
Abstract
Previous studies of acetate-promoted bioremediation of uranium-contaminated aquifers focused on Geobacter because no other microorganisms that can couple the oxidation of acetate with U(VI) reduction had been detected in situ. Monitoring the levels of methyl CoM reductase subunit A (mcrA) transcripts during an acetate-injection field experiment demonstrated that acetoclastic methanogens from the genus Methanosarcina were enriched after 40 days of acetate amendment. The increased abundance of Methanosarcina corresponded with an accumulation of methane in the groundwater. In order to determine whether Methanosarcina species could be participating in U(VI) reduction in the subsurface, cell suspensions of Methanosarcina barkeri were incubated in the presence of U(VI) with acetate provided as the electron donor. U(VI) was reduced by metabolically active M. barkeri cells; however, no U(VI) reduction was observed in inactive controls. These results demonstrate that Methanosarcina species could play an important role in the long-term bioremediation of uranium-contaminated aquifers after depletion of Fe(III) oxides limits the growth of Geobacter species. The results also suggest that Methanosarcina have the potential to influence uranium geochemistry in a diversity of anaerobic sedimentary environments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Injection of acetate into the groundwater of uranium-contaminated aquifers has been shown to be an effective way to stimulate microbially mediated reductive precipitation of soluble U(VI) to poorly soluble U(IV) [1,2,3]. A wide diversity of microorganisms are capable of U(VI) reduction [4,5,6,7,8,9] but only Geobacter species have been shown to reduce U(VI) with acetate as an electron donor. Although growth with acetate as the electron donor and U(VI) as the electron acceptor is possible [4], the low concentrations of U(VI), even in heavily contaminated subsurface environments requires that microbes use other forms of respiration as their primary means of energy conservation [10]. Geobacter species grow rapidly in the initial phases of subsurface uranium bioremediation with added acetate because Fe(III) oxides are typically abundant in subsurface environments [1, 11,12,13,14] and Geobacter species outcompete other Fe(III) reducers under conditions of high acetate availability [15, 16]. However, the potential for other microorganisms to contribute to acetate oxidation coupled to U(VI) reduction, especially after the Fe(III) oxides that support Geobacter growth are depleted, has not been intensively investigated. Sulfate reducers that can reduce U(VI) have been identified, but none of these are known to use acetate as an electron donor [5, 7, 9, 17]. Furthermore, relying on sulfate reducers to reduce U(VI) may not be a good long-term strategy because acetate additions can rapidly deplete sulfate from groundwater [18,19,20].
Unlike Fe(III)- and sulfate-reducers, methanogens can thrive for long periods of time in organic-rich environments without external inputs of electron acceptors because they can conserve energy either from acetate dismutation or from the reduction of carbon dioxide, an electron acceptor generated by fermentation in their environment. If methanogens were capable of U(VI) reduction then this would make long-term in situ bioremediation of U(VI) a more attractive practice. To our knowledge, U(VI) reduction by methanogens has not been previously described. Previous studies have shown that methanogens can transfer electrons to various Fe (III) forms [21,22,23,24,25,26], as well as vanadate [27], molecular sulfur [28] and quinones [22, 29]. However, acetate has not been shown to serve as an electron donor for these processes.
Evidence for methane production in response to acetate amendments during in situ uranium bioremediation [30] led us to investigate the potential for methanogens to further contribute to uranium bioremediation. The results suggest that Methanosarcina species that can couple the oxidation of acetate to the reduction of U(VI) might aid in the bioremediation process.
Materials and Methods
Description of Sampling Site
The Rifle 24-acre experimental site is located close to the Colorado River, on the premises of an earlier uranium ore processing facility. Uranium concentrations in the water table of the Rifle aquifer are 2–8 times higher than the drinking water contamination limit (0.126 μM) established by the uranium mill tailings remedial action (UMTRA). A detailed review of geochemical characteristics of the site has already been published [31] and in situ bioremediation of U(VI) has been intensely studied at this site [1,2,3]. Similar to previous years, acetate was injected into the subsurface at a concentration of ~ 15 mM between August and October, 2011 and monitored from six different wells [32]. Groundwater and sediments for this study were collected from well CD-01 (a down gradient well) and a background well (CU-01) that never received any acetate additions.
Nucleic Acid Extraction and cDNA Preparation
For nucleic acid extraction, it was first necessary to concentrate 50 L of groundwater by impact filtration on 293 mm diameter Supor membrane disc filters with pore sizes of 1.2 and 0.2 μm (Pall Life Sciences). All filters were placed into whirl-pack bags, flash frozen in a dry ice/ethanol bath, and shipped on dry ice back to the laboratory where they were stored at – 80 °C. RNA was extracted from the filters using a modified phenol–chloroform method, as previously described [12]. DNA was extracted from the filters with the FastDNA SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA) according to the manufacturer’s instructions.
Extracted RNA and DNA were quantified with a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and stored at – 80 °C until further analyses. A DuraScript enhanced avian RT single-strand synthesis kit (Sigma, Sigma-Aldrich, St Louis, MO, USA) was used to generate cDNA from RNA, as previously described [32]. In order to ensure that RNA samples were not contaminated with DNA, PCR with primers targeting the 16S rRNA gene was conducted on RNA samples that had not undergone reverse transcription.
PCR Amplification Parameters and Microbial Community Analysis
For clone library construction, fragments from the mcrA gene which codes for the large subunit of methyl CoM reductase and the 16S rRNA gene were amplified from cDNA with mcrAf/mcrAr primers [33] and with 344f/915r [34] (ESM 1: Supplementary Table S1). Amplicons were ligated into the pCR-TOPO2.1 TA cloning vector according to the manufacturer’s instructions (Invitrogen, the Netherlands). Inserts from the recombinant clones were directly amplified by PCR with M13 primers, purified and sequenced at the University of Massachusetts sequencing facility.
Quantification of Methanosarcina mcrA Transcript Abundance
The quantitative PCR primer set (msa_mcrA173f/271r) targeted mcrA genes from Methanosarcina species found in the Rifle subsurface and was designed according to the manufacturer’s specifications (Applied Biosystems) (ESM 1: Supplementary Table S1). Quantitative PCR amplification and detection was performed with the 7500 Real Time System (Applied Biosystems) using cDNA made by reverse transcription from total RNA extracted from groundwater collected during the bioremediation experiment. Each reaction mixture consisted of a total volume of 25 μl and contained 1.5 μl of the appropriate primers (stock concentration 1.5 μM), 5 ng cDNA, and 12.5 μl Power SYBR Green PCR Master Mix (Applied Biosystems). All qPCR experiments followed MIQE guidelines [35] and qPCR efficiencies were 98%. Optimal thermal cycling parameters consisted of an activation step at 50 °C for 2 min, an initial 10 min denaturation step at 95 °C, and 50 cycles of 95 °C for 15 s and 60 °C for 1 min. A dissociation curve generated by increasing the temperature from 58 to 95 °C at a ramp rate of 2% showed that the PCR amplification process yielded a single predominant peak, further supporting the specificity of the qPCR primer pair.
Phylogenetic Analysis
mcrA gene sequences were compared to Genbank nucleotide and protein databases with the BLASTn and BLASTx algorithms [36, 37]. Alignments were generated with MAFFT [38] and PRANK [39] algorithms. The phylogenetic tree was inferred with the Maximum Likelihood method using MEGA7 software [40]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (100 replicates) is shown next to the branches [41]. All positions with less than 95% coverage were eliminated and a total of 117 positions were considered in the final dataset.
Nucleotide sequences of mcrA genes used for phylogenetic analyses have been deposited in the Genbank database under accession numbers MF616623-MF616647.
U(VI) Reduction Studies
Methanosarcina barkeri (DSM 800) was selected for U(VI) reduction studies because sequences most similar to this strain were significant members of the Methanosarcina community (37% of the sequences). Although M. barkeri was isolated from an anaerobic sewage digester [42], it grows in freshwater medium and can utilize acetate as a substrate for methanogenesis, similar to methanogens likely to be enriched from the acetate-amended Rifle aquifer. In addition, the majority of studies examining reduction of extracellular electron acceptors by Methanosarcina have focused on M. barkeri [22,23,24,25].
Batch cultures of 500-mL M. barkeri were grown under strictly anaerobic conditions [42] on modified DSMZ medium 120 [43] with acetate (40 mM) as substrate, and incubated at 37 °C for ~ 3 weeks. Cultures were harvested when they reached an optical density at 600 nm of 0.19. All cell suspension preparations were performed in an anaerobic chamber to minimize oxygen exposure. Cells were pelleted by centrifugation for 10 min at 4000 × g in a Sorval RC 5B Plus centrifuge. These pellets were then washed twice in anoxic phosphate depleted buffer (PDB), which consisted of the following salts 0.2 g/L MgSO4 × 7H2O, 0.025 g/L CaCl2 × 2H2O, 1 g/L NaCl, and 2 g/L NaHCO3. Cell pellets were then resuspended in 10-mL anoxic PDB to a cell density of ~ 0.4–0.5 at 600 nm. To generate heat-killed cells, 3 mL of this suspension was autoclaved at 122 °C for 30 min. Six replicates were prepared by diluting 1 mL of the cell-suspension in 9-mL PDB buffer. For the heat-killed incubation, 1-mL autoclaved cell suspension was diluted in 9-mL PDB buffer. Sulfide (0.5 mM) was added to all inoculated tubes to ensure anoxic conditions. Acetate (40 mM) was also added to the tubes to fuel methanogenic activity. Triplicate live cell suspensions (active cells) and triplicate heat-killed controls (heat-killed cells) were incubated at 37 °C. The other three live cell suspensions were incubated at 4 °C (inactive cells). All cell suspensions were incubated with 0.2 mM U6+ prepared from a stock of uranyl-acetate (5 mM). Cell densities were determined with a bench top spectrophotometer, by absorbance measurements at 600 nm with mili-Q water as a blank.
The ability of Methanosarcina barkeri to reduce U(VI) was verified with U(VI) depletion measurements carried out on different cell suspensions over the course of 24 h. Samples (0.1 mL) were retrieved anaerobically and diluted in 14.9 mL anoxic bicarbonate (100 mM) and 14.9 mL Uraplex solution. Concentrations of U(VI) were then measured with a kinetic phosphorescence analyzer, as previously described [44].
Chemical Analyses
Groundwater samples for geochemical analyses were collected after purging 12 L of groundwater from the wells with a peristaltic pump. The phenanthroline method [45] was used to determine ferrous iron concentrations. Sulfate and thiosulfate concentrations were measured with an ion chromatograph (ICS-2100, Dionex, CA) equipped with an AS18 column under isocratic elution with 32 mM KOH as the eluent. Acetate concentrations were determined with a high performance liquid chromatograph equipped with an ion exclusion HPX-87H column (Biorad, Hercules, CA) using 8 mM sulfuric acid as eluent. In situ methane production was monitored as previously described [30]. Methane in the headspace of sediment/groundwater incubations was measured as previously described [43] using a gas chromatograph with a flame ionization detector (Shimadzu, GC-8A).
Results and Discussion
Evidence for Acetoclastic Methanogenic Activity During Acetate Amendments
Methanogens that utilize acetate are restricted to the order Methanosarcinales [46]. In order to determine whether the addition of acetate could promote the growth of acetoclastic methanogens in a uranium-contaminated aquifer, the activity of Methanosarcinales was investigated by monitoring Methanosarcina mcrA gene transcript abundance. Before day 39, fewer than two Methanosarcina mcrA mRNA transcripts were detected per mcrA gene copy number in the groundwater (Fig. 1a). However, by day 46, Methanosarcina mcrA transcripts increased by four orders of magnitude to 1.7 × 104 transcripts per gene copy. This increase in Methanosarcina coincided with a steep decline in groundwater sulfate concentrations (Fig. 1b). Although sulfate reducers and methanogens compete for acetate [47, 48], high concentrations of acetate in the groundwater (Fig. 1c) made it unlikely that growth of Methanosarcinales in the subsurface was being restricted by competition for acetate.

The injection of acetate into a uranium-contaminated aquifer, triggered acetate utilization coupled with iron reduction, sulfate reduction, and methanogenesis. a Quantitative RT-PCR of Methanosarcina mcrA mRNA transcripts normalized against Methanosarcina mcrA gene copy numbers recovered in the groundwater over the course of 100 days. b Concentrations of hydrogen sulfide (μM) and sulfate (mM) detected in the groundwater. c Concentrations of acetate (mM), Fe(II) (μM), and U(VI) (μM) detected in the groundwater. d Proportion of mcrA sequences from various methanogenic families found in cDNA clone libraries assembled from RNA extracted from groundwater at different points during the experiment. e Concentrations of methane and acetate in an active well (CD-01) and a background well (CU-01) on days 79, 89, and 100. For further reference to geochemical parameters and cDNA clone libraries, see Holmes et al. 2014
The increase in Methanosarcinales coincided with an increase in free sulfide in the groundwater, producing highly reducing conditions that favor the growth of methanogens. Another consideration is the slow growth rate of Methanosarcinales, which might have limited their growth after acetate injections even under the most favorable conditions. The lack of sufficient reducing conditions coupled with the slow growth rate of Methanosarcinales may explain the finding that although acetate concentrations were high during the Fe(III) reducing phase of the experiment (days 0–33) (Fig. 1c), the number of Methanosarcinales sequences stayed low until sulfate reduction became the primary subsurface metabolism (Fig. 1a, d). The increase in abundance of Methanosarcinales was later followed by a decline, which coincided with acetate limitation associated with the halt in acetate injections on day 68.
Measurements of methane concentrations in the groundwater were not initiated until day 79 (Fig. 1e). The high concentration of methane at this time demonstrated that methanogens had been active in the preceding days. Methane concentrations steeply declined over time coincident with the steep decline in acetate availability.
Phylogenetic Analysis of the In Situ Methanosarcina Community
Methanosarcinales accounted for the majority of methanogenic mcrA transcripts recovered from the groundwater on days 46 through 81 (Fig. 1d). The most abundant Methanosarcina mcrA cDNA sequences recovered from groundwater during this period clustered with M. horonobensis (48.2%) and M. barkeri (37% of the sequences) (Fig. 2). Other Methanosarcina mcrA cDNA sequences detected included sequences most similar to M. mazei (11.1% of the sequences), and M. acetivorans (3.7% of the sequences). More than half of these sequences clustered with acetoclastic Methanosarcina that are unable to use formate or hydrogen as substrates for growth (i.e., M. horonobensis and M. acetivorans) [49, 50] suggesting that they might be growing during the in situ U(VI) experiment via acetate dismutation.

Phylogenetic tree generated with the maximum likelihood algorithm comparing translated mcrA mRNA transcript sequences to McrA protein sequences from known methanogenic archaea. Bootstrap values were generated with 100 replicates and Methanobacterium formicum, Methanothermobacter marburgensis, and Methanobrevibacter ruminantium were used as outgroups
U(VI) Reduction by Metabolically Active Methanosarcina Cells
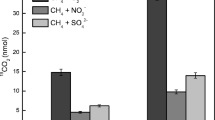
To evaluate whether Methanosarcina species might be capable of U(VI) reduction, cell suspensions of M. barkeri were incubated with acetate as the electron donor and 200-μM U(VI) as a potential electron acceptor. Within 1 day, the cells produced 1.6-mM methane while depleting 51% of the provided U(VI) (Fig. 3a). In contrast, cell suspensions incubated at 4 °C or autoclaved prior to incubation, did not produce methane or remove U(VI) (Fig. 3b, c). These results indicated that U(VI) removal could be attributed to U(VI) reduction by metabolically active cells.

Uranium U(VI) reduction by metabolically active Methanosarcina cells. Metabolically active cells which were defined as such because they were producing methane from acetate were able to convert 51% of U(VI) to U(IV) (a) whereas metabolically inactive cells kept at 4 °C in the same medium did not produce methane and also did not convert U(VI) to U(IV) (b), and neither did autoclaved cell suspensions from the same culture (c). The difference between original concentrations of U(VI) and the amount recovered in metabolically active cell suspensions after 24 h of exposure was statistically different (p = 0.0003)
Implications
Our findings that acetate additions during in situ uranium bioremediation promotes the growth of Methanosarcina and that a Methanosarcina can reduce U(VI) has important implications for the design of long-term in situ uranium bioremediation strategies. Previous interpretations of U(VI) reduction during acetate-amendment at the Rifle, Colorado study site have focused on the U(VI) reduction capacity of Geobacter species because of their prevalence at the site [1,2,3, 51,52,53,54] and because the sulfate-reducers that are enriched with acetate amendments [18, 19, 55, 56] are not likely to be effective U(VI) reducers. In fact, there has yet to be a description of an acetate-utilizing sulfate-reducing microorganism capable of U(VI) reduction. The results presented here suggest that Methanosarcina may also contribute to U(VI) reduction in the field experiments. Unlike Geobacter species, Methanosarcina do not require an external electron acceptor for acetate metabolism. Therefore, in long-term in situ uranium bioremediation, Methanosarcina may emerge as an important microbial catalyst for uranium removal.
Furthermore, microbial reduction of U(VI) may play an important role in the uranium geochemistry of a diversity of sedimentary environments [4]. Thus, the potential contribution of Methanosarcina to U(VI) reduction in anaerobic environments should be considered.
References
Anderson RT, Vrionis HA, Ortiz-Bernad I, Resch CT, Long PE, Dayvault R, Karp K, Marutzky S, Metzler DR, Peacock A, White DC, Lowe M, Lovley DR (2003) Stimulating the in situ activity of Geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer. Appl. Environ. Microbiol. 69:5884–5891. https://doi.org/10.1128/aem.69.10.5884-5891.2003
Williams KH, Long PE, Davis JA, Wilkins MJ, N'Guessan AL, Steefel CI, Yang L, Newcomer D, Spane FA, Kerkhof LJ, McGuinness L, Dayvault R, Lovley DR (2011) Acetate availability and its influence on sustainable bioremediation of uranium-contaminated groundwater. Geomicrobiol J. 28:519–539. https://doi.org/10.1080/01490451.2010.520074
Williams KH, Bargar JR, Lloyd JR, Lovley DR (2013) Bioremediation of uranium-contaminated groundwater: a systems approach to subsurface biogeochemistry. Curr. Opin. Biotechnol. 24:489–497. https://doi.org/10.1016/j.copbio.2012.10.008
Lovley DR, Phillips EJP, Gorby YA, Landa ER (1991) Microbial reduction of uranium. Nature 350:413–416. https://doi.org/10.1038/350413a0
Lovley DR, Roden EE, Phillips EJP, Woodward JC (1993) Enzymatic iron and uranium reduction by sulfate-reducing bacteria. Mar. Geol. 113:41–53. https://doi.org/10.1016/0025-3227(93)90148-O
Kashefi K, Lovley DR (2000) Reduction of Fe(III), Mn(IV), and toxic metals at 100 degrees C by Pyrobaculum islandicum. Appl Environ Microb 66:1050–1056. https://doi.org/10.1128/Aem.66.3.1050-1056.2000
Tebo BM, Obraztsova AY (1998) Sulfate-reducing bacterium grows with Cr(VI), U(VI), Mn(IV), and Fe(III) as electron acceptors. FEMS Microbiol. Lett. 162:193–198. https://doi.org/10.1111/j.1574-6968.1998.tb12998.x
Wu Q, Sanford RA, Loffler FE (2006) Uranium(VI) reduction by Anaeromyxobacter dehalogenans strain 2CP-C. Appl Environ Microb 72:3608–3614. https://doi.org/10.1128/Aem.72.5.3608-3614.2006
Wall JD, Krumholz LR (2006) Uranium reduction. Annu. Rev. Microbiol. 60:149–166. https://doi.org/10.1146/annurev.micro.59.030804.121357
Finneran KT, Anderson RT, Nevin KP, Lovley DR (2002) Potential for bioremediation of uranium-contaminated aquifers with microbial U(VI) reduction. Soil Sediment Contam 11:339–357. https://doi.org/10.1080/20025891106781
Holmes DE, Finneran KT, O'Neil RA, Lovley DR (2002) Enrichment of members of the family Geobacteraceae associated with stimulation of dissimilatory metal reduction in uranium-contaminated aquifer sediments. Appl. Environ. Microbiol. 68:2300–2306. https://doi.org/10.1128/Aem.68.5.2300-2306.2002
Holmes DE, Nevin KP, O'Neil RA, Ward JE, Adams LA, Woodard TL, Vrionis HA, Lovley DR (2005) Potential for quantifying expression of the Geobacteraceae citrate synthase gene to assess the activity of Geobacteraceae in the subsurface and on current-harvesting electrodes. Appl. Environ. Microbiol. 71:6870–6877. https://doi.org/10.1128/aem.71.11.6870-6877.2005
Holmes DE, O’Neil RA, Vrionis HA, N’Guessan LA, Ortiz-Bernad I, Larrahondo MJ, Adams LA, Ward JA, Nicoll JS, Nevin KP, Chavan MA, Johnson JP, Long PE, Lovley DR (2007) Subsurface clade of Geobacteraceae that predominates in a diversity of Fe(III)-reducing subsurface environments. ISME J 1:663–677. https://doi.org/10.1038/ismej.2007.85
Holmes DE, Giloteaux L, Barlett M, Chavan MA, Smith JA, Williams KH, Wilkins M, Long P, Lovley DR (2013) Molecular analysis of the in situ growth rates of subsurface Geobacter species. Appl. Environ. Microbiol. 79:1646–1653. https://doi.org/10.1128/AEM.03263-12
Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R, Lovley DR (2011) Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J 5:305–316. https://doi.org/10.1038/ismej.2010.117
Lovley DR, Ueki T, Zhang T, Malvankar NS, Shrestha PM, Flanagan KA, Aklujkar M, Butler JE, Giloteaux L, Rotaru AE, Holmes DE, Franks AE, Orellana R, Risso C, Nevin KP (2011) Geobacter: the microbe Electric’s physiology, ecology, and practical applications. In: Poole, RK (ed.) Adv microb Phys, pp. 1–100
Lovley DR, Phillips EJP (1992) Bioremediation of uranium contamination with enzymatic uranium reduction. Environ Sci Technol 26:2228–2234. https://doi.org/10.1021/es00035a023
Miletto M, Williams KH, N'Guessan AL, Lovley DR (2011) Molecular analysis of the metabolic rates of discrete subsurface populations of sulfate reducers. Appl. Environ. Microbiol. 77:6502–6509. https://doi.org/10.1128/aem.00576-11
Vrionis HA, Anderson RT, Ortiz-Bernad I, O'Neill KR, Resch CT, Peacock AD, Dayvault R, White DC, Long PE, Lovley DR (2005) Microbiological and geochemical heterogeneity in an in situ uranium bioremediation field site. Appl. Environ. Microbiol. 71:6308–6318. https://doi.org/10.1128/aem.71.10.6308-6318.2005
N'Guessan AL, Vrionis HA, Resch CT, Long PE, Lovley DR (2008) Sustained removal of uranium from contaminated groundwater following stimulation of dissimilatory metal reduction. Environ Sci Technol 42:2999–3004. https://doi.org/10.1021/es071960p
Vargas M, Kashefi K, Blunt-Harris EL, Lovley DR (1998) Microbiological evidence for Fe(III) reduction on early earth. Nature 395:65–67
Bond DR, Lovley DR (2002) Reduction of Fe(III) oxide by methanogens in the presence and absence of extracellular quinones. Environ. Microbiol. 4:115–124. https://doi.org/10.1046/j.1462-2920.2002.00279.x
Bodegom PM, Scholten JC, Stams AJ (2004) Direct inhibition of methanogenesis by ferric iron. FEMS Microbiol. Ecol. 49:261–268. https://doi.org/10.1016/j.femsec.2004.03.017
Sivan O, Shusta SS, Valentine DL (2016) Methanogens rapidly transition from methane production to iron reduction. Geobiology 14:190–203. https://doi.org/10.1111/gbi.12172
Liu D, Dong HL, Bishop ME, Wang HM, Agrawal A, Tritschler S, Eberl DD, Xie SC (2011) Reduction of structural Fe(III) in nontronite by methanogen Methanosarcina barkeri. Geochim Cosmochim Ac 75:1057–1071. https://doi.org/10.1016/j.gca.2010.11.009
Zhang J, Dong HL, Liu D, Fischer TB, Wang S, Huang LQ (2012) Microbial reduction of Fe(III) in illite-smectite minerals by methanogen Methanosarcina mazei. Chem. Geol. 292:35–44. https://doi.org/10.1016/j.chemgeo.2011.11.003
Zhang J, Dong HL, Zhao LD, McCarrick R, Agrawal A (2014) Microbial reduction and precipitation of vanadium by mesophilic and thermophilic methanogens. Chem. Geol. 370:29–39. https://doi.org/10.1016/j.chemgeo.2014.01.014
Stetter KO, Gaag G (1983) Reduction of molecular sulfur by methanogenic bacteria. Nature 305:309–311. https://doi.org/10.1038/305309a0
Cervantes FJ, de Bok FAM, Tuan DD, Stams AJM, Lettinga G, Field JA (2002) Reduction of humic substances by halorespiring, sulphate-reducing and methanogenic microorganisms. Environ. Microbiol. 4:51–57. https://doi.org/10.1046/j.1462-2920.2002.00258.x
Holmes DE, Giloteaux L, Orellana R, Williams KH, Robbins MJ, Lovley DR (2014) Methane production from protozoan endosymbionts following stimulation of microbial metabolism within subsurface sediments. Front. Microbiol. 5. doi: Artn 366 Doi https://doi.org/10.3389/Fmicb.2014.00366
Zachara JM, Long PE, Bargar J, Davis JA, Fox P, Fredrickson JK, Freshley MD, Konopka AE, Liu CX, McKinley JP, Rockhold ML, Williams KH, Yabusaki SB (2013) Persistence of uranium groundwater plumes: contrasting mechanisms at two DOE sites in the groundwater-river interaction zone. J. Contam. Hydrol. 147:45–72. https://doi.org/10.1016/j.jconhyd.2013.02.001
Giloteaux L, Holmes DE, Williams KH, Wrighton KC, Wilkins MJ, Montgomery AP, Smith JA, Orellana R, Thompson CA, Roper TJ, Long PE, Lovley DR (2013) Characterization and transcription of arsenic respiration and resistance genes during in situ uranium bioremediation. ISME J 7:370–383. https://doi.org/10.1038/ismej.2012.109
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiol 148:3521–3530
Casamayor EO, Massana R, Benlloch S, Ovreas L, Diez B, Goddard VJ, Gasol JM, Joint I, Rodriguez-Valera F, Pedros-Alio C (2002) Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4:338–348. https://doi.org/10.1046/J.1462-2920.2002.00297.X
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622. https://doi.org/10.1373/Clinchem.2008.112797
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J. Mol. Biol. 215:403–410. https://doi.org/10.1006/jmbi.1990.9999
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. https://doi.org/10.1093/molbev/mst010
Loytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. PNAS 102:10557–10562
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Felsenstein J (1985) Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.2307/2408678
Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS (1979) Methanogens: reevaluation of a unique biological group. Microbiol. Rev. 43:260–296
Rotaru AE, Shrestha PM, Liu F, Markovaite B, Chen S, Nevin KP, Lovley DR (2014) Direct interspecies electron transfer between Geobacter metallireducens and Methanosarcina barkeri. Appl. Environ. Microbiol. 80:4599–4605. https://doi.org/10.1128/AEM.00895-14
Orellana R, Leavitt JJ, Comolli LR, Csencsits R, Janot N, Flanagan KA, Gray AS, Leang C, Izallalen M, Mester T, Lovley DR (2013) U(VI) reduction by diverse outer surface c-type cytochromes of Geobacter sulfurreducens. Appl Environ Microb 79:6369–6374. https://doi.org/10.1128/Aem.02551-13
Muir MK, Andersen TN (1977) Determination of ferrous iron in copper-process metallurgical solutions by ortho-phenanthroline colorimetric method. Metall. Trans. B 8:517–518. https://doi.org/10.1007/Bf02696942
Kendall M, Boone D (2006) The order Methanosarcinales. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K, Stackebrandt E (eds) The prokaryotes: Vol 3: archaea bacteria: Firmicutes, Actinomycetes. Springer Science+Business Media, LLC, New York, pp 244–256
Lovley DR, Klug MJ (1983) Sulfate reducers can out-compete methanogens at fresh-water sulfate concentrations. Appl. Environ. Microbiol. 45:187–192
Oremland RS, Polcin S (1982) Methanogenesis and sulfate reduction: competitive and noncompetitive substrates in estuarine sediments. Appl. Environ. Microbiol. 44:1270–1276
Shimizu S, Upadhye R, Ishijima Y, Naganuma T (2011) Methanosarcina horonobensis sp. nov., a methanogenic archaeon isolated from a deep subsurface Miocene formation. Int. J. Syst. Evol. Microbiol. 61:2503–2507. https://doi.org/10.1099/ijs.0.028548-0
Sowers KR, Baron SF, Ferry JG (1984) Methanosarcina acetivorans sp. nov., an acetotrophic methane-producing bacterium isolated from marine sediments. Appl. Environ. Microbiol. 47:971–978
Wilkins MJ, VerBerkmoes NC, Williams KH, Callister SJ, Mouser PJ, Elifantz H, N’Guessan AL, Thomas BC, Nicora CD, Shah MB, Abraham P, Lipton MS, Lovley DR, Hettich RL, Long PE, Banfield JF (2009) Proteogenomic monitoring of Geobacter physiology during stimulated uranium bioremediation. Appl. Environ. Microbiol. 75:6591–6599. https://doi.org/10.1128/aem.01064-09
Wilkins MJ, Callister SJ, Miletto M, Williams KH, Nicora CD, Lovley DR, Long PE, Lipton MS (2011) Development of a biomarker for Geobacter activity and strain composition; Proteogenomic analysis of the citrate synthase protein during bioremediation of U(VI). Microb Biotech 4:55–63. https://doi.org/10.1111/j.1751-7915.2010.00194.x
Wilkins MJ, Wrighton KC, Nicora CD, Williams KH, McCue LA, Handley KM, Miller CS, Giloteaux L, Montgomery AP, Lovley DR, Banfield JF, Long PE, Lipton MS (2013) Fluctuations in species-level protein expression occur during element and nutrient cycling in the subsurface. PLoS One 8:e57819. https://doi.org/10.1371/journal.pone.0057819
Yun J, Ueki T, Miletto M, Lovley DR (2011) Monitoring the metabolic status of Geobacter species in contaminated groundwater by quantifying key metabolic proteins with Geobacter-specific antibodies. Appl. Environ. Microbiol. 77:4597–4602. https://doi.org/10.1128/aem.00114-11
Holmes DE, Giloteaux L, Williams KH, Wrighton KC, Wilkins MJ, Thompson CA, Roper TJ, Long PE, Lovley DR (2013) Enrichment of specific protozoan populations during in situ bioremediation of uranium-contaminated groundwater. ISME J 7:1286–1298. https://doi.org/10.1038/ismej.2013.20
Dar S, Tan H, Peacock A, Jaffe P, N’Guessan L, Williams K, Strycharz-Glaven S (2013) Spatial distribution of Geobacteraceae and sulfate-reducing bacteria during in situ bioremediation of uranium-contaminated groundwater. Remediation J 23:31–49
Funding
This work is a contribution to grant by the Office of Science (BER), US Department of Energy, Awards no. DE-SC0004080 and DE-SC0004814 and Cooperative Agreement no. DE-FC02-02ER63446. AER has been supported by grants from the Novo Nordisk Foundation, Innovationsfonden (grant no. 410600017) and Danish Research Council (418100203).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOCX 25 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Holmes, D.E., Orelana, R., Giloteaux, L. et al. Potential for Methanosarcina to Contribute to Uranium Reduction during Acetate-Promoted Groundwater Bioremediation. Microb Ecol 76, 660–667 (2018). https://doi.org/10.1007/s00248-018-1165-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1165-5