
Abstract
Phylogenetic reconstruction of ribosomal history suggests that the ribonucleoprotein complex originated in structures supporting RNA decoding and ribosomal mechanics. A recent study of accretion of ancestral expansion segments of rRNA, however, contends that the large subunit of the ribosome originated in its peptidyl transferase center (PTC). Here I re-analyze the rRNA insertion data that supports this claim. Analysis of a crucial three-way junction connecting the long-helical coaxial branch that supports the PTC to the L1 stalk and its translocation functions reveals an incorrect branch-to-trunk insertion assignment that is in conflict with the PTC-centered accretion model. Instead, the insertion supports the ancestral origin of translocation. Similarly, an insertion linking a terminal coaxial trunk that holds the L7–12 stalk and its GTPase center to a seven-way junction of the molecule again questions the early origin of the PTC. Unwarranted assumptions, dismissals of conflicting data, structural insertion ambiguities, and lack of phylogenetic information compromise the construction of an unequivocal insertion-based model of macromolecular accretion. Results prompt integration of phylogenetic and structure-based models to address RNA junction growth and evolutionary constraints acting on ribosomal structure.
Avoid common mistakes on your manuscript.
The structure of complex macromolecules must originate from simpler primordial substructures that gradually ‘transform’ into modern counterparts (Eck and Dayhoff 1966; Zuckerkandl 1975). However, establishing evolutionary origins is in itself a daring proposition. It demands the use of an ideographic (historical, retrodictive) conceptual framework that is typical of phylogenetic analysis. This framework generally involves: (i) identification of useful phylogenetic features (e.g., structural ‘characters’ with historical information), (ii) ‘transformation’ models describing character change, (iii) establishing directionality of transformations (time’s arrow) with suitable criteria (e.g., ontogenetic, paleontological, and stratigraphic) or using auxiliary assumptions (e.g., outgroups), and (iv) building rooted trees or networks that optimally portray data-driven retrodictions. Finding realistic evolutionary models is challenging (Kim et al. 2014). Transformations and rooting criteria must comply with background knowledge (e.g., known processes of structural change). Generating trees or networks from data and evolutionary models is computationally demanding. In contrast, the search for truth in the molecular sciences is for the most part nomothetic, seeking universal statements with general predictive utility. These explorations are both philosophically and operationally less complex. Since they are grounded on entities that are extant, they are more easily understood than phylogenetic retrodictions. Unfortunately and as I will describe below, the application of nomothetic thinking to problems in molecular evolution can be deceiving.
The ribosome is the most central ribonucleoprotein complex of the cell. Early research used phylogenetic methods widely used in morphometric analysis and considerations in statistical mechanics to study the origin and evolution of ribosomal RNA (rRNA) (Caetano-Anollés 2002). The study revealed that structural elements involved in ribosomal mechanics were primordial. Subsequent studies generated detailed phylogenetic models of macromolecular accretion, confirmed that ribosomal structures supporting processive readings of RNA originated earlier than the peptidyl transferase center (PTC) responsible for protein synthesis, and revealed tight coevolution between rRNA and associated proteins of the universally conserved ribosomal core (Caetano-Anollés et al. 2008; Sun and Caetano-Anollés 2009; Harish and Caetano-Anollés 2012). In contrast, a series of nomothetic studies simulated the evolution of the large subunit rRNA by dissecting helical-stack interactions in A-minor motifs and building up rRNA structures in concentric shells (e.g., Bokov and Steinberg 2009; Hsiao et al. 2009). Without using phylogenetic analysis and inspired by the ancient ‘RNA world’ hypothesis, studies assumed that the ribosome originated in the PTC and that extant molecular interactions and putative accretion shells were sufficient to portray evolutionary change. The origin of the ribosome was therefore established a priori and its validity left untested.
Petrov et al. (2014) recently extended the models of accretion of the large subunit structural core. They compared atomic structures of bacterial and eukaryotic ribosomes and found that new eukaryote-specific rRNA segments inserted into old common core regions without significantly perturbing local conformations. Assuming that the ancient core evolved via similar insertion and expansion processes, 64 putative insertions of ‘branch’ helices onto preexisting ‘trunk’ helices were identified that involved 59 ancestral expansion segments (AES). Trunk-branch directionality was claimed to reside in ‘insertion fingerprints’ defined by: (i) minimal distortion of trunk segments at branch sites, (ii) bases paired and stacked in trunks on either side of the branch point, (iii) linear trunk axis, (iv) acute deviation of trunk-branch helical axes, and (v) close apposition of trunk sugar and phosphate moieties. A succession of branch-to-trunk insertions delimited six phases of stepwise evolutionary build up of the ribosome that started in the PTC. However, this ‘backward’ extrapolation did not make use of ideographic thinking; no differential sharing of derived structural core features were studied in organisms. Instead, extrapolations demanded that rRNA “evolved by analogous processes throughout its history” and that some premises of temporal ordering of the AES be fulfilled (forcing inside-to-outside growth, late appearance of translocation), which appear unwarranted and problematic. Given questionable assumptions, here I studied each and every AES insertion and expansion site using the structural criteria employed by the authors (listed above). My analysis reveals crucial flaws that compromise the main conclusions of the study:
-
1.
The identification of putative insertion sites often appears subjective and trunk-branch directionality ambiguous. This was particularly severe for distorted AES and two-way junctions. For example, AES7 (composed of H94 and H97 helices) is considered trunk and AES30 (H95 and H96 helices) branch. However, both AES helices showed equal minimum helical distortion at the putative insertion site, similar base stacking, and equally curved axes (Fig. S1). Thus AES30, which holds the functionally important and highly conserved alpha-sarcin-ricin loop, could represent a terminal point of origin in the accretion model. Alternatively, the AES7-30 structure could simply depict the conformation of a standard ‘family cH’ four-way junction that is common in varied RNA species, including riboswitches and the IRES element of the Hepatitis C virus (Laing and Schlick 2009). In fact, many core insertion sites actually constitute well-characterized three-way junctions typical of natural folding structures (Lescoute and Westhof 2006). This questions the likely existence or ancestral identity of insertions.
-
2.
Trunk-branch directionalities of several insertions appear erroneous or have been disregarded. These directionality reversals crucially affect the insertion-based evolutionary model. The insertion spanning AES1 (which supports the P-site of the PTC) and AES39 (harboring coaxial H76 and H79 stems with translocation functions) forms a typical ‘family A’ three-way junction (Lescoute and Westhof 2006). However, I found that AES39 was the older trunk and not AES1 (Fig. 1a). The conflicting AES39 ancestrality crucially falsifies an origin of the ribosome in the PTC. Remarkably, our structural phylogenomic analysis of ribosomal evolution [unacknowledged by Petrov et al. (2014)] supports the origin of large subunit rRNA in H76 (i.e., AES39) and its ribosomal replicative/mechanic functions (Harish and Caetano-Anollés 2012). Similarly, analysis of the AES22-23 insertion that is part of a seven-way junction that supports the central protuberance and L7/12 stalk revealed a directionality reversal that makes the H41 and H45 coaxial stacks (AES23) older than H40 and the rest of the multi-loop structure (Fig. 1b). Again, the phylogenomic model shows that H41, which forms the base of the GTPase center, is the second oldest structure of the large subunit (Harish and Caetano-Anollés 2012). Together with AES1-39 and other insertions that are not discussed, AES22-23 also falsifies an origin of the ribosome in the PTC.
Fig. 1 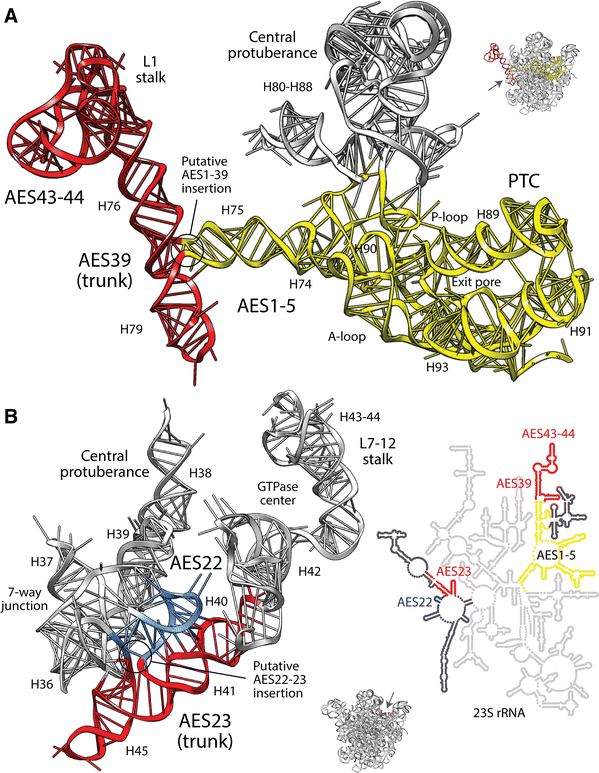
The trunk-branch directionality of putative ancestral insertions of expansion segments suggests an origin of the large rRNA molecule in ribosomal mechanics and not the PTC. a AES39 (red) supports translocation functions of the L1 stalk and AES1-5 (yellow) supports protein biosynthesis. Structural criteria and modeling by energy minimization indicates that the H75 branch of AES1 likely inserted into the H76-H79 trunk of AES39 that was originally stabilized by coaxial stacking. b Similarly, the preexistent AES 23 trunk (red) supports the L7-12 stalk and its GTPase center and the AES22 (blue) branch connects to the rest of the seven-way junction and to the bulk of the rRNA molecule, including the PTC (see detail with secondary structure model). Atomic features of insertions can be seen in Fig. S2. The red-yellow-blue colorings reflect a phylogenetically derived timeline, with red being old (Harish and Caetano-Anollés 2012) (Color figure online)
-
3.
The six-phase evolutionary model cannot be recovered unambiguously from ancestral insertion data. Dissecting pathways of accretion becomes increasingly complicated as trunk-branch relationships unfold from the PTC toward the increasingly branched molecule. Any attempt to fit insertion data into putative accretion shells (Hsiao et al. 2009) and A-minor interaction networks (Bokov and Steinberg 2009) centered on the PTC simply fail to test the a priori hypothesis of PTC-centered ribosomal origins. Malafide insertions and directionality reversals generate uncertainty and new origin scenarios.

In summary, unwarranted assumptions, dismissals of conflicting data, structural insertion ambiguities, and lack of phylogenetic information compromise the validity of the new nomothetic insertion-based model. Putative insertions, however, reveal puzzling matches to previous phylogenetic statements of origin, suggesting they may indeed hold ancient signatures of molecular accretion. My analysis highlights the limitations of structure-based inferences. However, they beg important questions. Do RNA junctions grow by insertions? Are they the natural outcome of folding? What is the role of structural stability and optimization in ribosomal evolution?
References
Bokov K, Steinberg SV (2009) A hierarchical model for evolution of 23S ribosomal RNA. Nature 457(7232):977–980
Caetano-Anollés G (2002) Tracing the evolution of RNA structure in ribosomes. Nucleic Acids Res 30:2575–2587
Caetano-Anollés G, Sun F-J, Wang M, Yafremava LS, Harish A, Kim HS, Knudsen V, Caetano-Anollés D, Mittenthal JE (2008) Origins and evolution of modern biochemistry: insights from genomes and molecular structures. Front Biosci 13:5212–5240
Eck RV, Dayhoff MO (1966) Evolution of the structure of ferredoxin based on living relics of primitive amino acids. Science 152:363–366
Harish A, Caetano-Anollés G (2012) Ribosomal history reveals origins of modern protein synthesis. PLoS ONE 7(3):e32776
Hsiao C, Mohan S, Kalahar BK, Williams LD (2009) Peeling the onion: ribosomes are ancient molecular fossils. Mol Biol Evol 26:2415–2425
Kim KM, Nasir A, Caetano-Anollés G (2014) The importance of using realistic evolutionary models for retrodicting proteomes. Biochimie 99:129–137
Laing C, Schlick T (2009) Analysis of four-way junctions in RNA structures. J Mol Biol 390:547–559
Lescoute A, Westhof E (2006) Topology of three-way junctions in folded RNAs. RNA 12:83–93
Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, Hud NV, Williams LD (2014) Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci USA 111(28):10251–10256
Sun F-J, Caetano-Anollés G (2009) The evolutionary history of the structure of 5S ribosomal RNA. J Mol Evol 69:430–443
Zuckerkandl E (1975) The appearance of new structures and functions in proteins during evolution. J Mol Evol 7:1–57
Acknowledgments
I wish to thank Dr. Niles Lehman for supporting this debate, especially in light of the unwillingness of PNAS to discuss subject matter related to one of their publications. I also thank Derek Caetano-Anollés for help in the analysis of insertion fingerprints and for suggestions. Computational biology is supported by grants from NSF (OISE-1172791 and DBI-1041233) and USDA (ILLU-802-909).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caetano-Anollés, G. Ancestral Insertions and Expansions of rRNA do not Support an Origin of the Ribosome in Its Peptidyl Transferase Center. J Mol Evol 80, 162–165 (2015). https://doi.org/10.1007/s00239-015-9677-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9677-9