroidal pore model, the pore is formed by the membrane itself, which becomes highly curved. In this model, the peptide stabilizes the pore by binding to regions of high curvature. While it has been generally assumed that peptides involved in toroidal pore formation were, like in the barrel stave model, highly ordered, helical and orientated perpendicular to the plane of the membrane, recent computational studies suggest that in fact the peptides may lie at oblique angles to the pore and be quite disordered (Leontiadou et al. 2006; Sengupta et al. 2008).
The interaction of aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1 with a range of zwitterionic and anionic bilayers has been studied using a wide variety of physical techniques (Gehman et al. 2008). Surface Plasmon spectroscopy performed on DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) bilayers suggests that at peptide concentrations of 1 to 20 μM, aurein 1.2 and citropin 1.1 induce the loss of lipid as a function of time (Gehman et al. 2008). However, maculatin 1.1 and caerin 1.1 do not lead to a net loss of lipid (Gehman et al. 2008). Furthermore, NMR relaxation data suggest that while aurein 1.2 and citropin 1.1 lead to an increase in the mobility of the phospholipid bilayers to which they are bound, maculatin 1.1 and caerin 1.1 affect more the long-range order (Gehman et al. 2008). Based on this and other data, it has been proposed that as aurein 1.2 and citropin 1.1 (13 and 16 amino acids, respectively) are not long enough to span the width of the membrane, they bind to but do not insert into the membrane. Instead, they disrupt the membrane in a detergent-like manner via what is known as a carpet mechanism. In contrast the behavior of maculatin 1.1 and caerin 1.1 would be consistent with peptide insertion and pore formation (Apponyi et al. 2004; Chia et al. 2002; Gehman et al. 2008).
The interpretation of the available experimental data is also heavily dependent on the structure of the different peptides in different environments. Circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopic studies suggest that all four peptides are largely unstructured in aqueous solution. They are, however, primarily α-helical in TFE (2,2,2-trifluoroethanol, 30–70 volume %) (Chia et al. 2000; Rozek et al. 2000; Wegener et al. 1999; Wong et al. 1997). The peptides have also been shown to adopt an α-helical structure in the presence of DPC (dodecylphosphocholine) micelles. For example, both NMR studies suggested that in the presence of DPC micelles maculatin 1.1 adopts a structure very similar to that observed in TFE/water (Chia et al. 2000). Caerin 1.1 appears also to be primarily helical when bound to DPC micelles (Wegener et al. 2003). In other environments the structure of the peptides is less certain. For example, CD spectra suggest that in the presence of DMPC small unilamellar vesicles (SUV), aurein 1.2 and citropin 1.1 primarily adopt an α-helical structure (Marcotte et al. 2003). However, in partially hydrated DMPC stacked bilayers, the absorbance at 222 nm of aurein 1.2 and citropin 1.1 is low compared to that observed in DMPC SUVs, suggesting a correspondingly low degree of helicity (Marcotte et al. 2003; Scholtz et al. 1991). Based on similar CD studies, caerin 1.1 appears to adopt a random-coil structure in the presence of fully hydrated DMPC SUVs, while at lower levels of hydration in orientated samples, both NMR and CD studies suggest that caerin 1.1 becomes more helical and perhaps adopts a transmembrane orientation (Marcotte et al. 2003). One difficulty, however, in the interpretation of these results in regard to the effect of hydration is that in the fully hydrated sample the fraction of unbound (unstructured) peptide will be increased, potentially biasing the CD spectra. Maculatin 1.1 shows intermediate behavior and has been suggested to be approximately 36% helical in the presence of egg phosphocholine SUVs (Niidome et al. 2004). Again it has been proposed to adopt a transmembrane orientation at high peptide-to-lipid ratios.
The question of whether the peptides have preference for binding to regions of high curvature or can insert directly into a lipid bilayer is critical to understanding the mechanism of action of antimicrobial peptides in atomic detail. For example, there is some evidence that amphipathic helices do not in general bind preferentially to regions of high curvature (Hatzakis et al. 2009). However, the lytic properties of peptides such as melittin and magainin have been show to be strongly correlated with the bending modulus of the bilayer (Allende et al. 2005; Lee et al. 2005; Matsuzaki et al. 1998). Experimentally it is not possible to probe the structural properties of individual peptides as they interact with lipid membranes. As a consequence a number of groups have turned to theoretical approaches, in particular molecular dynamics simulation techniques (Bond et al. 2008; Mihajlovic and Lazaridis 2010; Rzepiela et al. 2010). For example, Mihajlovic et al., using an explict model of the peptide combined with an implicit membrane model, attempted to compute the interaction energy between each of four peptides, alamethicin, melittin, magainin H2 and piscidin 1, and a membrane with different degrees of curvature ranging from a cylindrical hole to a parabolic pore in a flat bilayer (Mihajlovic and Lazaridis 2010). Based on these binding energies the authors concluded that the four peptides would preferentially bind at an oblique angle to a toroidal shaped pore. One difficulty in this work, however, is that the peptides were constrained to be an ideal helix, and both entropic and solvation effects were ignored. As noted above, atomistic simulations of the spontaneous formation of transmembrane pores by magainin and melittin have suggested that the peptides, at least initially after pore formation, are highly disordered and bind primarily to the rim as opposed to the middle of the pore (Leontiadou et al. 2006; Sengupta et al. 2008). Likewise Bond et al. performed extensive simulations on maculatin 1.1 using a coarse-grained force field, concluding that it inserted via the N-terminus (Bond et al. 2008). However, again the peptide was constrained to be primarily helical in all environments. While a helical conformation is appropriate in high concentrations of TFE and in the presence of DPC micelles, the percentage helicity in the presence of planar systems such as SUVs has been reported to be significantly lower (Niidome et al. 2004).
In this work, the structure of aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1 has been examined under different conditions using atomistic molecular dynamics simulation techniques. Specifically, the peptides have been simulated in pure water, in 50% by volume TFE in water, in the presence of a DPC micelle, in the presence of a POPC bilayer containing a preformed pore and in the presence of a fully hydrated lamellar DMPC bilayer. The primary aim was to investigate the effect of environment on the structure of the peptide at an atomic level and to investigate the effect of membrane curvature on structure and binding.
Methods
Simulation details
All simulations were performed under periodic conditions using the GROMACS simulation program version 3.3.3 (van der Spoel et al. 2005). The GROMOS 54a7 force field (Poger et al. 2010) was used to describe the peptide and lipids (Oostenbrink et al. 2004). The SPC (simple point charge) water model was used to describe the solvent water (Berendsen et al. 1981). The force field proposed by Fioroni et al. was used to describe TFE (Fioroni et al. 2000). Both the temperature and the pressure of the system were maintained close to the reference values using the Berendsen weak coupling method (Berendsen et al. 1984). Time constants of 0.1 and 1.0 ps were used for the temperature and pressure coupling, respectively. The LINCS (Hess et al. 1997) algorithm was used to constrain the length of all bonds within the lipids. The geometry of water was constrained using SETTLE (Miyamoto and Kollman 1992). To increase the time scale that could be simulated, polar hydrogens on the peptide that are normally treated explicitly in the force field were replaced by dummy atoms, the positions of which were calculated based on the heavy atoms to which they are attached (Feenstra et al. 1999). The masses of the hydrogen atoms that could not be treated as dummy atoms (the hydrogen atom in the –OH group of serine and in the –SH group of cysteine) were increased by 3 amu, and the masses of the heavy atoms to which those hydrogen atoms attached were decreased by 3 amu, so that the total mass of the systems was not changed. This eliminates high frequency librations and vibrations in the system, allowing a time step of 4 fs to be used to integrate the equations of motion without affecting thermodynamic properties of the system significantly (Anézo et al. 2003; Feenstra et al. 1999). Nonbonded interactions were evaluated using a twin-range cutoff scheme. Interactions within the short-range cutoff of 0.8 nm were calculated every time step, while interactions between 0.8 and 1.4 nm were updated every two steps together with the pair list. A reaction field correction (Tironi et al. 1995) was applied to correct for the truncation of the electrostatic interactions beyond 1.4 nm. The relative dielectric permittivity of the medium (mainly the SPC water) outside the cutoff of 1.4 nm was set to 62 (Heinz et al. 2001).
Initial structures
The initial structure for aurein 1.2 was taken from the Protein Data Bank (PDB ID 1VM5). The initial structures for citropin 1.1, maculatin 1.1 and caerin 1.1 were obtained using the ROBETTA protein structure prediction server (http://robetta.bakerlab.org/). Five systems were constructed for each of the four peptides. In the first system the peptide was placed in a periodic box (5.0 nm cube) and solvated with SPC water. In each case the distance between the peptide and the box wall was at least 1.4 nm. In the second system each peptide was placed in a periodic box (6.3 nm cube) and solvated with a 20 mol% solution of TFE in water (equivalent to 50% v/v TFE) and similar to the concentration used experimentally (Chia et al. 2000; Rozek et al. 2000; Wegener et al. 1999; Wong et al. 1997). In the third system, each peptide was placed in a periodic box (8.0 nm cube) containing a DPC micelle composed of 56 DPC molecules in water. The DPC micelle was generated by simulating a random mixture of 100 DPC lipids in a box of 15,000 SPC water molecules. The micelle formed spontaneously within about 20 ns. In the fourth system, each peptide was placed 1.0–1.5 nm above an equilibrated POPC bilayer in which a transmembrane pore had been created. The pore was formed by removing all the lipids within a radius of 3.0 nm of a chosen lipid from an equilibrated POPC bilayer containing 256 lipids/leaflet (12.0 × 12.0 nm2). The system was then equilibrated under constant area conditions to allow the head groups of the lipids surrounding the hole to reorientate, forming a toroidal pore. The final system contained a total of 315 lipids (approximately 157 lipids/leaflet). In the final system, the peptide was placed 1.0–2.0 nm above a fully hydrated DMPC bilayer (64 lipids/leaflet). Note, the minimum system required in the case of a membrane containing a pore is by necessity significantly larger than that required to model a planar membrane. The different system sizes were chosen for computational efficiency. In the present study, the DMPC bilayer was used as a flat bilayer model because a range of experimental data for the peptides is available for DMPC. The POPC bilayer for a toroidal pore model was used mainly for comparison with other peptides being studied in the same POPC bilayer. Both POPC and DMPC bilayers are widely used as model membranes. They share the same phosphocholine head group and are expected to bind the peptides in a similar manner. The peptides disrupt both DMPC and POPC bilayers, and thus the conclusions of the present study are not likely to be biased by the fact that two different bilayers were used.
The protonation state of titratable groups within the peptide was chosen as appropriate for pH 7. In order to achieve overall neutrality, 1 to 4 Cl− ions were added to each system. The simulation temperature was 303 K for all systems. The pressure coupling was isotropic in all cases not involving a bilayer. The POPC bilayer containing the pore was only allowed to relax along the direction normal to the bilayer (constant area). The pressure coupling was anisotropic in the case of the DMPC bilayer. For each system, at least two simulations starting from different random initial velocities were performed. The time scale of each simulation was in the range of 150–200 ns.
Data analysis
The secondary structure for each peptide was assigned based on the DSSP criteria (Kabsch and Sander 1983). The fraction of helix in a peptide was calculated as the number of amino acids adopting a helical conformation (including α-helix, 310-helix and π-helix) divided by the total number of residues in the peptide. The C-terminal NH2 group was not considered to be a residue. To validate the results of the simulations, the upper bound distance between specific pairs of hydrogens as inferred from nuclear Overhauser effect (NOE) NMR intensities was compared to average distances observed during the simulations. The average distance between each pair of hydrogens was calculated as <r −6>−1/6, where the angular brackets denote a time average (Zagrovic and van Gunsteren 2006). Only NOEs between hydrogen atoms that are separated by at least two amino acids were used in the comparison. A violation of the NOE upper bound (v ij ) for a hydrogen atom pair i and j was calculated as:
where NOE ij denotes the corresponding NOE upper bound for the hydrogen pair ij derived from the NMR spectrum (Zagrovic and van Gunsteren 2006). The upper bound distances derived based on the experimental NOE intensities were taken from references (Rozek et al. 2000; Wegener et al. 1999, 2003). Graphic images were produced using VMD (Humphrey et al. 1996).
Results and discussion
Water and 50% v/v trifluoroethanol
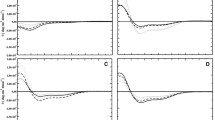
Figure 1 shows the fraction of helical residues for each of the four peptides simulated in different environments as a function of time. The peptides were simulated initially in both water and 50% v/v TFE to validate the simulation model. Figure 1 panels A1–D1 show the helix fraction in water. Although all four peptides were initially placed in an α-helical conformation, almost all traces of the original structure were lost within 200 ns. Only in the case of citropin 1.1 did the molecule remain partially helical with approximately 20% of residues remaining helical. The evolution of the secondary structure as a function of time in the case of citropin 1.1 and maculatin 1.1 is illustrated in Figs. 2 and 3, respectively. As can be seen in Fig. 2a for citropin 1.1, all traces of helicity were lost within 50 ns in the simulation shown. Nevertheless, the N-terminal residues again adopt a helical conformation after 130 ns. All the peptides sampled a wide range of conformations during the simulations, consistent with the experimental observation that all four peptides are unstructured in solution (Chia et al. 2000; Rozek et al. 2000; Wegener et al. 1999; Wong et al. 1997). In contrast, in the presence of 50% v/v TFE the peptides remained primarily helical (Fig. 1A2–D2). Aurein 1.2 and maculatin 1.1 remained over 60% helical. In the case of citropin 1.1 and caerin 1.1, the fraction of helix varied during or between the simulations, falling to 30% in the case of citropin 1.1 and to 20% for one of the simulations of caerin 1.1. Again the results closely match experimental studies that suggest that the peptides are primarily helical under these conditions (Chia et al. 2000; Rozek et al. 2000; Wegener et al. 1999; Wong et al. 1997). For example, Niidome et al. estimated maculatin 1.1 to be 43% helical in 98% TFE/water at 298 K (Niidome et al. 2004). Upper bound inter-proton distances derived from experimental NOE intensities in TFE for aurein 1.2 and citropin 1.1 were also compared to average distances extracted from the simulations. In the case of aurein 1.2, only 2 of the 31 long-range NOEs were violated by >0.1 nm and none by >0.2 nm. For citropin 1.1, 6 of the 33 long-range NOEs were violated by between 0.1 and 0.2 nm and 3 by >0.2 nm. The maximum violation was 0.36 nm between hydrogens located in the side chains of the residues 13 and 16 in the unstructured C-terminal region of the peptide (see Fig. 2b).

The helix fraction (HelixF) as a function of simulation time for the peptides aurein 1.2 (a), citropin 1.1 (b), maculatin 1.1 (c) and caerin 1.1 (d) in different environments. The environments were: water (1), 50% v/v trifluroethanol (TFE) (2), in the presence of a DPC micelle (3), in the presence of a POPC bilayer containing a pore (4) and in the presence of a planar DMPC bilayer (5). The triangles, circles and stars denote three independent simulations

The secondary structure of citropin 1.1 as a function of simulation time in: water (a), TFE (b), bound to a DPC micelle (c), bound to a POPC bilayers containing a pore (d) and bound to a planar DMPC bilayer (e). The structure on the right is the final conformation of the peptide after 200 ns of simulation in the corresponding environment

The secondary structure of maculatin 1.1 as a function of simulation time in: water (a), TFE (b), bound to a DPC micelle (c), bound to a POPC bilayers containing a pore (d) and bound to a planar DMPC bilayer (e). The structure on the right is the final conformation of the peptide after 200 ns of simulation in the corresponding environment
Binding to a DPC micelle
Having verified that the simulations could reproduce the differences in the structure of the peptides in water and 50% v/v TFE observed experimentally, the four peptides were then simulated in the presence of a DPC micelle containing 56 lipids. In each case the peptide was placed randomly in the simulation box and allowed to bind spontaneously to the micelle. The fraction of helix is shown in Fig. 1 panels A3–D3. As has been found experimentally the degree of helicity in the presence of a micelle was very similar to that observed in TFE. Both aurein 1.2 and citropin 1.1 were between 50 to 60% helical in two of the three simulations performed. As can be seen from Fig. 2c, the N-terminal region of the peptide, which is the most conserved region between the four peptides, is the most helical. Maculatin 1.1 was found to be even more helical when bound to the micelle than in 50% v/v TFE. In fact, in one of the two simulations maculatin 1.1 is over 80% α-helical. From the final structure shown on the right hand side of Fig. 3c, it can also be seen that maculatin 1.1 has lost the disordered region in what would be the helical turn preceding Pro15, and the peptide, while still bent, forms a continuous helix. This is in good agreement with the results of Chia et al., who using NMR have shown that maculatin 1.1 bound to DPC micelles is fully helical (Chia et al. 2000). Figure 4a and b shows the structure of citropin 1.1 and maculatin 1.1, respectively, bound to a DPC micelle after 200 ns of simulation. As can be seen in both cases, the peptide has not only become partially buried within the micelle with several lipids lying over the top of the peptide, but extends across the width of the micelle. In the case of citropin 1.1, this causes the peptide to partially unfold at the C-terminus. However, in the case of maculatin 1.1, the length and degree of curvature in the peptide closely match the width and degree of curvature of the micelle helping stabilize the peptide in a fully helical conformation. In contrast to the other peptides, caerin 1.1 was found to have a comparatively low fraction of helix when bound to the DPC micelle in the simulation. This was unexpected as NMR studies had suggested that, like maculatin 1.1, helix formation in caerin 1.1 was promoted by binding to a DPC micelle. Ten out of 43 NOE-derived distances for caerin 1.1 bound to a DPC micelle were violated in the simulations by >0.2 nm. However, all violations >0.2 nm involved residues 12 to 17, which correspond to the residues preceding Pro15 and Pro19, which due to the presence of the proline residues cannot readily form a helical structure. The maximum violation was 0.53 nm involving hydrogens from residues 13 and 16. While it is possible that the violations in this region simply reflect insufficient sampling, the large difference between the results obtained for maculatin and caerin observed in multiple simulations suggests that the failure of caerin to fold in this case reflects the fact that the DPC micelle used in these simulations was not an appropriate size for caerin 1.1 to adopt a helical conformation; specifically, the width of the micelle is too small to accommodate a fully helical peptide. Note, under experimental conditions, the size of the micelle would in part be determined by the interaction of the lipids with the peptide. It should also be noted that although the overall structure is similar in TFE and when bound to a DPC micelle, the factors that determine the structure of the peptide in these two environments differ significantly. TFE is not a substitute for a membrane environment.

The structures of selected peptides in specific lipid environments after 200 ns of simulation: citropin 1.1 bound to a DPC micelle (a), maculatin 1.1 bound to a DPC micelle (b); citropin 1.1 bound to a toroidal pore within a POPC bilayer (c); caerin 1.1 bound to a toroidal pore within a POPC bilayer (d). The peptide backbone is shown in magenta (cartoon representation); the molecular surface of hydrophobic and hydrophilic residues is shown in orange and blue, respectively. Lipid head groups are in blue and lipid tails in grey. In a and b the tails of lipids that cover the peptide are highlighted in orange
Binding to a curved or planar bilayer
The final two environments considered in this study were a highly curved membrane and a planar membrane. The aim was to determine if the peptides bound preferentially to regions of positive curvature and whether the structure of the peptide was dependent on the degree of local curvature. To generate a system with high positive curvature a hole was created within a POPC bilayer and the system equilibrated while maintaining the area of the membrane constant in order to form a toroidal shaped pore. Note, although in these simulations the pore was generated artificially, such pores are believed to form spontaneously in lipid bilayers and to remain metastable on time scales of milliseconds or longer (Heimburg 2010). The planar membrane was an equilibrated DMPC bilayer without a pore. The peptide was placed in a random orientation at 1.5 nm above the membrane and allowed to bind spontaneously. Figure 1 panels A4–D4 and panels A5–D5 show the fraction helicity for the four peptides in the presence of a pore or a planar membrane, respectively. Although there is considerable variability between the individual simulations, the peptides are on average significantly more helical in the presence of the pore than when bound to the planar surface. The effect is the least evident in the case of aurein 1.2, which remained helical in two of the three simulations when bound to the planar membrane. Citropin 1.1 showed increased helicity when bound to a region of high curvature associated with the pore (Fig. 1B4), but a rapid loss of helicity when bound to the planar membrane (Fig. 1B5). As is evident from Fig. 2e, the binding to the planar membrane is in fact associated with a complete loss of secondary structure. In contrast, as seen in Fig. 4c, when bound within the pore, the peptide is able to adopt a curved helical structure in which the hydrophobic side chains shown in orange are able to embed between the lipid head groups that line the pore. Note, in all cases the peptides lie at an angle to the plane of the membrane binding toward the rim of the pore and embedded only into the head group region. The peptides did not penetrate into the core of the membrane as some studies have proposed (Bond et al. 2008).
In the case of maculatin 1.1, three simulations were performed in the presence of the POPC bilayer containing a pore. In two of these simulations, the peptide bound to a region of high curvature near the middle of the pore, while in the third it bound to a planar region of the membrane. As can be seen from Fig. 1C4, in the two cases where the peptide bound to the pore, it remained helical, whereas in the case where it bound to the planar region of the membrane, it unfolded. Three simulations were also performed in the presence of a planar membrane. In one the peptide completely unfolded within 50 ns. In the other two the peptide remained between 20 and 40% helical. However, as is evident from Fig. 3e, the value for the overall helicity masks a high degree of fluctuation within the structure. Caerin 1.1 also shows a higher degree of helicity in the presence of the pore compared to when it is bound to a planar membrane, although the effect is less pronounced than for the other peptides. In two of the three simulations in the presence of the pore, the peptide remains approximately 30% helical (Fig. 1D4). In the third simulation all secondary structure was lost within 50 ns. In both simulations where the peptide bound to a planar membrane, all helicity was lost within 100 ns. Figure 4d shows the final configuration after 180 ns of one of the simulations of caerin 1.1 bound to the POPC membrane containing a pore. As can be seen, the N-terminal helical region bound toward the rim of the pore at a similar angle and in a similar position to that adopted by citropin 1.1 (Fig. 4c). However, being longer, caerin 1.1 was able to penetrate through the pore, and the C-terminal region, which shows some helical character, is interacting with the rim of the pore on the opposite side of the membrane.
Conclusions
In this work, molecular dynamics simulations have been used to investigate the structure of four selected antimicrobial peptides, namely aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1, in solution as well as in a variety of membrane environments. In line with the experiment, the simulations predict that the peptides are unstructured in aqueous solution, but are predominantly α-helical in 50% (v/v) TFE. The simulations also predict the peptides have a strong propensity to form helical structures when bound to an appropriately sized DPC micelle, again in line with the experiment (Chia et al. 2000; Marcotte et al. 2003; Niidome et al. 2004; Rozek et al. 2000; Wegener et al. 1999, 2003; Wong et al. 1997). This suggests that the simulation protocol is capable of reproducing the structural properties of the peptide in different environments. Simulations of the peptides in the presence of a bilayer containing a transmembrane pore suggest that the peptides have a strong preference to bind to regions of high curvature. In fact, the simulations show that dependent on length, the peptides are not stable in an α-helical conformation when bound to a planar membrane. Overall the simulations suggest that the shorter peptides aurein 1.2 and citropin 1.1 act via a detergent-like mechanism because they can induce high local, but not long-range curvature, whereas the longer peptides maculatin 1.1 and caerin 1.1 require longer range curvature to fold and thus bind to and stabilize transmembrane pores. This said, the simulation studies described in this work have involved relatively low peptide to lipid ratios, whereas aurein 1.2, citropin 1.1, maculatin 1.1 and caerin 1.1 are all believed to be most active at high peptide to lipid ratios (close to saturation). The effect of high peptide concentrations and the relationship between the elastic properties of the membrane and peptide activity are the focus of ongoing work.
Abbreviations
- AMP:
-
Antimicrobial peptide
- CD:
-
Circular dichroism
- DMPC:
-
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DPC:
-
Dodecylphosphocholine
- GUV:
-
Giant unilamellar vesicle
- NOE:
-
Nuclear Overhauser effect
- POPC:
-
2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine
- SPC:
-
Simple point charge
- SUV:
-
Small unilamellar vesicle
- TFE:
-
2,2,2-trifluoroethanol
References
Allende D, Simon SA, McIntosh TJ (2005) Melittin-induced bilayer leakage depends on lipid material properties: evidence for toroidal pores. Biophys J 88:1828–1837
Almeida PF, Pokorny A (2009) Mechanism of antimicrobial, cytolytic, and cell-penetrating peptides: from kinetics to thermodynamics. Biochemistry 48:8083–8093
Ambroggio EE, Separovic F, Bowie JH, Fidelio GD, Bagatolli LA (2005) Direct visualization of membrane leakage induced by the antibiotic peptides: maculatin, citropin, and aurein. Biophys J 89:1874–1881
Anézo C, de Vries AH, Höltje HD, Tieleman DP, Marrink S-J (2003) Methodological issues in lipid bilayer simulations. J Phys Chem B 107:9424–9433
Apponyi MA, Pukala TL, Brinkworth CS, Maselli VM, Bowie JH, Tyler MJ, Booker GW, Wallace JC, Carver JA, Separovic F, Doyle J, Llewellyn LE (2004) Host-defence peptides of Australian anurans: structure, mechanism of action and evolutionary significance. Peptides 25:1035–1054
Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. In: Pullman B (ed) Intermolecular forces. Dordrecht, The Netherlands, pp 331–342
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690
Boland MP, Separovic F (2006) Membrane interactions of antimicrobial peptides from Australian tree frogs. Biochim Biophys Acta 1758:1178–1183
Bond PJ, Parton DL, Clark JF, Sansom MS (2008) Coarse-grained simulations of the membrane-active antimicrobial peptide maculatin 1.1. Biophys J 95:3802–3815
Castro MS, Fontes W (2005) Plant defense and antimicrobial peptides. Protein Pept Lett 12:11–16
Chia BC, Carver JA, Mulhern TD, Bowie JH (2000) Maculatin 1.1, an anti-microbial peptide from the Australian tree frog, Litoria genimaculata. Solution structure and biological activity. Eur J Biochem 267:1894–1908
Chia CSB, Torres J, Cooper MA, Arkin IT, Bowie JH (2002) The orientation of the antibiotic peptide maculatin 1.1 in DMPG and DMPC lipid bilayers. Support for a pore-forming mechanism. FEBS Lett 512:47–51
Feenstra KA, Hess B, Berendsen HJC (1999) Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J Comput Chem 20:786–798
Fioroni M, Burger K, Mark AE, Roccatano D (2000) A new 2, 2, 2-trifluoroethanol model for molecular dynamics simulations. J Phys Chem B 104:12347–12354
Gehman JD, Luc F, Hall K, Lee TH, Boland MP, Pukala TL, Bowie JH, Aguilar MI, Separovic F (2008) Effect of antimicrobial peptides from Australian tree frogs on anionic phospholipid membranes. Biochemistry 47:8557–8565
Hancock REW, Sahl HG (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 12:1551–1557
Hatzakis NS, Bhatia VK, Larsen J, Madsen KL, Bolinger PY, Kunding AH, Castillo J, Gether U, Hedegård P, Stamou D (2009) How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat Chem Biol 5:835–841
Heimburg T (2010) Lipid ion channels. Biophys Chem 150:2–22
Heinz TN, van Gunsteren WF, Hünenberger PH (2001) Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J Chem Phys 115:1125–1136
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Lee MT, Hung WC, Chen FY, Huang HW (2005) Many-body effect of antimicrobial peptides: on the correlation between lipid’s spontaneous curvature and pore formation. Biophys J 89:4006–4016
Leontiadou H, Mark AE, Marrink S-J (2006) Antimicrobial peptides in action. J Am Chem Soc 128:12156–12161
Marcotte I, Wegener KL, Lam YH, Chia BC, de Planque MR, Bowie JH, Auger M, Separovic F (2003) Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem Phys Lipids 122:107–120
Matsuzaki K, Sugishita K, Ishibe N, Ueha M, Nakata S, Miyajima K, Epand RM (1998) Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 37:11856–11863
Mihajlovic M, Lazaridis T (2010) Antimicrobial peptides bind more strongly to membrane pores. Biochim Biophys Acta 1798:1494–1502
Miyamoto S, Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13:952–962
Niidome T, Kobayashi K, Arakawa H, Hatakeyama T, Aoyagi H (2004) Structure-activity relationship of an antibacterial peptide, maculatin 1.1, from the skin glands of the tree frog, Litoria genimaculata. J Pept Sci 10:414–422
Oostenbrink C, Villa A, Mark AE, van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25:1656–1676
Poger D, van Gunsteren WF, Mark AE (2010) A new force field for simulating phosphatidylcholine bilayers. J Comput Chem 31:1117–1125
Rozek T, Wegener KL, Bowie JH, Olver IN, Carver JA, Wallace JC, Tyler MJ (2000) The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis. The solution structure of aurein 1.2. Eur J Biochem 267:5330–5341
Rzepiela AJ, Sengupta D, Goga N, Marrink SJ (2010) Membrane poration by antimicrobial peptides combining atomistic and coarse-grained descriptions. Faraday Discuss 144:431–443
Scholtz JM, Qian H, York EJ, Stewart JM, Baldwin RL (1991) Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers 31:1463–1470
Sengupta D, Leontiadou H, Mark AE, Marrink S-J (2008) Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim Biophys Acta 1778:2308–2317
Tironi IG, Sperb R, Smith PE, van Gunsteren WF (1995) A generalized reaction field method for molecular dynamics simulations. J Chem Phys 102:5451–5459
van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718
Wegener KL, Wabnitz PA, Carver JA, Bowie JH, Chia BC, Wallace JC, Tyler MJ (1999) Host defence peptides from the skin glands of the Australian blue mountains tree-frog Litoria citropa. Solution structure of the antibacterial peptide citropin 1.1. Eur J Biochem 265:627–637
Wegener KL, Carver JA, Bowie JH (2003) The solution structures and activity of caerin 1.1 and caerin 1.4 in aqueous trifluoroethanol and dodecylphosphocholine micelles. Biopolymers 69:42–59
Wong H, Bowie JH, Carver JA (1997) The solution structure and activity of caerin 1.1, an antimicrobial peptide from the Australian green tree frog, Litoria splendida. Eur J Biochem 247:545–557
Zagrovic B, van Gunsteren WF (2006) Comparing atomistic simulation data with the NMR experiment: how much can NOEs actually tell us? Proteins 63:210–218
Acknowledgments
This study was in part supported by the award of an Australian Research Council (ARC) Federation Fellowship to A.E.M. R.C. is a recipient of a PhD tuition fee waiver scholarship from The University of Queensland. The access to high-performance computing resources of The University of Queensland and the National Computational Infrastructure (NCI) National Facility is gratefully acknowledged. We would also like to thank F. Separovic and M.I. Aguilar for helpful discussions, and K. Wegener, J.H. Bowie and F. Separovic for providing NMR data.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Membrane-active peptides: 455th WE-Heraeus-Seminar and AMP 2010 Workshop.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chen, R., Mark, A.E. The effect of membrane curvature on the conformation of antimicrobial peptides: implications for binding and the mechanism of action. Eur Biophys J 40, 545–553 (2011). https://doi.org/10.1007/s00249-011-0677-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-011-0677-4