
Abstract
Objective
To study the impact of the 79A>C polymorphism in the cytidine deaminase (CDA) gene on the pharmacokinetics of gemcitabine and its metabolite 2′,2′-difluorodeoxyuridine (dFdU) in non-small-cell lung cancer (NSCLC) patients.
Patients and methods
Patients (n = 20) received gemcitabine 1,125 mg/m2 as a 30 min i.v. infusion as part of treatment for NSCLC. Plasma samples were collected during 0–6 h after gemcitabine administration. Gemcitabine and dFdU were quantified by high performance liquid chromatography with ultraviolet detection. The CDA 79A>C genotype was determined with PCR and DNA sequencing.
Results
Gemcitabine was rapidly cleared from plasma and undetectable after 3 h. The allele frequency of the 79A>C polymorphism was 0.40. Diplotypes were distributed as A/A n = 8, A/C n = 8 ,and C/C n = 4. No significant differences were found between the different CDA genotypes and gemcitabine or dFdU AUC, clearance, or half-life.
Conclusion
The 79A>C polymorphism in the CDA gene does not have a major consistent and signficant impact on gemcitabine pharmacokinetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the last decade, several new chemotherapeutic agents have been evaluated for their beneficial effect in patients with non-small-cell lung cancer (NSCLC). So far, gemcitabine plus cisplatin has emerged as one of the standard regimens for the treatment of advanced NSCLC [1]. Nowadays, this regimen is mainly used in patients with squamous cell histology [2].
Gemcitabine (2′,2′-difluorodeoxycytidine; dFdC) is a fluoropyrimidine antimetabolite that is transported into cells by equilibrative nucleoside transporters [3]. Within the cell, gemcitabine is phosphorylated into gemcitabine monophosphate (dFdCMP) by deoxycytidine kinase (dCK) and thereafter into di- and triphosphates [4]. Gemcitabine triphosphate (dFdCTP) is incorporated in DNA, and subsequently only one deoxynucleotide molecule more can be inserted, which leads to the halt of DNA synthesis [5]. This process is amplified through inhibition of the enzyme ribonucleotide reductase (RNR) by gemcitabine diphosphate (dFdCDP). Inhibition of RNR leads to intracellular depletion of dCDP and dCTP, which favours the incorporation of dFdCTP into DNA [6, 7].
Only a small part of the gemcitabine dose is responsible for the cytotoxic effects, since more than 90% of the dose is inactivated by the enzyme cytidine deaminase (CDA) into 2′,2′-difluorodeoxyuridine (dFdU). CDA is ubiquitous in the human body, catalyzing the hydrolytic deamination of (deoxy-)cytidine to (deoxy-)uridine. The total enzyme capacity of CDA in all organs and tissues determines the biotransformation rate of gemcitabine into dFdU and thereby indirectly the duration of exposure to gemcitabine. Interestingly, cloning of human CDA has revealed a 79A > C nonsynonymous coding single nucleotide polymorphism (cSNP) in exon 1 of the CDA gene corresponding with two protein variants with more than twofold difference in vitro deamination rates [8, 9]. The A genotype corresponds to the wild type Lys-carrying enzyme and the C genotype to a Gln-carrying variant (Gln27 CDA). The wild type CDA enzyme has been reported to exert a 1.3- to 3.3-fold higher deamination rate of cytarabine than Gln27 CDA [8]. Thus, the pharmacokinetics of gemcitabine may also be affected by this genetic polymorphism.
The aim of the current study was to evaluate the impact of the common 79A > C cSNP in the CDA gene on the pharmacokinetics of gemcitabine and its metabolite dFdU in gemcitabine-treated NSCLC patients.
Patients and methods
Patient selection
This study was performed as a site study of a phase III trial of gemcitabine plus epirubicin versus gemcitabine plus cisplatin in advanced NSCLC patients [10]. Patients had to meet the inclusion and exclusion criteria of the main study. In short, patients were included if they had stage III/IV NSCLC. No prior chemotherapy was allowed. Radiotherapy should have been completed at least 4 weeks before inclusion, and patients should have recovered from any toxic side effect. All patients had to have a performance status ≤ 2 according to the Eastern Cooperative Oncology Group (ECOG) scale and a life expectancy of at least 12 weeks. An adequate bone marrow reserve and normal renal (creatinine clearance ≥ 60 ml/min) and liver function were required. The glomerular filtration rate (GFR) was calculated according to the formula of Cockcroft and Gault [11]. Patients were excluded if they had active infections, second primary malignancies, uncorrected hypercalcemia, or an LVEF ≤ 45% measured by multiple gated acquisition (MUGA) scan. A detailed description of the inclusion and exclusion criteria is published elsewhere [10]. The local medical ethics committee of the hospital approved the protocol. All patients gave written informed consent before study entry including genotyping procedures.
Treatment and sample collection
Gemcitabine (Gemzar®, Lilly, Nieuwegein, the Netherlands) in a dose of 1,125 mg/m2 in 250 mL 0.9% NaCl solution was administered intravenously as a 30 min infusion. Patients co-treated with epirubicin received the epirubicin dose after completion of the gemcitabine infusion. The cisplatin dose was administered the day after the gemcitabine infusion.
Blood sampling
Blood sampling was carried out on the first day of the first chemotherapy cycle. For pharmacokinetic sampling, a cannula was placed intravenously in the arm of the patient contralateral to the side of drug administration. Blood samples of 9 mL were collected in heparinized tubes containing 0.25 mg tetrahydrouridine (THU) in 50 μL water, just before chemotherapy and at t = 25, 40, 50, 60, 75, 90, 105, 120, 150, 180, 270, and 360 min after the start of the gemcitabine infusion. Blood samples were immediately placed on ice and centrifuged at 2,500 g for 10 min within 1 h after collection. Plasma samples were stored at −80°C until further analysis.
Genotyping material
Harvesting of oral mucosa cells was performed before chemotherapy by thorough mouth rinsing for 30 s with 5 mL 0.9 % NaCl solution. The cell suspension was collected in plastic cups and subsequently placed on ice. Within 1 h after collection, the cell suspension was centrifuged for 10 min at 190 g at 4°C. The supernatant was discarded and cells were washed in 10 mL ice cold phosphate buffered saline, pH = 7.40 (PBS) and centrifuged for 10 min at 190 g at 4°C. The pellet was resuspended in 1 mL ice cold PBS. The remaining suspension was transferred to a microcentrifuge cup and centrifuged briefly (15 s) at 10,000 g. The supernatant was discarded, and the pellet was kept frozen at −80°C until further analysis.
Pharmacokinetic analysis
Gemcitabine hydrochloride and dFdU were obtained from Eli Lilly (Indianapolis, IN). Tetrahydrouridine was purchased from Calbiochem (La Jolla, CA). All other chemicals were of standard analytical grade. The analysis of gemcitabine and dFdU in plasma was carried out by high performance liquid chromatography as described by Freeman et al. [12].
Cytidine deaminase genotyping
The CDA exon 1 polymorphism (C/A) at codon position 27 was genotyped by direct sequencing, in both directions, of PCR-amplified genomic DNA.
First, the CDA region flanking the polymorphic site was amplified using PCR with forward primer 5′- AGTAGCTTCCCCTTCCAGTAGC and reverse primer 5′-CCTCTTCCTGTACATCTTCCTCTG. The 25 μL reactions contained 2.5 units Taq polymerase (Amersham Biosciences, Uppsala, Sweden); 0.5 mM dNTP mix (Roche Diagnostics, Mannheim, Germany); 1x PCR buffer (Roche Diagnostics), 0.05 mM MgCl2; 0.2 μM of each primer; and approximately 50 ng genomic DNA. The amplification was performed on a PTC-225 thermal cycler (MJ Research, Waltham, MA), using a stepdown protocol. The first five cycles were carried out at 94°C, 65°C, and 72°C, each for 30 s. The next five cyles were carried out at 94°C, 63°C, and 72°C, each for 30 s. The last 25 cyles were at 94°C, 60°C, and 72°C, each for 30 s. Following cycling the PCR products were purified with the Qiagen Qiaquick PCR purification kit (Westburg, Leusden, the Netherlands). Subsequently, 100 ng of the purified PCR product was cycle sequenced with a Dyeterminator kit (US81090, Amersham Biosciences, Roosendaal, the Netherlands) in a thermal cycler (MJ Research), using 0.05 mM sequencing primer. For the reverse reaction, the same primer was used as in the PCR, but for the forward reaction an internal primer was used (5′-GGTACCAACATGGCCCAGAAG). After the cycle reaction the sequencing products were cleaned on a Sephadex plate (Amersham Biosciences) by centrifugation for 5 min at 910 g. The eluted sample was vacuum dried for 45 min at 65°C. Finally, 20 μL of loading solution (Amersham Biosciences) was added to dissolve the sequencing products. The samples were analyzed on a MegaBACE 1000 capillary sequencer (Amersham Biosciences) by injecting the samples for 45 s at 3 kV and running them for 5 h at 4 kV. The data were processed with Sequence Analyzer 3.0 (Amersham Biosciences) and Seqman II (DNASTAR, Madison, WI).
Pharmacokinetic modelling
Pharmacokinetic data were analyzed with the Mw\Pharm software package (version 3.5; MediWare, Groningen, the Netherlands) using the KinFit module. For gemcitabine and its metabolite dFdU, the AUC (using the trapezoid rule), clearance, distribution volume, and elimination half-lives were calculated by non-compartmental analysis.
Statistical analysis
Patient data were grouped according to genotype. Differences in laboratory and demographic data between groups were tested using the Mann-Whitney test.
CDA genotype-related differences in pharmacokinetic data were investigated by analyzing the wild type AA genotype patient group against the combined heterozygote AC and homozygote CC genotype patient group using the Mann-Whitney test. We assumed an effect of 30% of the alternative genotype on the gemcitabine AUC as clinically relevant. From the literature, the allele frequency of the wild type genotype was estimated at 0.7, and therefore an almost equal distribution of patients in AA wild type (49%) and combined homozygote CC plus heterozygote AC (51%) patient groups was expected. We calculated that group sample sizes of 10 and 10 were needed to achieve 92% power to detect a difference of 30% between the null hypothesis that both group means are equal and the alternative hypothesis that the mean AUC of the mutant group is 30% larger, with group standard deviations of 20% and with a significance level of 0.05 using a two-side Mann-Whitney test assuming that the actual distribution is logistic.
Additionally, differences between uncombined CDA genotype groups regarding gemcitabine AUC, clearance, and half-life were also analyzed using the Kruskall-Wallis test. Statistical analyses were performed using the SPSS 14.0 statistical package (SPSS 2005, Chicago, IL). Power calculations were performed using NCSS 2004 Statistical & Power Analysis Software (NCSS, Kaysville, UT).
Results
Patients
A total number of 20 patients were included. Patients were grouped according to CDA genotype. Patient characteristics are presented in Table 1.
The groups were comparable with respect to demographic parameters, renal function, and liver enzymes. All patients, except one, had AST levels below the upper limit of normal. Two patients had a slightly reduced creatinine clearance (CrCL), between 50 and 60 ml/min. Co-administration of epirubicin and cisplatin was equally distributed within and between groups.
Pharmacokinetics
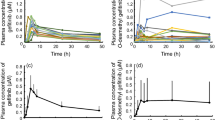
Mean pharmacokinetic curves of gemcitabine and dFdU in plasma are presented in Fig. 1 according to the different genotypes. Gemcitabine was rapidly cleared from plasma and undetectable after 3 h in all patients. The pharmacokinetic data are presented in Table 2.

Pharmacokinetics of gemcitabine and its metabolite dFdU after a 30 min infusion of 1,125 mg/m2 gemcitabine grouped according to genotype of the cytidine deaminase 79A>C polymorphism
The influence of the presence of epirubine on gemcitabine pharmacokinetics was checked by grouping the patients according to treatment schedule (gemcitabine plus cisplatin or gemcitabine plus epirubicine). No difference in gemcitabine pharmacokinetics was measured between the two groups using the Mann-Whitney U-test.
Pharmacogenetic analysis
The gemcitabine pharmacokinetic data of all 20 patients were analyzed with respect to the impact of the 79A > C polymorphism in the CDA gene. The frequencies of the A/A, A/C, and C/C genotypes were 0.40, 0.40, and 0.20 respectively. The allele frequency of the 79A > C polymorphism was 0.40 in our study. Detailed data are presented in Table 2. The mean pharmacokinetic curves of gemcitabine and dFdU according to CDA genotype are presented in Fig. 1. No differences in pharmacokinetic parameters were observed between the wild type AA genotype group and the combined heterozygote AC and homozygote CC genotype group, using the Mann-Whitney test. Additionally, in the Kruskal-Wallis analysis, we found no significant differences in gemcitabine AUC, clearance, or serum half-life among the different CDA genotypes (see Table 2).
Discussion
In this study, we investigated the impact of the common 79A > C nonsynonymous single nucleotide polymorphism in the cytidine deaminase gene on gemcitabine and dFdU pharmacokinetics in NSCLC patients. In all patients gemcitabine was rapidly cleared from plasma, as was expected from previous reports [13].
The most important factor that determines the clearance rate of gemcitabine is the activity of CDA. This enzyme rapidly catabolizes gemcitabine. The total enzyme capacity of CDA in all organs and tissues determines the biotransformation rate of gemcitabine into dFdU and thus the duration of exposure to gemcitabine. Despite increasing interest in DNA polymorphisms related to gemcitabine metabolism, so far only a few reports have been published on the relation between CDA polymorphisms and gemcitabine pharmacokinetics.
The 79A > C cSNP is the most common CDA polymorphism with previously reported allele frequencies of 0.20 in Japanese, 0.11 in African Americans, 0.04 in Africans, 0.30 in Caucasian Americans, and 0.36 in European Caucasians [8, 14–16]. The polymorphism at nucleotide position 79 corresponds to codon27, which is located within an N-terminal core domain and might be involved in binding pocket loops that cooperate with zinc ligands [15, 17]. The A genotype corresponds to the wild type Lys-carrying enzyme and the C genotype to a Gln-carrying variant (Gln27 CDA). Kirch et al. found that the deamination rate of cytarabine by bacterial wild type CDA enzyme was a 1.3–3.3 fold higher than by Gln27 CDA [7]. Gilbert et al. recently reported in a functional genomics study in mammalian cells that the activity of human recombinant Gln27 CDA was about 66% of the wild type CDA activity for gemcitabine [8]. Thus, assuming highest CDA activity in A/A, intermediate activity in A/C, and lowest in C/C diplotypes, we studied the consequences of this polymorphism on gemcitabine AUC, clearance, and serum half-life.
All patients in our study were Caucasian Europeans, and in this group we found an allele frequency of 0.40, which is in line with the allele frequencies of 0.30 and 0.36 earlier reported for Caucasians [8, 16]. We did, however, not observe an impact of the CDA 79A > C genotype on gemcitabine pharmacokinetics. Obviously, our study was powered to detect differences of at least 30% in population means and smaller differences in pharmacokinetic parameters may have gone unnoticed. In the post-hoc statistical analysis, we found a much larger variation in gemcitabine AUC values, ranging from 30–50%, than the initially expected 20%. As a result we achieved only 25% power to detect a difference. We reasoned however that, due to the fact that the data in both groups largely overlap, it seems very unlikely that dose adjustment according to CDA genotype will become clinically feasible, if a difference might exist.
Interestingly, this matter has recently been elucidated, as, in parallel with our study, Sugiyama et al. investigated the 79A > C polymorphism in a population of 251 Japanese patients. In line with our data, they could not detect any effect of the 79A > C polymorphism on the gemcitabine pharmacokinetics after a 30 min 800–1,000 mg/m2 infusion [14]. It is important to realize that the two studies differ with respect to administered gemcitabine dose and patient ethnicity. The mean dose in our study was 38% higher as a result of both a higher standard dose (1,125 mg/m2) and generally higher body surface area (BSA) values in Caucasians. Obviously, these factors do not change or modulate the observed lack of impact on pharmacokinetic outcomes of the 79A > C polymorphism.
One explanation for our findings may be a far more complex regulation of CDA gene transcription, leading to substantial interindividual differences in quantities of CDA enzyme, unrelated to the 79A > C polymorphism. Although Kirch et al. [8] and Gilbert et al. [9] demonstrated reduced enzyme activity with the 79A > C polymorphism using in vitro deamination of ara-C and dFdC, respectively, it is possible that additional polymorphisms within CDA also influence enzyme activity or that variants in other enzymes in gemcitabine metabolism play a greater than expected role. Other studies have shown that the CDA ‘5 flanking region contains numerous potential transcription factor binding sites that can cause variations in transcription [18]. Unfortunately, we did not perform direct measurement of CDA activity in our patients and thus could not check the genotype-phenotype relation at the enzyme level.
Moreover, we hypothesized that possibly the total capacity of CDA in the body may far exceed the amount needed to cope with plasma levels resulting from 800–1,125 mg/m2 gemcitabine doses. In that case, differences in pharmacokinetics may only become manifest at much higher plasma concentrations when Michaelis-Menten enzyme saturation is becoming relevant.
Finally, the pharmacokinetic results might have been dispersed as a result of co-administration of other chemotherapeutic agents. Indeed, all patients in our study received combined chemotherapy, but cisplatin was administered on day 2 and therefore did not interact with our results. We analyzed that co-administration of epirubicin did not influence the pharmacokinetics of gemcitabine, which is in line with previously reported findings regarding the pharmacokinetics of both drugs when combined in treatment schedules [19–21].
Although we could not demonstrate an effect of the CDA 79A > C polymorphism on gemcitabine pharmacokinetics, the polymorphism still may be relevant at the cellular level. Bathla et al. recently demonstrated that the post-induction treatment-related mortality in children treated for acute myeloid leukemia with high dose cytarabine was significantly higher in children bearing the CC compared to the AC or AA genotype [22]. Cytarabine and gemcitabine share a common metabolic pathway. Furthermore, Tibaldi et al. showed that the wild type CDA Lys27Lys (AA) genotype was associated with better clinical outcome, longer time to progression, and overall survival [23]. Interestingly, they also found an increased CDA enzymatic activity in red blood cells associated with the CDA Gln/Gln (CC) genotype. Although these findings contradict previous results from in-vitro functional genomics experiments by Kirch et al. and Gilbert et al., this clinical study illustrates that the association between genotype and treatment efficacy may be quite complex.
Recently two new polymorphisms in the CDA gene were reported. These concern the 208G>A and 435T>C polymorphism with allele frequencies of 0.04 and 0.30 in Japanese patients, respectively [15]. The 208G>A polymorphism produces an alanine-to-threonine substitution (Ala70Thr) within the conserved catalytic domain. Introduction of this gene in yeast null mutants resulted in a 20% reduction of the 50% inhibitory concentration value for cytarabine [15]. Sugiyama et al. also studied both variants and found a decreased gemcitabine clearance in patients with the 208G>A polymorphism [14]. However, the 208G>A polymorphism has not been detected so far in Caucasians and was therefore not included in our study. The 435T>C polymorphism (Thr145Thr) does not encode for an amino acid change. Since an impact of this silent 425T>C polymorphism on CDA enzyme activity was expected to be less probable, we did not include this polymorphism in our investigations.
Conclusions
We conclude that the common 79A>C CDA polymorphism does not have a major consistent, significant effect on gemcitabine plasma clearance. The full pharmacogenetic picture of CDA in relation to gemcitabine clearance and gemcitabine pharmacodynamics may nevertheless be quite complex and can only be elucidated in large clinical trials.
Abbreviations
- AUC:
-
Area under the curve
- BSA:
-
Body surface area
- CDA:
-
Cytidine deaminase
- CrCL:
-
Creatinine clearance
- dCDP:
-
Deoxycytidine diphosphate
- dCTP:
-
Deoxycytidine triphosphate
- dFdC:
-
Gemcitabine
- dFdCMP:
-
Gemcitabine monophosphate
- dFdCDP:
-
Gemcitabine diphosphate
- dFdCTP:
-
Gemcitabine triphosphate
- dFdU:
-
2′,2′-Difluorodeoxyuridine
- GFR:
-
Glomerular filtration rate
- HPLC:
-
High performance liquid chromatography
- NSCLC:
-
Non-small-cell lung cancer
- RNR:
-
Ribonucleotide reductase
- THU:
-
Tetrahydrouridine
References
Schiller JH et al (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346:92–98
Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, Serwatowski P et al (2008) Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 26:3543–3551
Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA et al (1998) Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58:4349–4357
Heinemann V, Hertel LW, Grindey GB, Plunkett W (1988) Comparison of the cellular pharmacokinetics and toxicity of 2′, 2′-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res 48:4024–4031
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W (1991) Action of 2′, 2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 51:6110–6117
Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W (1990) Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′, 2′-difluorodeoxycytidine. Mol Pharmacol 38:567–572
Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V (1995) Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 22(4 Suppl 11):3–10
Kirch HC, Schroder J, Hoppe H, Esche H, Seeber S, Schutte J (1998) Recombinant gene products of two natural variants of the human cytidine deaminase gene confer different deamination rates of cytarabine in vitro. Exp Hematol 26:421–425
Gilbert JA et al (2006) Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res 12:1794–1803
Wachters FM et al (2003) First-line gemcitabine with cisplatin or epirubicin in advanced non-small-cell lung cancer: a phase III trial. Br J Cancer 89:1192–1199
Cockcroft DW, Gault MH (1976) Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41
Freeman KB et al (1995) Validated assays for the determination of gemcitabine in human plasma and urine using high-performance liquid chromatography with ultraviolet detection. J Chromatogr B Biomed Appl 665:171–181
Abbruzzese JL et al (1991) A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9:491–498
Sugiyama E et al (2007) Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J Clin Oncol 25:32–42
Yue L, Saikawa Y, Ota K, Tanaka M, Nishimura R, Uehara T, Maeba H et al (2003) A functional single-nucleotide polymorphism in the human cytidine deaminase gene contributing to ara-C sensitivity. Pharmacogenetics 13:29–38
Fukunaga AK, Marsh S, Murry DJ, Hurley TD, McLeod HL (2004) Identification and analysis of single-nucleotide polymorphisms in the gemcitabine pharmacologic pathway. Pharmacogenom J 4:307–314
Betts L, Xiang S, Short SA, Wolfenden R, Carter CW (1994) Cytidine deaminase. The 2.3A crystal structure of an enzyme: transition-state analog complex. J Mol Biol 235:635–656
Demontis S, Terao M, Brivio M, Zanotta S, Bruschi M, Garattini E (1998) Isolation and characterization of the gene coding for human cytidine deaminase. Biochim Biophys Acta 1443:323–333
Perez-Manga G et al (2002) Gemcitabine in combination with doxorubicin in advanced breast cancer: final results of a phase II pharmacokinetic trial. J Clin Oncol 18:2545–2552
Fogli S, Danesi R, Gennari A, Donati S, Conte PF, Del Tacca M (2002) Gemcitabine, epirubicin and paclitaxel: pharmacokinetic and pharmacodynamic interactions in advanced breast cancer. Ann Oncol 13:919–927
Conte PF et al (2001) Gemcitabine plus epirubicin plus taxol (GET) in advanced breast cancer: a phase II study. Breast Cancer Res Treat 68:171–179
Bathla D, Gerbing RB, Alonzo TA, Conner H, Ross JA, Meshinchi S, Zhai X et al (2008) Cytidine deaminase genotype and toxicity of cytosine arabinoside therapy in children with acue myeloid leukemia. Br J Haematol 144:388–394
Tibaldi C, Giovannetti E, Vasile E, Mey V, Laan A, Nannizzi S et al (2008) Correlation of CDA, ERCC1, and XPD polymorphisms with response and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res 14:1797–1803
Conflict of interest
An unrestricted research grant from Eli Lilly, Nieuwegein, the Netherlands, was received for this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Maring, J.G., Wachters, F.M., Slijfer, M. et al. Pharmacokinetics of gemcitabine in non-small-cell lung cancer patients: impact of the 79A>C cytidine deaminase polymorphism. Eur J Clin Pharmacol 66, 611–617 (2010). https://doi.org/10.1007/s00228-010-0799-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-010-0799-0