
Abstract
Background and Objective
Dacomitinib is a kinase inhibitor indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR)-activating mutations. To evaluate the effect of hepatic impairment on the pharmacokinetics of dacomitinib, two dedicated studies were conducted to inform optimal dosing.
Methods
Study 1 (NCT01571388) evaluated the effect of mild and moderate hepatic impairment on the plasma pharmacokinetics, safety, and tolerability after a single oral dose of dacomitinib 30 mg, and Study 2 (NCT03865446) evaluated the same endpoints in a severe hepatic impairment population. Both studies were phase I, open-label, parallel-group studies. A one-way analysis of variance (ANOVA) with unequal variance assumption and hepatic impairment group as a fixed effect was used to compare the natural log of area under the plasma concentration-time curve extrapolated to infinite time (AUCinf), AUC from time zero to the last quantifiable concentration (AUClast), and maximum plasma concentration (Cmax) for each hepatic impairment group to the respective normal hepatic function group. Since dacomitinib is a cytochrome P450 (CYP) 2D6 substrate, only participants with extensive or intermediate CYP2D6 phenotypes were included in the primary analysis.
Results
The AUCinf for participants with mild, moderate, or severe hepatic impairment decreased by 6%, decreased by 23%, and increased by 4%, respectively, compared with normal hepatic function, while the Cmax for participants with mild, moderate, or severe hepatic impairment increased by 3%, decreased by 20%, and increased by 31%, respectively, compared with normal hepatic function. A single oral dose of dacomitinib 30 mg was well tolerated in all participants.
Conclusion
Based on these pharmacokinetic results, dacomitinib pharmacokinetics of participants with mild, moderate, or severe hepatic impairment were not statistically different relative to participants with normal hepatic function based on the ANOVA analysis. No dacomitinib dose adjustments for patients with hepatic impairment are recommended.
Clinical Trial Registration
ClinicalTrials.gov NCT01571388, registered 5 April 2012; ClinicalTrials.gov NCT03865446, registered 6 March 2019.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dacomitinib 30 mg was tolerated in all participants across Study 1 and Study 2. |
Changes in area under the curve and maximum plasma concentration for participants across all hepatic impairment groups were not considered to be clinically significant. |
No dacomitinib dosing adjustments are necessary for patients with mild, moderate, or severe hepatic impairment. |
1 Introduction
Epidermal growth factor receptor (EGFR)-activating mutations in non-small cell lung cancer (NSCLC) are well-known driver mutations for oncogene addicted survival and proliferation of NSCLC that occur in pulmonary adenocarcinomas. In NSCLC, the use of EGFR tyrosine kinase inhibitors (TKIs) demonstrated improvement over standard of care platinum-based doublet chemotherapies [1,2,3,4,5,6,7]. Evidence strongly implicates the EGFR-activating mutations (exon 19 deletions or L858R point mutation in exon 21) as a mechanism of tumorigenesis and the determinant of sensitivity to the EGFR TKIs dacomitinib, gefitinib, erlotinib, afatinib, and osimertinib.
Dacomitinib is a selective, adenosine triphosphate (ATP)-competitive, irreversible, small-molecule inhibitor of the ErbB human epidermal growth factor receptor (HER) family of receptor tyrosine kinases (RTKs) [8]. When used as first-line treatment in patients with EGFR mutation-positive NSCLC, dacomitinib was found to significantly improve progression-free survival (PFS) and overall survival (OS) over gefitinib, a first-generation EGFR TKI [9,10,11]. Based on these data, dacomitinib was approved for the first-line treatment of patients with metastatic NSCLC containing EGFR exon 19 deletion or exon 21 L858R substitution [12].
Hepatic metabolism is the main route of clearance of dacomitinib, with oxidation and glutathione conjugation as the major pathways. The most abundant circulating metabolite was O-desmethyl dacomitinib (PF-05199265), which had similar in vitro pharmacologic activity as dacomitinib. Cytochrome P450 (CYP) 2D6 was the major isozyme involved in the formation of O-desmethyl dacomitinib, while CYP3A4 contributed to the formation of other minor oxidative metabolites [12]. Following a single oral 45 mg dose of [14C] radiolabeled dacomitinib, 79% of the radioactivity was recovered in feces (20% as dacomitinib) and 3% in urine (<1% as dacomitinib) [13].
Liver impairment is expected to affect the pharmacokinetics of drugs metabolized by the liver; however, depending on the characteristics of the drug investigated, the extent of effect that liver impairment can have on the pharmacokinetics of the drug can vary. In a review by Rodighiero, the author states that liver disease can (1) reduce the activity of drug metabolizing enzymes; (2) reduce the synthesis of plasma proteins; or (3) change liver blood flow [14]. Because hepatic metabolism is the main route of clearance for dacomitinib, it is expected that hepatic impairment may have an effect on dacomitinib clearance and thus dacomitinib area under the concentration-time curve (AUC).
Study 2 (NCT03865446) was a dedicated hepatic impairment study that evaluated the effect of severe hepatic impairment on the pharmacokinetics of dacomitinib, as a follow up to Study 1 (NCT01571388, Giri et al. [15]) in which the effect of mild and moderate hepatic impairment was evaluated on the pharmacokinetics of dacomitinib. These two studies were conducted to inform the drug label to examine any need to dose reduce dacomitinib in the presence of hepatic impairment. In this publication, the effect of mild, moderate, and severe hepatic impairment on dacomitinib pharmacokinetics was evaluated for participants with normal CYP2D6 metabolic function, using the data from Study 1 and Study 2, to inform the appropriate clinical use and dose regimen of dacomitinib in patients with NSCLC with underlying hepatic dysfunction. Dacomitinib (VIZIMPRO) is distributed by Pfizer Laboratories, New York, NY, USA.
2 Methods
This analysis pooled the data from across two studies and aligned the PGX criteria across the participant data. Study 1 was a phase I, open-label, non-randomized, parallel group study assessing the pharmacokinetic profile of a single 30 mg oral dose of dacomitinib in healthy participants with normal hepatic function or participants with mild and moderate hepatic impairment. The study design has been previously described by Giri et al. [15]. Two subjects from the original publication have been removed in this analysis to only include participants who were extensive metabolizers (EMs) or intermediate metabolizers (IMs) of CYP2D6. By removing participants who are poor metabolizers (PMs) or ultrarapid metabolizers (UM), the analysis can control for the effect that polymorphisms may have on the pharmacokinetics of dacomitinib and align the Study 1 population with the Study 2 population.
The remainder of the Methods section will focus on Study 2, which was a phase I, open-label, non-randomized, parallel-group study that assessed the pharmacokinetic profile of a single 30 mg oral dose of dacomitinib in healthy participants with normal hepatic function or participants with severe hepatic impairment.
2.1 Eligibility of Participants
Eligible participants for both studies were male and/or female of non-childbearing potential, aged 18–75 years inclusive, body weight of >50 kg, and a body mass index (BMI) of 17.5–40 kg/m2. Participants with an estimated glomerular filtration rate (eGFR) of ≤60 mL/min/1.73 m2 based on the Modification of Diet in Renal Disease (MDRD) equation were excluded from the trial. Participants with hepatic impairment were required to have hepatic dysfunction due to hepatocellular disease and not secondary to any acute ongoing hepatocellular process, with documented stable hepatic impairment (Child–Pugh class A, B, or C) with no clinically significant changes 30 days prior to dacomitinib dosing, and healthy participants were matched for age (±5 years for Study 1 and ±10 years for Study 2) and body weight (±10 kg for Study 1 and ±15 kg for Study 2) and sex.
Key exclusion criteria included the following: (1) for all participants, any condition possibly affecting drug absorption (e.g., gastrectomy), previous administration with an investigational drug within 30 days (or as determined by the local requirement), or five half-lives preceding the first dose of investigational drug used in this study (whichever was longer); (2) for healthy participants, use of prescription or non-prescription drugs and dietary or herbal supplements within 7 days or five half-lives (whichever was longer) prior to the first dose of study medication (except acetaminophen ≤1 g/day); (3) for participants with severe hepatic impairment, use of CYP2D6 substrates with a narrow therapeutic index, CYP2D6 inhibitors within 7 days or five half-lives (whichever was longer) prior to the first dose of dacomitinib until the completion of the last pharmacokinetic sample collection, and proton pump inhibitors (PPIs) within 7 days prior to the first dose of dacomitinib until the completion of the last pharmacokinetic sample collection; hepatic carcinoma and hepatorenal syndrome; and/or any clinically significant disease that contraindicated study drug or that may affect the pharmacokinetics of dacomitinib.
2.2 Dacomitinib Administration
A 30 mg single oral dose of dacomitinib was administered after a >8-h fast with 180 (Study 1) or 240 mL (Study 2) of water. In order to standardize the conditions on pharmacokinetic sampling days, all participants were required to refrain from lying down (except when required for blood pressure [BP], pulse rate, and electrocardiogram [ECG] measurements), eating, and drinking beverages other than water during the first 4 h after dosing. Participants were required to stay in-house at least until the collection of the 144 h (Day 7) pharmacokinetic sample following dacomitinib dosing, and return for follow-up visits on Days 8–10 and Day 12.
2.3 Evaluation of Pharmacokinetics and Protein Binding
Blood samples of approximately 3 mL were collected into appropriately labeled tubes containing dipotassium ethylenediamine tetra-acetic acid (K2EDTA) at each specified time point for measurement of plasma concentrations of dacomitinib and its metabolite PF-05199265. Serial blood samples for pharmacokinetics were collected predose and at 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, 216, and 264 h following dosing. Separate blood samples (10 mL) were collected into tubes containing K2EDTA to measure plasma protein binding of dacomitinib and PF-05199265 at predose and 8 and 48 h postdose in Study 1; and at predose and 6 and 8 h postdose in Study 2 [15].
The pharmacokinetic parameters determined for dacomitinib and its metabolite PF-05199265, using non-compartmental analysis (using openNCA, a Pfizer proprietary validated software) of the observed plasma concentration–time data, are reported in Table 1. The actual sample collection times were used for the pharmacokinetic parameter analysis. Parameters AUC extrapolate to infinite time (AUCinf), terminal elimination half-life (t½), apparent clearance after oral dose (CL/F), and apparent volume of distribution after oral dose (Vz/F) were reported only when a well characterized terminal phase was observed, defined as one with at least three data points, r2 ≥ 0.9, and the extrapolated AUC (AUCextrap) % ≤20; however, based on extensive sampling and a good regression (r2 ≥ 0.9), the acceptance criteria for dacomitinib was expanded to include additional profiles with an AUCexrap% ≤40. Concentrations that were below the limit of quantification (BLQ) were set to zero for analysis.
2.4 Bioanalytical Assay
Plasma samples were analyzed for dacomitinib and PF-05199265 concentrations at Intertek Pharmaceutical Services (San Diego, CA, USA) using a validated, sensitive, and specific high-performance liquid chromatography (HPLC) atmospheric pressure ionization tandem mass spectrometry (MS/MS) method. Plasma protein binding samples were analyzed for the extent of the protein binding of dacomitinib and its metabolite PF-05199265 at Intertek Pharmaceutical Services. The samples were dialyzed using a Rapid Equilibrium Dialysis (RED) method. The dialyzed phosphate buffered saline was analyzed for unbound concentrations of dacomitinib and PF-05199265 using the same validated method above.
Calibration standard responses were linear over the range of 0.100–20.0 ng/mL for both analytes using a weighted (L/concentration2) linear least-squares regression. Those samples with concentrations above the upper limits of quantification were adequately diluted into the calibration range. The lower limit of quantification (LLOQ) for dacomitinib and PF-05199265 was 0.100 ng/mL. Clinical specimens with plasma dacomitinib and PF05199265 concentrations below the LLOQ are reported as BLQ. Additional information on the dacomitinib assay can be found in the study by Geri et al. [15].
2.5 Assessment of Safety
The overall safety profile was characterized by laboratory test abnormalities, physical examination, vital signs, ECGs, and adverse events (AEs). Similar to Study 1, in Study 2 BP and pulse rate were measured at screening, predose on Day 1, 24 h, 144 h, and follow up. A single standard 12-lead ECG was obtained following a 10-min supine-rest at screening, predose on Day 1, 6 h, 144 h, and at follow up for Study 2, and at screening, predose on Day 1, and at 6, 48, and 264 h postdose for Study 1. An AE is any untoward medical occurrence in a patient or clinical study participant, temporally associated with the use of study intervention, whether or not considered related to the study intervention. The investigator makes assessment of intensity for each AE as follows.
-
Mild: An event that is easily tolerated by the participant, causing minimal discomfort and not interfering with everyday activities.
-
Moderate: An event that causes sufficient discomfort and interferes with normal everyday activities.
-
Severe: An event that prevents normal everyday activities.
An SAE is defined as any untoward medical occurrence that results in death, is life threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent disability/incapacity, is a congenital anomaly/birth defect, or in other cases where an event may jeopardize the participant or may require medical or surgical intervention to prevent one of the other outcomes listed above.
2.6 Cytochrome P450 (CYP) 2D6 Genotyping
A 3 mL blood sample was collected prior to Day 1 from each participant for genotyping CYP2D6 polymorphism at Pfizer Clinical Pharmacogenomics Laboratory (Groton, CT, USA). Sixteen polymorphisms including 15 single nucleotide polymorphisms [*2(2850), *2(4180), *3, *4, *5, *6, *7, *8, *9, *10, *14, *17, *18, *21, *36, *41] and 1 gene duplication (XN) were evaluated (Table 2). The allelic nomenclature is based on the ‘star’ nomenclature (https://www.pharmvar.org/) [16]. The inference of phenotypes from genotypes was based on the guidelines on the Pharmacogenomics Knowledge Base (PharmGKB) website (https://www.pharmgkb.org/) [17].
Only participants identified as a CYP2D6 EM or CYP2D6 IM were included in the primary statistical analysis to evaluate the impact of severe hepatic impairment on dacomitinib plasma pharmacokinetics.
2.7 Statistics
Sample size was based on recommendations from US FDA guidance on the assessment of the influence of hepatic impairment on drug pharmacokinetics (FDA guidance for impaired hepatic function, 2003), and eight participants were planned to be enrolled into each group. One-way analysis of variance (ANOVA) was used to compare the natural log-transformed AUCinf, AUClast, Cmax, AUCinf,u, AUClast,u and Cmax,u of dacomitinib and PF-05199265 for the hepatic impairment cohort to the normal hepatic function cohort. This analysis was conducted in RStudio version 3.6.1 (RStudio, PBC, Boston, MA, USA) for Study 1 and SAS version 8 (SAS Institute, Inc., Cary, NC, USA) for Study 2. Estimates of the adjusted mean differences and corresponding 90% confidence intervals (CIs) were obtained from the ANOVA analysis. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the adjusted geometric mean ratios (GMRs) [test/reference] and 90% CIs for the ratios as a percentage. The ANOVA analysis was conducted in accordance with the FDA Guidance for Industry: Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling [18].
3 Results
3.1 Participant Disposition and Baseline Characteristics
Twenty-five participants were included in Study 1; eight participants had normal hepatic function, eight had mild hepatic impairment, and nine had moderate hepatic impairment. Study 2 included a total of 16 participants; eight of the participants were healthy and eight had severe hepatic impairment. All 16 participants were treated with dacomitinib and all completed the study.
Participant demographics across both studies at baseline are summarized in Table 3. In both studies, all participants were male and the participant group with normal hepatic function was comparable with the hepatic impairment groups with respect to weight, age, and sex.
3.2 CYP2D6 Genotypic Results
To diminish the potential influence of CYP2D6 polymorphisms on dacomitinib pharmacokinetics, only participants identified as a CYP2D6 EM or CYP2D6 IM were included in the primary statistical analysis. All 16 treated participants in Study 2 were identified as an EM or IM. In Study 1, 23 participants were identified as an EM or IM, with one UM in the moderate metabolizer group and one PM in the normal hepatic impairment group (Table 3). The pharmacokinetic data for the UM and PM participants were reported individually and were not included in the primary statistical analysis (electronic supplementary Table S1).
3.3 Pharmacokinetic Results
The observed plasma concentration versus time profiles and pharmacokinetic parameters of dacomitinib in participants with normal hepatic function, or mild, moderate, or severe hepatic impairment are presented in Fig. 1 and Table 4, respectively. The dacomitinib pharmacokinetics for both healthy volunteer groups were similar to what has been described in the FDA-approved dacomitinib label [12].

Median concentrations for dacomitinib and PF-05199265 for Study 1 and Study 2 (linear and semi-log). Concentrations that were below the LLOQ were set as LLOQ/2, 0.05 ng/mL. The vertical dashed error bars represent the 5th and 95th percentiles around the median concentrations. LLOQ lower limit of quantification
3.3.1 Dacomitinib Pharmacokinetics
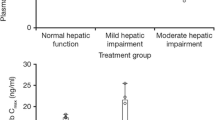
Following administration of a single oral dose of dacomitinib 30 mg in the fasted state, there was no significant effect of severe hepatic impairment on AUCinf based on the adjusted GMR, however Cmax was increased by 31% when compared with the healthy participant group. Similar to the results of Study 1, the CL/F was only slightly different between the severe hepatic impairment group and the normal hepatic function group, with values of 40.83 and 42.61 L/h, respectively (Table 4). Vz/F stayed relatively the same between the two treatment groups but the half-life of the severe hepatic function group was higher (75.63 h) than that of the normal hepatic function group (63.90 h) (Table 4).
Results for Study 1 demonstrated that dacomitinib AUCinf and Cmax were unchanged in participants with mild hepatic impairment and decreased by 23% and 20%, respectively, in participants with moderate hepatic impairment when compared with participants with normal hepatic function (Table 4).
3.3.2 PF-05199265 Pharmacokinetics
Regarding PF-05199265 exposure, concentrations were slightly lower among participants with mild hepatic impairment compared with participants with normal hepatic function (Fig. 2). This trend was more pronounced for the total exposure in the moderate and severe hepatic impairment groups, indicated by decreases in AUCinf and Cmax of 43% and 58%, respectively, for the moderate hepatic impairment group and 19% and 66%, respectively, for the severe hepatic impairment group compared with the normal liver function group (Table 4). Additionally, the half-life of PF-05199265 was greatly increased in the severe hepatic impairment group (113.9 h) compared with the normal hepatic function group (72.79 h). The metabolite-to-dacomitinib ratios were larger in the normal hepatic function group than in the severe hepatic impairment group, with MRAUCinf and MRCmax values of 0.4460 and 0.5443 versus 0.3117 and 0.1671, respectively (Table 4).

Boxplots for AUCinf and Cmax for Study 1 and Study 2. The red diamond represents the geometric mean value for each respective group; the black line represents the median value; black circles represent the actual parameters; and the upper and lower hinges represent the 25th and 75th percentiles. The lower whisker extends to the smallest value, no less than 1.5 times the interquartile range, and the upper whisker extends to the largest value, no greater than 1.5 times the interquartile range. Black dots outside of the whiskers represent outliers. AUCinf area under the plasma concentration–time curve from time zero extrapolated to infinite time, Cmax maximum plasma concentration
3.4 Protein-Binding and Unbound Dacomitinib Pharmacokinetic Parameters
High plasma protein binding of dacomitinib and PF-05199265 were observed in participants with normal hepatic function as well as mild, moderate, or severe hepatic impairment. In Study 1, the fraction unbound (fu) was generally similar across normal hepatic function, mild hepatic impairment, and moderate hepatic impairment (0.016, 0.020, and 0.018 for dacomitinib; and 0.00011, 0.00015, and 0.00022 for PF-05199265, respectively) (Table 5). In Study 2, while the fu was slightly increased in hepatic impairment for both dacomitinib and its metabolite, it was generally comparable between participants with normal hepatic function and participants with severe hepatic function (0.022 vs. 0.029 for the dacomitinib parent, and 0.00018 vs. 0.00080 for PF-05199265) (Table 5).
In Study 1, the adjusted GMR for AUCinf,u of dacomitinib in participants with mild or moderate hepatic impairment compared with normal hepatic function was 1.14 (90% CI 0.81–1.63) and 0.87 (90% CI 0.62–1.22), respectively. The adjusted GMR for Cmax,u of dacomitinib in participants with mild or moderate hepatic impairment compared with normal hepatic function was 1.00 (90% CI 0.69–1.44) and 0.77 (90% CI 0.54–1.08), respectively (Table 5).
In Study 2, dacomitinib unbound exposure based on geometric mean AUCinf,u and Cmax,u values increased by 32% and approximately 69%, respectively, for the severe hepatic impairment group compared with the normal hepatic function group (adjusted GMR for dacomitinib for AUCinf,u and Cmax,u was 1.32 [90% CI 0.88–1.96] and 1.69 [90% CI 1.08–2.63], respectively) [Table 5].
3.5 Safety
Dacomitinib administered as a single oral dose of 30 mg was well tolerated in healthy participants and participants with mild, moderate, or severe hepatic impairment. There were no serious AEs, discontinuations due to study treatment, or treatment-related deaths in either Study 1 or Study 2. In Study 2, no AEs were reported in any participants in the normal hepatic function or severe hepatic impairment groups, and in Study 1, no AEs were reported in any participants in the normal hepatic function or mild hepatic impairment groups. One participant in the moderate hepatic impairment group died in a motor vehicle accident. This death was considered a serious AE but it was deemed not to be treatment-related by the investigator [15].
No clinically significant changes in vital signs, laboratory values, or ECG measurements were observed during either of the studies.
4 Discussion
4.1 Mechanisms for No Effect with Hepatic Impairment
The effect of hepatic impairment (mild, moderate, or severe) on dacomitinib pharmacokinetics was evaluated following a single dose of dacomitinib 30 mg in participants with normal CYP2D6 activity (i.e., CYP2D6 EM or IM). As mentioned in the Methods section, the purpose of excluding participants without normal CYP2D6 activity was to ensure that any observed changes in dacomitinib pharmacokinetics could be attributed to hepatic impairment and not confounded by unbalanced CYP2D6 activity between groups, as well as to align the patient populations from Study 1 with Study 2. Plasma dacomitinib AUCinf and Cmax were similar in participants with mild hepatic impairment and were reduced marginally (by approximately 23% and 20% for AUCinf and Cmax, respectively) for participants with moderate hepatic impairment relative to participants with normal hepatic function. In participants with severe hepatic impairment, the total dacomitinib exposure was similar for AUCinf and increased by approximately 31% for Cmax in participants with severe hepatic impairment when compared with participants with normal hepatic function, following a 30 mg dose. One potential limitation of these studies was that they were conducted as two separate studies and have two different control groups. It would have been easier to compare across treatment groups had this been a full hepatic impairment study.
The greatest dacomitinib exposure increase observed across all hepatic impairment groups compared with normal, for both Study 1 and Study 2, was in severe hepatic impairment participants, with a 4.4% increase in AUCinf (virtually no increase) and a 31% increase in Cmax. The clinical relevance of this observed dacomitinib exposure increase with severe hepatic impairment should be considered in the context of known exposure–response relationships for dacomitinib efficacy and safety. Completed exposure–response analyses have demonstrated that higher AUC of dacomitinib was associated with larger tumor shrinkage [19] and higher probabilities of experiencing AEs of skin toxicity (data on file). While dacomitinib Cmax increased by 31% in severe hepatic impaired participants, this change is not expected to be clinically relevant as the exposure-response analysis for safety shows that overall exposure (i.e., AUC or average concentration at steady state), not the Cmax, is associated with certain AEs (data on file, Pfizer Inc.), and as mentioned, AUCinf was similar between normal and severe hepatic impaired participants. Moreover, dacomitinib concentrations are not found to correlate with changes in corrected QT interval [20]. While both Study 1 and Study 2 were conducted in hepatic impaired but otherwise healthy participants, because dacomitinib has linear pharmacokinetics, the results observed after a single dose of dacomitinib 30 mg in these participants can be readily extrapolated to dacomitinib 45 mg at steady state after long-term once-daily dosing in cancer patients. Thus, the clinical findings in Study 1 and Study 2, combined with knowledge of the dacomitinib exposure–response relationship, support no starting dose adjustment of dacomitinib is required in cancer patients with mild, moderate, or severe hepatic impairment.
Hepatic metabolism is the main route of clearance of dacomitinib, with oxidation and glutathione conjugation as the major pathways. CYP2D6 was identified to be the major isozyme involved in the formation of O-desmethyl dacomitinib, PF-05199265, while CYP3A4 contributed to the formation of other minor oxidative metabolites as determined through in vitro studies [12]. The minimal effect on dacomitinib CL/F may be explained by a significant contribution of extrahepatic glutathione conjugation to the dacomitinib elimination pathway in participants with hepatic impairment. Additionally, PF-05199265 exposure decreased with hepatic impairment despite no change in parent exposure. This suggests that while there is a decrease in CYP2D6 activity (consistent with hepatic impairment), leading to a decreased formation of the metabolite PF-05199265, extrahepatic (non-CYP2D6) pathways are compensating for the decrease in CYP2D6 metabolism of dacomitinib, resulting in similar parent exposure.
Both dacomitinib and PF-05199265 are highly bound to plasma proteins. In vitro binding of dacomitinib to human plasma proteins is approximately 98% and is independent of drug concentrations from 250 to 1000 ng/mL [12], and therefore, consistent with recommendations in the FDA guidance [18], the fu was estimated for both analytes. The fu values of dacomitinib and PF-05199265 did not appear comparable between hepatic impaired participants and matched healthy participants. As plasma concentrations of albumin are reduced with hepatic impairment, it is expected that plasma protein binding would meaningfully decrease in the presence of hepatic impairment [21]. The unexpected fu observed in participants with hepatic impairments is not clear, but the extremely high protein binding for dacomitinib may be associated with challenges in measuring the small free fraction of both analytes with high precision in the clinical study.
The effect of hepatic impairment on the unbound pharmacokinetic parameters generally followed a similar direction of change as the total pharmacokinetic parameters. In Study 1, the unbound exposure for dacomitinib parent (AUCinf,u, AUClast,u, and Cmax,u) was comparable between normal hepatic function participants and mild hepatic impairment participants, with the highest unbound dacomitinib parent exposure increase of approximately 26% for Cmax,u. Similarly, the unbound dacomitinib parent exposure was comparable between normal hepatic function participants and moderate hepatic impairment participants. In Study 2, there was an increase in unbound dacomitinib exposure (AUCinf,u, AUClast,u, and Cmax,u) in severe hepatic impaired participants compared with normal hepatic function participants, with the highest unbound dacomitinib parent exposure increase of approximately 69% in Cmax,u. These results suggest that severe hepatic impairment may have a minor effect on unbound dacomitinib parent exposure.
A comparison of dacomitinib with other approved EGFR TKIs may help to provide context around the results seen in these dacomitinib hepatic impairment trials, how hepatic impairment has affected similar medications, and the similarity and differences between these drugs. Currently, the following EGFR TKIs are approved for use in advanced NSCLC: gefitinib, erlotinib, afatinib, dacomitinib, and osimertinib [22]. Like dacomitinib, gefitinib, erlotinib, and osimertinib are primarily metabolized in the liver, while afatinib mainly undergoes renal excretion with minimal hepatic metabolism. Dedicated pharmacokinetic studies in patients with hepatic impairment have been conducted for gefitinib, erlotinib, and osimertinib that informed the dosing recommendations on the respective drug labels regarding hepatic impairment.
Gefitinib is extensively metabolized in the liver, predominately by CYP3A4 [23]. In addition, there is also evidence that gefitinib is metabolized by CYP1A1, which is mainly expressed in extrahepatic tissue [24]. A dedicated hepatic impairment study indicated a significant increase in gefitinib exposure was found in patients with a Child–Pugh score of B or C due to cirrhosis. In this study, increases in mean AUCinf by 40%, 263%, and 166% were observed for participants with mild, moderate, or severe hepatic impairment due to cirrhosis, respectively, compared with participants with normal hepatic function [23, 25]. In another study, cancer patients with liver metastases were assigned to one of three groups (normal, moderate, or severe hepatic impairment) based on alanine aminotransferase and bilirubin levels [25]. No clinically relevant differences in exposure for patients with moderate or severe hepatic impairment due to liver metastases was observed. No gefitinib dose adjustments for liver dysfunction were recommended, however it is advised to monitor adverse reactions when gefitinib is administered to patients with moderate or severe hepatic impairment.
Erlotinib was metabolized primarily by CYP3A4 and to a lesser extent by CYP1A2, as well as the extrahepatic isoform CYP1A1 [26]. Erlotinib exposure was similar in cancer patients with moderately impaired hepatic function (Child–Pugh B) compared with patients with adequate hepatic function, including patients with primary liver cancer or hepatic metastases [26, 27]. Although there does not appear to be an effect of hepatic impairment on erlotinib pharmacokinetics, there are safety concerns with worsening hepatic function. As a result, there are dosing recommendations due to these aforementioned safety concerns with worsening hepatic function but not due to pharmacokinetic considerations.
Osimertinib is metabolized via CYP3A [28]. A dedicated study evaluating cancer patients with a mild or moderate Child–Pugh classification of hepatic impairment on osimertinib pharmacokinetics showed geometric mean osimertinib plasma concentrations were lower in patients with mild (n = 7) or moderate (n = 5) hepatic impairment versus normal hepatic function (n = 10), with a decrease in AUC by 63% and 68%, respectively [29]. Based on these pharmacokinetic findings, it was recommended that no dose adjustments be made for mild and moderate hepatic impairment. The unexpected lower exposure of osimertinib in hepatic impairment is attributed to the reduced absorption associated with a decrease in osimertinib Cmax, although the biological reason is unclear [29].
Overall, there are some similarities between dacomitinib and these other EGFR TKIs. Much like dacomitinib, erlotinib, osimertinib, and gefitinib are all EGFR TKIs that undergo primarily hepatic metabolism. Also like dacomitinib, the observed pharmacokinetic changes of these drugs observed with hepatic impairment was not as expected. One possible explanation for this phenomenon could be the potential extrahepatic routes of metabolism and elimination. For erlotinib, extrahepatic metabolism by CYP3A4 in the intestine, CYP1A1 in the lung, and CYP1B1 in tumor tissue may potentially contribute to its metabolic clearance [30]. Similarly, gefitinib is also metabolized by CYP1A1 in lung cancer cells [31]. There is evidence that osimertinib also undergoes extrahepatic elimination via direct glutathione conjugation [32]. Similar to osimertinib, dacomitinib also undergoes elimination via glutathione conjugation [13], which may potentially contribute to extrahepatic elimination of dacomitinib. As liver microsome and hepatocyte studies may not be able to accurately characterize the contribution of hepatic versus extrahepatic routes of metabolism and elimination, this could be a plausible explanation for what was observed in all of these drugs.
5 Conclusions
In summary, hepatic metabolism is the main route of clearance of dacomitinib, with oxidation and glutathione conjugation as the major pathways. As a result, dedicated studies are warranted to understand the pharmacokinetics in participants with liver dysfunction. Two separate studies were subsequently conducted to assess the effect of mild, moderate, and severe hepatic impairment, as determined by Child–Pugh classification, on dacomitinib pharmacokinetics following a single 30 mg oral dose. Compared with participants with normal hepatic function, there was no clinically meaningful change in dacomitinib exposure in participants with all levels of liver dysfunction. Single doses of dacomitinib 30 mg were well tolerated across all groups of both studies. Based on the findings from Study 1 and Study 2, the current VIZIMPRO drug label recommends no dosing adjustment for dacomitinib is needed for cancer patients with mild, moderate, or severe hepatic impairment (Child–Pugh Class A, B, or C) [12]. This phenomenon may be described by extrahepatic routes of dacomitinib metabolism and elimination as seen with other EGFR tyrosine kinase inhibitors.
References
Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29:2866–74. https://doi.org/10.1200/JCO.2010.33.4235.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. https://doi.org/10.1056/NEJMoa0909530.
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. https://doi.org/10.1016/S1470-2045(09)70364-X.
Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34. https://doi.org/10.1200/JCO.2012.44.2806.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–22. https://doi.org/10.1016/S1470-2045(13)70604-1.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42. https://doi.org/10.1016/s1470-2045(11)70184-x.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. https://doi.org/10.1056/NEJMoa0810699.
Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–32. https://doi.org/10.1158/0008-5472.CAN-07-1885.
Wu Y-L, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:1454–66. https://doi.org/10.1016/s1470-2045(17)30608-3.
Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J Clin Oncol. 2018;36:2244–50. https://doi.org/10.1200/JCO.2018.78.7994.
Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Updated overall survival in a randomized study comparing dacomitinib with gefitinib as first-Line treatment in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. Drugs. 2021;81:257–66. https://doi.org/10.1007/s40265-020-01441-6.
Food and Drug Administration (FDA). VIZIMPRO® (dacomitinib) prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211288s000lbl.pdf. Accessed 10 Dec 2021.
Bello CL, Smith E, Ruiz-Garcia A, Ni G, Alvey C, Loi CM. A phase I, open-label, mass balance study of [(14)C] dacomitinib (PF-00299804) in healthy male volunteers. Cancer Chemother Pharmacol. 2013;72:379–85. https://doi.org/10.1007/s00280-013-2207-9.
Rodighiero V. Effects of liver disease on pharmacokinetics. An update. Clin Pharmacokinet. 1999;37:399–431. https://doi.org/10.2165/00003088-199937050-00004.
Giri N, Masters JC, Plotka A, Liang Y, Boutros T, Pardo P, et al. Investigation of the impact of hepatic impairment on the pharmacokinetics of dacomitinib. Invest New Drugs. 2015;33:931–41. https://doi.org/10.1007/s10637-015-0256-0.
Gaedigk A, Ingelman-Sundberg M, Miller NA, Leeder JS, Whirl-Carrillo M, Klein TE, et al. The Pharmacogene Variation (PharmVar) Consortium: incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin Pharmacol Ther. 2018;103:399–401. https://doi.org/10.1002/cpt.910.
Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92:414–7. https://doi.org/10.1038/clpt.2012.96.
US Food and Drug Administration (FDA). Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. 2003. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf. Accessed 10 Dec 2021.
Fostvedt L, Nickens D, Tan W. Abstract 2103: Application of longitudinal exposure-response modelling to support dacomitinib starting dose in patients with EGFR mutation-positive NSCLC. Worldwide Virtual Event. World Conference on Lung Cancer; 28–31 January 2021.
Tan W, Giri N, Quinn S, Wilner K, Parivar K. Evaluation of the potential effect of dacomitinib, an EGFR tyrosine kinase inhibitor, on ECG parameters in patients with advanced non-small cell lung cancer. Invest New Drugs. 2020;38:874–84. https://doi.org/10.1007/s10637-019-00887-0.
Blaschke TF. Protein binding and kinetics of drugs in liver diseases. Clin Pharmacokinet. 1977;2:32–44. https://doi.org/10.2165/00003088-197702010-00003.
Shah R, Lester JF. Tyrosine kinase inhibitors for the treatment of EGFR mutation-positive non-small-cell lung cancer: a clash of the generations. Clin Lung Cancer. 2020;21:e216–28. https://doi.org/10.1016/j.cllc.2019.12.003.
US Food and Drug Administration (FDA). IRESSA® (gefitinib) prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206995s003lbl.pdf. Accessed 10 Dec 2021.
Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–7. https://doi.org/10.1158/1078-0432.CCR-07-0088.
Horak J, White J, Harris AL, Verrill M, Carmichael J, Holt A, et al. The effect of different etiologies of hepatic impairment on the pharmacokinetics of gefitinib. Cancer Chemother Pharmacol. 2011;68:1485–95. https://doi.org/10.1007/s00280-011-1611-2.
US Food and Drug Administration (FDA). TARCEVA® (erlotinib) prescribing information. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed 10 Dec 2021.
O’Bryant CL, Haluska P, Rosen L, Ramanathan RK, Venugopal B, Leong S, et al. An open-label study to describe pharmacokinetic parameters of erlotinib in patients with advanced solid tumors with adequate and moderately impaired hepatic function. Cancer Chemother Pharmacol. 2012;69:605–12. https://doi.org/10.1007/s00280-011-1733-6.
US Food and Drug Administration (FDA). TAGRISSO™ (osimertinib) prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208065s000lbl.pdf. Accessed 10 Dec 2021.
Grande E, Harvey RD, You B, Batlle JF, Galbraith H, Sarantopoulos J, et al. Pharmacokinetic study of osimertinib in cancer patients with mild or moderate hepatic impairment. J Pharmacol Exp Ther. 2019;369:291–9. https://doi.org/10.1124/jpet.118.255919.
Peters S, Zimmermann S, Adjei AA. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: comparative pharmacokinetics and drug-drug interactions. Cancer Treat Rev. 2014;40:917–26. https://doi.org/10.1016/j.ctrv.2014.06.010.
Alfieri RR, Galetti M, Tramonti S, Andreoli R, Mozzoni P, Cavazzoni A, et al. Metabolism of the EGFR tyrosin kinase inhibitor gefitinib by cytochrome P450 1A1 enzyme in EGFR-wild type non small cell lung cancer cell lines. Mol Cancer. 2011;10:143. https://doi.org/10.1186/1476-4598-10-143.
Dickinson PA, Cantarini MV, Collier J, Frewer P, Martin S, Pickup K, et al. Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab Dispos. 2016;44:1201–12. https://doi.org/10.1124/dmd.115.069203.
Acknowledgements
The authors would like to thank Haihong Shi for her contribution to the conduct of these studies. They also thank the study participants as well as the research personnel at the study sites.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Pfizer Inc.
Conflicts of interest
This research was sponsored by Pfizer Inc. Joseph Chen, Robert R. LaBadie, Joanne Salageanu, Chin-Hee Chung, and Weiwei Tan are employees of Pfizer Inc. and may own Pfizer stock. Joseph Piscitelli (Postdoctoral Fellow) was an unpaid contractor to Pfizer; the fellowship program with the University of California San Diego was supported by an educational grant from Pfizer.
Data sharing statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Code availability
Not applicable.
Author contributions
JP: acquisition and interpretation of data. JC: study design, acquisition and interpretation of data. RRL: study design, interpretation of data. JS: interpretation of data. CC: study design, acquisition and interpretation of data. WT: study design, acquisition and interpretation of data. All authors reviewed and provided intellectual contributions on previous versions of the manuscript and read and approved the final manuscript.
Ethical standards
These studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization (ICH) Good Clinical Practice (GCP) Guidelines. In addition, all local regulatory requirements were followed, in particular those affording greater protection to the safety of trial participants. An Institutional Review Board approved the protocols and all participants gave written, informed consent. These trials were registered on ClinicalTrials.gov: NCT01571388 (Study 1); NCT03865446 (Study 2).
Consent to participate
All participants provided written informed consent before undergoing any study procedures.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Piscitelli, J., Chen, J., LaBadie, R.R. et al. The Effect of Hepatic Impairment on the Pharmacokinetics of Dacomitinib. Clin Drug Investig 42, 221–235 (2022). https://doi.org/10.1007/s40261-022-01125-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-022-01125-x