
Abstract
The bacterial strain SECRCQ15T was isolated from seeds of Chenopodium quinoa in Spain. Phylogenetic, chemotaxonomic, and phenotypic analyses, as well as genome similarity indices, support the classification of the strain into a novel species of the genus Ferdinandcohnia, for which we propose the name Ferdinandcohnia quinoae sp. nov. To dig deep into the speciation features of the strain SECRCQ15T, we performed a comparative genomic analysis of the genome of this strain and those of the type strains of species from the genus Ferdinandcohnia. We found several genes related with plant growth-promoting mechanisms within the SECRCQ15T genome. We also found that singletons of F. quinoae SECRCQ15T are mainly related to the use of carbohydrates, which is a common trait of plant-associated bacteria. To further reveal speciation events in this strain, we revealed genes undergoing diversifying selection (e.g., genes encoding ribosomal proteins) and functions likely lost due to pseudogenization. Also, we found that this novel species contains 138 plant-associated gene-cluster functions that are unique within the genus Ferdinandcohnia. These features may explain both the ecological and taxonomical differentiation of this new taxon.
Similar content being viewed by others


Introduction
Each earth ecosystem, including animals and plants, has their own distinctive microbial communities, which sometimes have evolved to develop important ecological functions (Nayfach et al. 2021; Debray et al. 2022; Hartmann and Six 2023). For instance, the plant microbiome is capable to benefit its host by providing access to nutrients and through the protection against biotic and abiotic factors (Ali et al. 2022; Chialva et al. 2022). Specially, endophytic microbes usually share a beneficial relation with their plant hosts (Flores-Félix et al. 2015; García-Fraile et al. 2015; Poveda et al. 2022). Indeed, some microbes have been adapted to the lifestyle in some specific plant tissues, and furthermore, some of them are vertically transmitted to descendants within seeds (Abdelfattah et al. 2022; Simonin et al. 2022). These seed symbionts may have been co-evolved to interact intimately with the host, and their study is of utmost importance to understand biological and ecological processes as well as to inspire advances in the development of biofertilizers (Dubey et al. 2021; Abdelfattah et al. 2022; Laranjeira et al. 2022; Simonin et al. 2022).
The evolution and adaptation into a new ecological niche are usually responsible of the bacterial speciation events (Baquero et al. 2021), and this evolution can be investigated through the analysis of the genomes and their gene content. For instance, it is known that many host-associated bacteria share common genes that facilitate competition, colonization, and evasion of host immune system (Wiesmann et al. 2022). In the case of the bacterial adaptation to the plant niche, there are few studies that provide insights into the processes (i.e., gain of genes, mutations) that lead the adaptation to this habitat (Levy et al. 2018a, b; Li et al. 2021a, b). However, it is not completely understood, not only how a bacterium evolves to gain fitness to the plant environment, but neither how a novel species arises in a differentiate niche.
Here we characterized an endophytic strain of quinoa seeds named SECRCQ15T phylogenetically related to the Ferdinandcohnia species and whose genetic, chemotaxonomic, and phenotypic characteristics showed that it is a novel species of this genus which we propose to name as Ferdinandcohnia quinoae sp. nov. Through genome analyses, we characterize this species based on its putative roles in its plant host. Then, we show a comparative genomic study where we highlight the genomic features distinctive of the novel species F. quinoae with respect to the remaining species of genus Ferdinandcohnia, which were isolated from different ecosystems, such as air -F. onubensis (Dominguez-Moñino et al., 2018)-, compost -'F. nitroreducens' (Guo et al. 2016)-, human stools -'F. sinesaloumensis' and 'F. timonensis' (Kokcha et al. 2012; Senghor et al. 2017)- and soil -F. humi (Heyrman et al. 2005), F. salidurans (Son et al. 2019) and F. aciditolerans (Ding et al. 2019)-. Our findings provide novel insights into the speciation features of a novel species occupying a different niche than the remaining species of the genus Ferdinandcohnia.
Methods
Strain Isolation
The strain SECRCQ15T was isolated from Chenopodium quinoa seeds harvested from plants cultivated in Ciudad Rodrigo (Salamanca, Spain, 40° 35′ 02.6′′ N 6° 31′ 56.5′′ W). Quinoa seeds were surface sterilized with ethanol (70%) for 3 min and sodium hypochlorite (2%) for 2 min and were washed 5 times in sterile distilled water. An aliquot of the last wash water was plated on tryptic soy agar (TSA) and incubated at 28 °C for 48 h as a disinfection control, where no bacterial growth was observed. Surface sterilized seeds were crushed in a sterile mortar and resuspended in sterile water. Decimal dilutions from the suspension were obtained to isolate the endophytic bacteria and 100 μL of each suspension was spread on TSA plates (Sigma Co.) which were incubated at 28 °C for 48 h. Despite we have isolated other strains (data not shown), here we focus on the analysis of the strain SECRCQ15T due to its taxonomic novelty. The strain was cryopreserved (− 80 °C; 25% glycerol solution) and plated when needed.
Whole Genome Sequencing
We extracted DNA of the strain SECRCQ15T after 2 days of growth in TSA (28 °C) using the Quick DNA Fungal/Bacterial Miniprep kit (Zymo Research, Irvine, CA, USA). The draft genome was sequenced on an Illumina NextSeq 500 Platform (75 pb Paired End). The genome contigs were assembled with SPAdes (Bankevich et al. 2012). The completeness and contamination levels were measured as previously detailed (Saati-Santamaría et al. 2022a) with BUSCO (Simão et al. 2015), and CheckM (Parks et al. 2015), respectively. We made both the structural and the functional genome annotation with RAST (v2.0) (Aziz et al. 2008).
Phylogenetic Analyses
Amplification and sequencing of 16S rRNA gene were performed according to Carro et al. (2012) with some modifications. Briefly, 16S rDNA amplification was done with primers 27F (5′-AGAGTTTGATCTGGCTCAG-3′) (Weisburg et al. 1991) and 1522R (5′-AAGGAGGTGATCCANCC-3′) (Carro et al. 2012), using a REDTaq® ReadyMix™ (Sigma, USA) in 50 μl reaction volume following the manufacturer’s instructions. PCR products were purified directly from the gel with the GeneJET Gel Extraction and a DNA Cleanup Micro Kit (Thermo Scientific™, Göteborg, Sweden). Afterward, 16S rDNA PCR products were bidirectionally sequenced using a BigDye™ Terminator (v3.1) at the Sequencing DNA Service (NUCLEUS; University of Salamanca, Spain). Reads were aligned with SeqMan Pro software (DNASTART Inc., USA) to obtain the SECRCQ15T 16S rDNA consensus sequence. This consensus sequence was compared to public sequences using BlastN (Altschul et al. 1990) (against the GenBank database) and EzTaxon-e (Kim et al. 2012) programs. The most closely related sequences from type strains were aligned with the Clustal_X software (Thompson et al. 1997) and distances were calculated according to Kimura´s two-parameter model (Kimura 1980) The phylogenetic trees were inferred using the neighbor joining (NJ) and maximum likelihood (ML) models (Saitou & Nei 1987; Rogers & Swofford, 1998). MEGA7 package (Kumar et al. 2016) was used for all analyses. We used all available gene and genomic sequences as available from the type strains of the Ferdinandcohnia species.
Genome Analyses and Comparative Genomics
We measured the genomes relatedness through genome similarity indexes as detailed before (Saati-Santamaría et al. 2021). Briefly, we used PYANI software (v0.2.10) (Pritchard et al. 2016) to measure the ANIb values and the Genome-to-Genome Distance Calculator (GGDC v2.1) (Auch et al. 2010; Meier-Kolthof et al. 2013) to measure the digital DNA-DNA hybridization (dDDH). Average amino acid identity (AAI) was calculated with the online tool ANI/AAI-Matrix from Enveomics toolbox (Rodriguez-R and Konstantinidis 2016) with default settings, which uses MMSeqs2 for protein comparisons, a minimum query coverage of 50% and a minimum identity of 40% for AAI calculations. The phylogenomic tree was created with the UBCG (v3) tool (default settings) (Na et al. 2018) which created codon alignments (with MAFFT) and trees based on 92 housekeeping genes. These are universal bacterial core genes that have been proven to be valuable to infer phylogenomic relationship of bacteria (Na et al. 2018). Then, the tree was visualized and edited in the interactive tree of life (iTOL) tool (v5) (Letunic and Bork 2021).
We annotated the functions of the genes/proteins with KofamKOALA (Aramaki et al. 2020), PLaBAse (Patz et al. 2021) and antiSMASH (v6.0) (Blin et al. 2021). The comparative genomic analysis was done with Anvi’o (Eren et al. 2015). The Horizontal Gene Transfer (HGT) events were searched with HGTector2 (Zhu et al. 2014).
We used the pseudofinder.py command (Syberg-Olsen et al. 2022) to search pseudogenes in the SECRCQ15T. We used the whole Swissprot database as a reference and we retained the results with e value < 1e-4 (‘diamond’ search). As the input for this analysis, we used the GBK (gene bank format) annotation file provided by Prokka (Seemann 2014). This annotation was done with the ‘compliant’ and ‘rfam’ flags activated to enable the annotation of non-coding RNAs (ncRNAs) to eliminate false positive 'pseudogene' candidates. The genome the analysis of dN/dS was also done with pseudofinder, by using the ‘sleuth’ command. We used as a test the SECRCQ15T genome, and the genomes of other Ferdinandcohnia species as a reference (run separately) (Ferdinandcohnia aciditolerans YN-1T, GCF_003640645.1; Ferdinandcohnia humi DSM 16318T, GCF_001439915.1; Ferdinandcohnia onubensis 0911MAR22V3T, GCF_002734215.1; ‘Ferdinandcohnia timonensis’ MM10403188T, GCF_000285535.1). The results were classified as follows: positive selection: dN/dS > 1; neutral selection: dN/dS = 1; purifying selection: dN/dS < 1.
Phenotypic and Chemotaxonomic Analyses
The strain was grown on nutrient agar (NA, Sigma Co.) for 48 h at 22 °C to check for motility by phase-contrast microscopy using the hanging drop method. Gram staining was carried out by the procedure described by Doetsch (1981) after 24 h of incubation at 28 °C. The flagellation type was determined by electron microscopy after 48 h of incubation of strain SECRCQ15T on nutrient agar at 22 °C. The cells were gently suspended in sterile water and then stained with 2% uranyl acetate and examined at 80 kV with a Tecnai Spirit Twin transmission electron microscope.
The cellular fatty acids were analyzed by using the Microbial Identification System (MIDI; Microbial ID) Sherlock 6.1 and the library RTSBA6 according to the technical instructions provided by this system (Sasse 1990). The strains F. humi LMG 22167 T and ‘F. timonensis’ DSM 25372 T were included as reference, which were obtained from their corresponding culture collections: BCCM/LMG (https://bccm.belspo.be/about-us/bccm-lmg), and the German Collection of Microorganisms and Cell Cultures (DSMZ; https://www.dsmz.de/), respectively. The strains were grown on TSA plates (Becton Dikinson, BBL) for 48 h at 28 °C. Other chemotaxonomic analyses were carried out by the Identification Service of Leibniz Institute DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) for which the strain SECRCQ15T was cultivated in TSB (Becton Dickinson, BBL) for 48 h at 28 °C and 180 rpm. The respiratory quinones and polar lipids were analyzed as described by Tindall (1990). To perform the analysis of peptidoglycan the whole cells of strain SECRCQ15T were hydrolyzed with HCl at 100 °C during 15 h. The hydrolysates were subjected to thin-layer chromatography on cellulose plates using the solvent system of Rhuland et al. (1995).
The phenotypic characterization included the characteristics recommended in the minimal standards for aerobic endospore-forming bacteria (Logan et al. 2009) and was performed according to the methods described by Claus and Berkeley (1986) and by using API 20NE and API32GN systems (bioMerieux, France) according to the manufacturer’s instructions but adding MgSO4 to the media supplied by the kit, since this salt improves the growth of Ferdinadcohnia strains in these systems. The strain was unreactive in API 50CHB systems as occurred with its closest related taxon F. humi (Heyrman et al. 2005). The anaerobic growth was tested in fluid tetrathionate medium (Sigma, USA). Acetoin production, ability to grow in the presence of 2, 5, and 7% NaCl, nitrate reduction, phenylalanine deaminase, catalase, caseinase, gelatinase, amylase, and oxidase were analyzed as was described elsewhere (Claus and Berkeley 1986). Acid production from D-glucose, D-xylose, D-mannitol and L-arabinose and gas from glucose were analyzed in liquid medium as described previously (Claus and Berkeley 1986). Growth was determined at 4, 15, 25, 28, 37, 40, and 45 °C in TSA medium (Difco, BBL). The growth at pH 7 to 8 was tested in nutrient broth (Difco, BBL) containing 200 mM of Na2HPO4/NaH2PO4 and the growth at pH 9 and 10 was tested in the same medium containing 200 mM of NaHCO3/Na2CO3. The strain F. humi LMG 22167T was included in the phenotypic study as reference.
Results
Genome Assembly
The assembly of the SECRCQ15T genome yielded 265 contigs (L50 = 15; N50 = 89,801), and a total genome size of 4,443,130 bp, with 4351 coding region sequences (CDS) and 44 RNAs. The genomic G + C content is 36.0%. The genome contains 100% of complete BUSCO genes and 1.8 × contamination.
Phylogenetic and Phylogenomic Location of the Strain SECRCQ15T
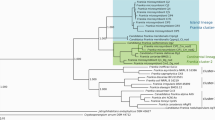
First, we aimed to ensure the taxonomic placement of the strain SECRCQ15T. The comparison of the 16S rRNA gene sequence of this strain (1500 nucleotides) against those of type strains held in EzTaxon-e database indicated that it belongs to the genus Ferdinandcohnia, with F. humi DSM 16318T and “F. timonensis” 10403023T sharing the highest similarity, 98.1%, The remaining species shared similarity values lower than 98%; “F. nitroreducens” GSS08T 97.8%, “F. sinesaloumensis,” F. onubensis and F. salidurans 97.4% and F. aciditolerans 96.9%. The phylogenetic analyses based on the 16S rRNA gene (both NJ and ML analyses), showed that the strain SECRCQ15T formed a branch phylogenetically divergent from the remaining Ferdinandcohnia species (Fig. 1a). Similarly, the phylogenomic tree place the strain in a separate branch (Fig. 1b).

Phylogeny of the SECRCQ15T strain. a Maximum likelihood phylogenetic tree based on nearly complete 16S rRNA gene sequence (1500 nucleotides) of Ferdinandcohnia quinoae SECRCQ15T and the remaining species of the genus Ferdinandcohnia. Bacillus subtilis IAM 12118T was used as outgroup. The significance of each branch is indicated by a bootstrap value calculated in percentage for 1000 subsets. Bar, 5 nt substitutions per 1000 nt. b Phylogenomic tree based on the consensus of 92 housekeeping gene phylogenies built with the UBCG pipeline. Bar, 100 nt substitutions per 1000 nt
The genomes of the Ferdinandcohnia species showed less than 75% of ANIb values with respect to the genome of the strain SECRCQ15T (Table 1) and the dDDH values were lower than 24% in all cases (Table 1). They are below threshold values used for bacterial species differentiation (Chun et al. 2018; Jain et al. 2018; Peral-Aranega et al. 2020; González-Dominici et al. 2021). The phylogenetic analyses and the phylogenomic tree based on 92 housekeeping genes confirmed that strain SECRCQ15T represents a new species of genus Ferdinandcohnia for which we propose the name Ferdinandcohnia quinoae sp. nov. with SECRCQ15T as type strain (Fig. 1). In addition, the chemotaxonomic and phenotypic features of this strain (shown in Supplementary material) support its classification as a different Ferdinandcohnia species.
Novelty in the Gene Content Within the Ferdinandcohnia pangenome
To shed light on the novel genomic/metabolic features of F. quinoae we performed a comparative genomic analysis of the strain SECRCQ15T and the genomes of the closest related type strains. This analysis allowed to identify 1254 gene singletons in the SECRCQ15T genome (genes unique to this strain) (Fig. 2). Out of these singletons, 862 were assigned to a COG (Cluster of Orthologous Groups of proteins) category, which are mainly related with signal transduction, carbohydrate metabolism, and transcription (Fig. 2).

Comparative analyses of the Ferdinandcohnia genomes. The 7 inner circles show the presence/absence of Gene Clusters (GC) (homologous genes across genomes). Both the ANIb matrix and the GC presence/absence patterns support the differentiation of the F. quinoae species. We show below the functional categories of the SECRCQ15T singletons, which serves as a summary of the metabolism that drove the ecological speciation of this new taxa
We compared SECRCQ15T gene clusters annotations (COGs) with those suggested to be related with plant-bacteria interactions in a previous comparative genomic study (Levy et al. 2018a, b). We used a list of Bacillus COGs that were significantly more abundant in plant isolates than in soil isolates. This search returned 138 of the SECRCQ15T gene clusters (11.15%) as plant-associated COGs. In contrast, the core-genome of the Ferdinancohnia genus (Fig. 2) just contains 35 plant-associated COGs (8.64% of 405 core gene clusters). This enrichment of PA-functions of F. quinoae suggest an adaptative mechanism to live within the plant environment.
Finally, we aimed to unveil what functions have been horizontally acquired by this strain and which microorganisms acted as donors. We found 422 HGT events within the SECRCQ15T genome, which mainly come from closely related taxa (Fig. 3a), with a few exceptions, such as genes potentially acquired from the Sporosarcina genus (Caryophanales order), or from the Peptococcaceae family (Clostridia class). Much of these HGT are related with the metabolism of carbohydrates (84 genes) (Fig. 3b), or signal transduction (46 genes), but also many of them (66 genes) belong to COGs with no described function. Of these HGTs, 174 belonged to the singletons of the SECRCQ15T strain, which represent 41% of the HGTs, and 13.8% of these singletons. This may indicate that most of the HGT events, since are unique for this strain, likely occurred recently and/or may be related with its speciation process, but still about 85% of the unique genes of the strain in the genus have an uncertain origin, such as gene diversification.

Putative horizontal gene transfer events found in the Ferdinandcohnia quinoae SECRCQ15T genome. a Summary of the taxonomy of the donor strains for the transferred genes. b Summary of the metabolism categories of the horizontally acquired genes
Gene-Wide Evolution of Ferdinandcohnia quinoae sp. nov. SECRCQ15T
We explored the degree of selection acting on protein-coding genes by means of the dN/dS ratio, which represents the non-synonymous (dN) to synonymous (dS) nucleotide substitution rate. We found 4–9 genes (depending on the reference species used for this analysis) that are under positive (diversifying) selection (dN/dS > 1), of which 1–3 show values > 2 (Fig. 4). Interestingly, we found a high positive selection acting on genes encoding ribosomal proteins, which help to structure the ribosomal RNA. We also found other functions being diversified, such as the Cold shock-like protein CspLA, a thiosulfate sulfotransferase (GlpE), and an arsenate reductase (arsC). The evolution of these genes might be related with some selective pressure related with the SECRCQ15T plant niche.

Gene-wide evolution analysis of the strain SECRCQ15T. dN/dS ratio of SECRCQ15T genes using as a reference the genomes of other species in the genus: a Ferdinandcohnia aciditolerans YN-1T (GCF_003640645.1); b Ferdinandcohnia humi DSM 16318T (GCF_001439915.1); c Ferdinandcohnia onubensis 0911MAR22V3T (GCF_002734215.1); d ‘Ferdinandcohnia timonensis’ MM10403188T (GCF_000285535.1)
Beyond diversification, we also investigated to what extend some functions have been lost in the SECRCQ15T genome due to pseudogenization, which might reveal functions that are not needed within this strain and that can help to understand their speciation events. We found 219 pseudogenes (5% of the total open reading frames) (Table 2). Most of these lost functions were unknown, related with signal transduction mechanism, transcription or to cell wall biogenesis, among others (Fig. S3).
Ecological Features of the SECRCQ15T Strain
To unveil potential ecological functions of the new proposed species, we analyzed its genome to find genes likely related with the interaction with its host plant and/or the surrounding microbiome. We found enzymes related with plant hormone biosynthesis, such as an Indole-3-acetamide amidohydrolase and an Indole-3-acetaldehyde:NAD + oxidoreductase, both proteins involved in indolacetate (IAA) synthesis. There are also proteins encoded that are related with the production of dimethylallyl diphosphate and geranyl-PP from D-Glyceraldehyde 3-phosphate (glycolysis product). These two metabolites are substrates for the biosynthesis of zeatin and monoterpenes, respectively, and could be incorporated into the plant biosynthetic pathways. Also, this strain could boost plant systemic response due to the presence of the microbial associated molecular patterns (MAMPs) flagellin (Flg22) and Elongation Factor-TU (Ef-TU). The presence of genes encoding an acetolactate synthase, responsible for the synthesis of acetoin represents, not only a putative role in ISR elicitation, but also another plant growth-promoting mechanism. Additionally, the genome encodes alkaline phosphatases (EC 3.1.3.1) and diverse pyrophosphatases (EC 3.6.1.1), which, together with the action of acids likely produced by this strain (i.e., there are genome evidences of malic acid biosynthesis) can help to release phosphorous accessible for the plant absorption. We also found complete pathways involved in vitamin biosynthesis, such as thiamine (vit B1), riboflavin (vit B2), nicotinate and nicotinamide (vit B3), pantothenate (vit B5), folate (vit B9), and menaquinone (vit K2). Finally, this strain may have a role in the sulfur cycle, since it encodes the proteins Sat, CysC, CysH, and CysII, responsible of the complete assimilatory sulfate reduction to sulfide. The genome does not show any putative role of this strain in atmospheric carbon (CO2) or methane (CH4) fixation.
We found that that strain SECRCQ15T has a total of 2287 putative genes associated with bacterial plant growth-promoting traits “PGPTs,” according to the PLaBAse database. Of these, 70.4% are coincident with indirect mechanisms such as colonization, competition in the rhizosphere or resistance to stresses. While 29.6% corresponds to direct plant promotion mechanisms such as the genes already detected associated with auxin production, vitamin biosynthesis or the production of organic acids from different sugars. We also detected the miaA and miaB genes, isopentenyltransferase and methylpentenyltransferase, respectively, that are essential for the biosynthesis of cytokinins. Also, the genes gabD and gabT, which catabolise the degradation of gamma-aminobutyrate (GABA) on succinate were annotated on its genome. In addition, this analysis revealed an extensive genetic background for resistance and adaptation to the environment. This adaptation skill is supported by 204 genes involved in the degradation of xenobiotics such as the paa cluster for phenylacetate degradation, or the degradation of dichloropropene (adhP-propanol dehydrogenase); and resistance to metals such as arsenic and antimony (arsA, arsB, arsC, arsR), copper (cusR), zinc (znuABC) or lead (prbB), among others.
We searched for biosynthetic gene clusters (BGCs) that may produce any known specialized metabolite, but the search just allowed to detect already undescribed BGCs with no similarity against those available in the MiBIG and antiSMASH databases: a lasso peptide, a type III PKS, a hybrid linear azol(in)e-containing peptide (LAP) and a ribosomally synthesized and post-translationally modified peptide product (RiPP). Due to its novelty, these BGCs may produce novel secondary metabolites with ecological or biotechnological interest.
Discussion
In this work we have characterized one endophytic strain isolated from quinoa seeds obtained from a local farmer (Salamanca, Spain). The accurate assignment of this strain to a taxon was addressed by diverse phylogenetic and phylogenomic analyses and by physiological, phenotypic and chemotaxonomic approaches. The 16S rRNA gene analysis continues to be essential to place the bacterial isolates into a genus and to know their closest related species, which in the case of strain SECRCQ15T were those of genus Ferdinandcohnia. The species of this genus were initially included within the genus Bacillus, which was subject to a deep reclassification recently (Gupta et al. 2020), being the new species described in this work, F. quinoae sp. nov., the first Ferdinandcohnia species described after this reclassification, which was based on phylogenomic analyses.
To deeper analyze both the taxonomic assignment of strain SECRCQ15T and its potential role as a quinoa seed endophyte, we obtained its genome sequence, which is the first available genome of a quinoa endophytic strain. The analysis of this genome confirmed that this strain belongs to a new species of genus Ferdinandcohnia, being its closest phylogenetically related type strains those of F. humi DSM 16318T, which was isolated from an agricultural soil (Heyrman et al. 2005), and “F. timonensis” 10403023T, which was isolated from human stools (Kokcha et al. 2012). Several phenotypic and chemotaxonomic differences between these strains support that they are different species, and thus, we propose the description of Ferdinandcohnia quinoae sp. nov.
Considering that the proposed novel species expand the host-range of the genus Ferdinandcohnia to include plant seeds, we discussed its genome innovations and features that may fulfill the evolutionary process to fulfill its adaptation to that environment and its ecological roles in there. We found a large proportion of singletons within the F. quinoae SECRCQ15T sp. nov. genome that are related with the carbohydrate metabolism. This functional expansion can be related with the broad availability of sugars and complex carbohydrates found within the seed and plant environment (Sasse et al. 2018; McLaughlin et al. 2023). Similarly, this set of genes encompass many plant-associated genes (Levy et al. 2018a, b), which also support its drift toward the plant lifestyle. Also, the enrichment of singletons related with transcription and signal transduction mechanisms may reflect a deeper speciation, not only in coding-sequences, but also in the regulation of cell functions, which outcomes could be investigated trough transcriptomics or other -omic data.
We also found several functions undergoing positive selection, such as ribosomal proteins. These mutations may have been driven by the need to interact with other biomolecules, and may affect ribosome assembly, leading to extensive alterations in both transcriptomic and proteomic profiles (Gómez et al. 2017). Diversifying selection on arsenate reductase and thiosulfate sulfurtransferase might be related with an improved metabolism of arsenate and thiosulfate. In the context of plant-bacteria interactions, some bacteria activate arsenic detoxification mechanisms. This can be relevant in environments where plants are exposed to arsenic contamination, as bacteria may contribute to the transformation of arsenate into a less toxic form, potentially influencing the overall arsenic bioavailability to plants (Cavalca et al. 2010). Thiosulfate can serve as a sulfur source for both plants and bacteria. Some bacteria may produce thiosulfate as a byproduct. In turn, plants can take up thiosulfate as a sulfur nutrient. Hence, thiosulfate sulfurtransferase diversification might lead to a enhanced sulfur metabolism, which might influence plant health and growth (Nakajima et al. 2019; Ranadev et al. 2023).
Beyond their speciation events, the complete genome of F. quinoae SECRCQ15T revealed ecological features related with host-microbe interactions, concretely with beneficial plant-bacteria interactions. Further experiments will serve to elucidate its functions within quinoa seeds or developed plants. Similarly, the novelty of the BGCs related with the production of specialized metabolites, not only uncover ecological features (Saati-Santamaría, 2023), but also suggests this strain as a promising one for natural product research (Kalkreuter et al. 2020; Hemmerling & Piel 2022; Saati-Santamaría et al. 2022b).
Conclusion
In sum, we provide an indepth-analyses of the speciation features of the strain SECRCQ15T. Both the phylogenies and the functional differentiation of this strain support its classification as a novel species. The ecological functions encoded within the F. quinoae sp. nov. genome will help to further understand the bacterial communities of quinoa plants. Also, this work expands the host range of the Ferdinandcohnia genus. The genomic analyses revealed signs of functional adaptation to the plant environment in the F. quinoae type strain and explain its adaptation within this niche.
Protologue
Description of Ferdinandcohnia quinoae gen. nov. sp. nov.
Ferdinandcohnia quinoae (qui.no'ae. N.L. gen. n. quinoae, of quinoa).
Cells of the strain SECRCQ15T were straight, aerobic, motile Gram-stain positive rods (width 0.6–0.8 µm, length 3.0–5.0 µm). Oval subterminal endospores were formed in swollen sporangia. Catalase and oxidase positive. Colonies of this strain on nutrient agar medium are opaque, white cream colored, raised with entire margins and smooth surfaces. Anaerobic growth was negative. It grows from pH 6 to pH 9 (optimal pH is 7). It can grow in the presence of 4% NaCl. It grows from 12 to 44 °C (optimal temperature is 30 °C). Nitrate is reduced to nitrite. Production of β-galactosidase and hydrolysis of aesculin are positive. Production of indole, urease, arginine dehydrolase, and H2S is negative. Production of gelatinase is weak. In API 20NE Assimilation of glucose, maltose, mannose, and gluconate is positive and that of L-arabinose, mannitol, N-acetyl-glucosamine, caprate, adipate, malate, citrate, and phenylacetate is negative. In API ID32GN glucose, melibiose, D,L-lactate, glycogen, maltose, and 3-hydroxi-butyrate are assimilated, but the assimilation of L-rhamnose, N-acetyl-glucosamine, D-ribose, inositol, sucrose, mannitol, salicin, L-fucose, D-sorbitol, L-arabinose, itaconate, suberate, propionate, caprate, valerate, citrate, malonate, acetate, 2 and 5 keto-gluconate, 3 and 4 hydroxi-benzoate, L-serine, L-alanine, L-histidine, and L-proline is negative. The major quinone is MK-7. Mesodiaminopimelic acid was not detected in the peptidoglycan. The lipid profile consists of diphosphatidylglycerol, phosphatidylglycerol, phosphatidylethanolamine, one unidentified aminophospholipid, and one unidentified phospholipid. The major fatty acids are iso-C15:0 and anteiso-C15:0. The G + C content of the strain SECRCQ15T is 35.96 mol%. The type strain SECRCQ15T (= LMG 32511 T = CECT 30513T) was isolated from seeds of quinoa (Chenopodium quinoa) in Spain. The 16S rRNA gene and genome sequence were deposited at DDBJ/EMBL/GenBank under accession numbers OM791795 and JAKTTI000000000, respectively.
Data Availability
The genome sequence is available under the NCBI (https://www.ncbi.nlm.nih.gov/) bioproject PRJNA807880 (BioSample: SAMN26011952; Assembly: GCA_022427965.1; Genome nucleotide sequences: JAKTTI000000000). The 16S rRNA gene sequence is available through the following accession number: OM791795.
References
Abdelfattah A, Tack AJ, Lobato C, Wassermann B, Berg G (2022) From seed to seed: the role of microbial inheritance in the assembly of the plant microbiome. Trends Microbiol 31(4):346–355
Ali S, Tyagi A, Park S, Mir RA, Mushtaq M, Bhat B et al (2022) Deciphering the plant microbiome to improve drought tolerance: mechanisms and perspectives. Environ Exp Bot 201:104933
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, Ogata H (2020) KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36(7):2251–2252
Auch AF, von Jan M, Klenk HP, Göker M (2010) Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2:117–134
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T et al (2008) The RAST server: rapid annotations using subsystems technology. BMC Genom 9:75
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Baquero F, Coque TM, Galán JC, Martinez JL (2021) The origin of niches and species in the bacterial world. Front Microbiol 12:657986
Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, Van Wezel GP, Medema MH, Weber T (2021) antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res 49(W1):W29–W35
Carro L, Spröer C, Alonso P, Trujillo ME (2012) Diversity of Micromonospora strains isolated from nitrogen fixing nodules and rhizosphere of Pisum sativum analyzed by multilocus sequence analysis. Syst Appl Microbiol 35:73–80
Cavalca L, Zanchi R, Corsini A, Colombo M, Romagnoli C, Canzi E, Andreoni V (2010) Arsenic-resistant bacteria associated with roots of the wild Cirsium arvense (L.) plant from an arsenic polluted soil, and screening of potential plant growth-promoting characteristics. Syst Appl Microbiol 33(3):154–164
Chialva M, Lanfranco L, Bonfante P (2022) The plant microbiota: composition, functions, and engineering. Curr Opin Biotechnol 73:135–142
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS et al (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68(1):461–466
Claus D, Berkeley RCW (1986) Genus Bacillus Cohn 1872, 174AL. In: Sneath PHA, Mair NS, Sharpe ME, Holt JG (eds) Bergey’s manual of systematic bacteriology, vol 2. Williams Wilkins, Baltimore, pp 1105–1139
Debray R, Herbert RA, Jaffe AL, Crits-Christoph A, Power ME, Koskella B (2022) Priority effects in microbiome assembly. Nat Rev Microbiol 20(2):109–121
Ding MJ, Shang NJ, Xiao ZX, Shao F, Liu L et al (2019) Bacillus aciditolerans sp. nov, isolated from paddy soil. Int J Syst Evol Microbiol 69:1155–1161
Doetsch RN (1981) Determinative methods of light microscopy. In: Gerdhardt P, Murray RGE, Costilow RN, Nester EW, Wood WA, Krieg NR, Phillips GB (eds) Manual of methods for general bacteriology. American Society for Microbiology, Washington, pp 21–33
Dominguez-Moñino I, Jurado V, Gonzalez-Pimentel JL, Miller AZ, Hermosin B et al (2018) Bacillus onubensis sp. nov, isolated from the air of two Andalusian caves. Syst Appl Microbiol 41:167–172
Dubey A, Kumar A, Malla MA, Chowdhary K, Singh G, Gudasalamani-Ravikanth H et al (2021) Approaches for the amelioration of adverse effects of drought stress on crop plants. Front Biosci (landmark Ed) 26(10):928–947
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO (2015) Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319
Flores-Félix JD, Silva LR, Rivera LP, Marcos-García M, García-Fraile P et al (2015) Plants probiotics as a tool to produce highly functional fruits: the case of Phyllobacterium and vitamin C in strawberries. PLoS ONE 10(4):e0122281
García-Fraile P, Menéndez E, Rivas R (2015) Role of bacterial biofertilizers in agriculture and forestry. AIMS Bioeng 2(3):183–205
Gomez JE, Kaufmann-Malaga BB, Wivagg CN, Kim PB, Silvis MR et al (2017) Ribosomal mutations promote the evolution of antibiotic resistance in a multidrug environment. Elife 6:e20420
González-Dominici LI, Saati-Santamaría Z, García-Fraile P (2021) Genome analysis and genomic comparison of the novel species Arthrobacter ipsi reveal its potential protective role in its bark beetle host. Microb Ecol 81(2):471–482
Guo J, Wang YQ, Yang G, Chen Y, Zhou S, Zhao Y, Zhuang L (2016) Bacillus nitroreducens sp. nov, a humus-reducing bacterium isolated from a compost. Arch Microbiol 198:347–352
Gupta RS, Patel S, Saini N, Chen S (2020) Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int J Syst Evol Microbiol 70:5753–5798
Hartmann M, Six J (2023) Soil structure and microbiome functions in agroecosystems. Nat Rev Earth Environ 4(1):4–18
Hemmerling F, Piel J (2022) Strategies to access biosynthetic novelty in bacterial genomes for drug discovery. Nat Rev Drug Discov 21(5):359–378
Heyrman J, Rodriguez-Diaz M, Devos J, Felske A, Logan NA et al (2005) Bacillus arenosi sp. nov, Bacillus arvi sp. nov. and Bacillus humi sp. nov, isolated from soil. Int J Syst Evol Microbiol 55:111–117
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9(1):1–8
Kalkreuter E, Pan G, Cepeda AJ, Shen B (2020) Targeting bacterial genomes for natural product discovery. Trends Pharmacol Sci 41(1):13–26
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M et al (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA Gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kokcha S, Mishra AK, Lagie JC, Million M, Leroy Q, Raoult D, Fournier PE (2012) Non contiguous-finished genome sequence and description of Bacillus timonensis sp. nov. Stand in Genomic Sci 6:346–355. https://doi.org/10.4056/sigs.2776064
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 3:1870–1874
Laranjeira SS, Alves IG, Marques G (2022) Chickpea (Cicer arietinum L.) seeds as a reservoir of endophytic plant growth-promoting bacteria. Curr Microbiol 79(9):277. https://doi.org/10.1007/s00284-022-02942-1
Letunic I, Bork P (2021) Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49(W1):W293–W296
Levy A, Conway JM, Dangl JL, Woyke T (2018a) Elucidating bacterial gene functions in the plant microbiome. Cell Host Microbe 24(4):475–485
Levy A, Salas Gonzalez I, Mittelviefhaus M, Clingenpeel S, Herrera Paredes S, Miao J et al (2018b) Genomic features of bacterial adaptation to plants. Nat Genet 50(1):138–150
Li E, de Jonge R, Liu C, Jiang H, Friman VP, Pieterse CM et al (2021a) Rapid evolution of bacterial mutualism in the plant rhizosphere. Nat Commun 12(1):3829
Li E, Zhang H, Jiang H, Pieterse CM, Jousset A, Bakker PA, de Jonge R (2021b) Experimental-evolution-driven identification of Arabidopsis rhizosphere competence genes in Pseudomonas protegens. Mbio 12(3):e00927-e1021
Logan NA, Berge O, Bishop AH, Busse HJ, De Vos P et al (2009) Proposed minimal standards for describing new taxa of aerobic, endospore-forming bacteria. Int J Syst Evol Microbiol 59:2114–2121
McLaughlin S, Zhalnina K, Kosina S, Northen TR, Sasse J (2023) The core metabolome and root exudation dynamics of three phylogenetically distinct plant species. Nat Commun 14(1):1649
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf 14:60
Na SI, Kim YO, Yoon SH, Ha SM, Baek I, Chun J (2018) UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J Microbiol 56:280–285
Nakajima T, Kawano Y, Ohtsu I, Maruyuama-Nakashita A, Allahham A et al (2019) Effects of thiosulfate as a sulfur source on plant growth, metabolites accumulation and gene expression in Arabidopsis and rice. Plant Cell Physiol 60(8):1683–1701
Nayfach S, Roux S, Seshadri R, Udwary D, Varghese N, Schulz F et al (2021) A genomic catalog of Earth’s microbiomes. Nat Biotechnol 39(4):499–509
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25(7):1043–1055
Patz S, Gautam A, Becker M, Ruppel S, Rodríguez-Palenzuela P, Huson DH (2021) PLaBAse: a comprehensive web resource for analyzing the plant growth-promoting potential of plant-associated bacteria. BioRxiv. https://doi.org/10.1101/2021.12.13.472471
Peral-Aranega E, Saati-Santamaría Z, Kolařik M, Rivas R, García-Fraile P (2020) Bacteria belonging to Pseudomonas typographi sp. nov. from the bark beetle Ips typographus have genomic potential to aid in the host ecology. InSects 11(9):593. https://doi.org/10.3390/insects11090593
Poveda J, Rodríguez VM, Díaz-Urbano M, Sklenář F, Saati-Santamaría Z et al (2022) Endophytic fungi from kale (Brassica oleracea var. acephala) modify roots-glucosinolate profile and promote plant growth in cultivated Brassica species. First description of Pyrenophora gallaeciana. Front Microbiol 13:981507
Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK (2016) Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8:12–24
Ranadev P, Revanna A, Bagyaraj DJ, Shinde AH (2023) Sulfur oxidizing bacteria in agro ecosystem and its role in plant productivity—a review. J Appl Microbiol 134(8):lxad161
Rhuland LE, Work E, Denman RF, Hoare DS (1995) The behaviour of the isomers of α, ε-diaminopimelic acid on paper chromatograms. J Am Chem Soc 77:4844–4846
Rodriguez-R LM, Konstantinidis KT (2016) The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 4:e1900v1
Rogers JS, Swofford DL (1998) A fast method for approximating maximum likelihoods of phylogenetic trees from nucleotide sequences. Syst Biol 47:77–89
Saati-Santamaría Z (2023) Global map of specialized metabolites encoded in prokaryotic plasmids. Microbiol Spectr 11(4):e01523-e1623
Saati-Santamaría Z, Peral-Aranega E, Velázquez E, Rivas R, García-Fraile P (2021) Phylogenomic analyses of the genus Pseudomonas lead to the rearrangement of several species and the definition of new genera. Biology 10:782
Saati-Santamaría Z, Baroncelli R, Rivas R, García-Fraile P (2022a) Comparative genomics of the genus pseudomonas reveals host-and environment-specific evolution. Microbiol Spectr 10(6):e02370-e2422
Saati-Santamaría Z, Selem-Mojica N, Peral-Aranega E, Rivas R, García-Fraile P (2022b) Unveiling the genomic potential of Pseudomonas type strains for discovering new natural products. Microb Genom 8(2):000758
Saitou N, Nei M (1987) A neighbour-joining method: a new method for reconstructing phylogenetics trees. Mol Biol Evol 44:406–425
Sasse M (1990) Identification of bacteria by gas chromatography of cellular fatty acids, MIDI Technical Note 101. MIDI Inc, Newark
Sasse J, Martinoia E, Northen T (2018) Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci 23(1):25–41
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069
Senghor B, Seck EH, Khelaifia S, Bassene H, Sokhna C, Fournier PE, Raoult D, Lagier JC (2017) Description of ’Bacillus dakarensis’ sp. nov, ’Bacillus sinesaloumensis’ sp. nov, ’Gracilibacillus timonensis’ sp. nov, 'Halobacillus massiliensis’ sp. nov, ’Lentibacillus massiliensis’ sp. nov, ’Oceanobacillus senegalensis’ sp. nov, ’Oceanobacillus timonensis’ sp. nov, ’Virgibacillus dakarensis’ sp. nov. and ’Virgibacillus marseillensis’ sp. nov, nine halophilic new species isolated from human stool. New Microb New Infect 17:45–51
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212
Simonin M, Briand M, Chesneau G, Rochefort A, Marais C, Sarniguet A, Barret M (2022) Seed microbiota revealed by a large-scale meta-analysis including 50 plant species. New Phytol 234(4):1448–1463
Son JS, Hwang YJ, Lee SY, Ghim SY (2019) Bacillus salidurans sp. nov, isolated from salt-accumulated pepper rhizospheric soil. Int J Syst Evol Microbiol 69:116–122
Syberg-Olsen MJ, Garber AI, Keeling PJ, McCutcheon JP, Husnik F (2022) Pseudofinder: detection of pseudogenes in prokaryotic genomes. Mol Biol Evol 39(7):153
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The clustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Tindall BJ (1990) Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol Lett 66:199–202
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173(2):697–703
Wiesmann CL, Wang NR, Zhang Y, Liu Z, Haney CH (2022) Origins of symbiosis: shared mechanisms underlying microbial pathogenesis, commensalism and mutualism of plants and animals. FEMS Microbiol Rev 47(6):fuac048
Zhu Q, Kosoy M, Dittmar K (2014) HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom 15:717
Acknowledgements
The authors thank to Dr Aharon Oren for his valuable help with the naming of the new species. The authors thank the Strategic Research Programs for Units of Excellence from Junta de Castilla y León (Escalera de Excelencia CLU-2O18-04 and CLU-2019-05) co-funded by the Junta de Castilla y León and European Union (ERDF “Europe drives our growth”). ZSS acknowledge a Grant co-financed by the European NextGenerationEU, Spanish “Plan de Recuperación, Transformación y Resiliencia”, Spanish Ministry of Universities, and University of Salamanca (“Ayudas para la recualificación del sistema universitario español 2021–2022”). JDFF acknowledge of a postdoctoral fellowshing funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 101034371.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was supported by Junta de Castilla y León (Spanish Regional Government) Grant SA293P18.
Author information
Authors and Affiliations
Contributions
Conceptualization: EMM, EV; Data curation: PGF, ZSS; Formal Analysis: PGF, ZSS, JDFF, EV; Funding acquisition: EMM; Investigation: PGF, ZSS, JDFF, EV, JMI; Methodology: PGF, ZSS, JDFF, EV, JMI; Project administration: EMM; Supervision: EMM, EV, PGF, Writing—original draft: PGF, ZSS Writing—review and editing: JDFF, EV, JMI, EMM.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Handling editor: David Alvarez-Ponce.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saati-Santamaría, Z., Flores-Félix, J.D., Igual, J.M. et al. Speciation Features of Ferdinandcohnia quinoae sp. nov to Adapt to the Plant Host. J Mol Evol 92, 169–180 (2024). https://doi.org/10.1007/s00239-024-10164-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10164-1