
Abstract
Coastal marine ecosystems are highly productive and important for global fisheries. To mitigate over exploitation and to establish efficient conservation management plans for species of economic interest, it is necessary to identify the oceanographic barriers that condition divergence and gene flow between populations with those species, and that determine their relative amounts of genetic variability. Here, we present the first population genomic study of an Octopus species, Octopus insularis, which was described in 2008 and is distributed in coastal and oceanic island habitats in the tropical Atlantic Ocean. Using genomic data, we identify the South Equatorial current as the main barrier to gene flow between southern and northern parts of the range, followed by discontinuities in the habitat associated with depth. We find that genetic diversity of insular populations significantly decreases after colonization from the continental shelf, also reflecting low habitat availability. Using demographic modelling, we find signatures of a stronger population expansion for coastal relative to insular populations, consistent with estimated increases in habitat availability since the Last Glacial Maximum. The direction of gene flow is coincident with unidirectional currents and bidirectional eddies between otherwise isolated populations. Together, our results show that oceanic currents and habitat breaks are determinant in the diversification of coastal marine species where adults have a sedentary behavior but paralarvae are dispersed passively, shaping standing genetic variability within populations. Lower genetic diversity within insular populations implies that these are particularly vulnerable to current human exploitation and selective pressures, calling for the revision of their protection status.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Marine coastal ecosystems and the organisms inhabiting them play a fundamental role in human consumption (Lotze et al. 2006; Pauly and Zeller 2016; Watson and Tidd 2018). While coastal habitats (0–200 m depth) only cover 3% of the ocean’s area, they are the source of 50% of marine fisheries and aquaculture production (Palomares and Pauly 2019). Moreover, coastal systems are more prone to pollution (Waycott et al. 2009). In addition to these human-induced selective pressures, coastal habitats are also under natural selection, due to habitat dynamics influenced by rain, evaporation, storms and sea-level changes (Burkett et al. 2008). Genetic diversity within populations is the main target of selection, and, therefore, quantifying the relative amount of genetic diversity within populations is determinant for assessing species adaptability and vulnerability (Frankham 1995; Lande 1995; DeWoody et al. 2021). Understanding how genetic diversity is conditioned on demographic responses to past environmental change, such as change in effective population size and gene flow, has been a major task in evolutionary biology as it provides insights into how species might react to future environmental change (Hoffmann and Sgrò 2011; Moritz and Agudo 2013). Consequently, understanding what drives differentiation in coastal oceanic habitats is paramount not only for deducing population structure, connectivity, and dispersal, but also for developing efficient conservation strategies and informing local stakeholders.
Genetic and, more recently, genomic studies of marine species are shifting our understanding on diversification in the marine environment (Selkoe et al. 2016). Many morphologically conserved taxa that were identified as single widely distributed species have been recognized as multiple cryptic species (Duda et al. 2009; Amor et al. 2017a, b), often composed by differentiated populations (Peijnenburg et al. 2006), suggesting that strong barriers to gene flow can operate in marine taxa (Johannesson et al. 2018; Volk et al. 2021; Filatov et al. 2021). Genomic studies in economically important fish species revealed that genetic diversity within populations, i.e. effective population size, varies between and within species (Barry et al. 2022; Sodeland et al. 2022), suggesting that genetic drift can be a strong driver of diversification in the ocean. Such studies also show that genetic barriers between populations of the same species are coincident with temperature and salinity gradients (Jørgensen et al. 2005; Guo et al. 2015, 2016; Fisher et al. 2022), suggesting that local adaptation can further drive divergence in the presence of gene flow. Coastal species, particularly those with a sedentary behavior, where dispersal is restricted to early developmental stages, are good study organisms to investigate how oceanographic barriers influence a species’ demographic history (i.e. drift and gene flow over time).
The cephalopod Octopus insularis Leite & Haimovici, 2008 inhabits tropical shallow reefs, from the intertidal to depths of 40 m (Leite et al. 2008, 2009a; Bouth et al. 2011), where it acts as an opportunistic predator (Leite et al 2009b, 2016; da Silva et al. 2018). It is distributed along the eastern continental shelf of the American continent, from Florida, several Caribbean islands (O’Brien et al. 2021), over the Gulf of Mexico (Flores-Valle et al. 2018) to the south of Brazil and in several oceanic islands in the south Atlantic (Leite et al. 2008, 2009a; Amor et al. 2017a; Lima et al. 2023). The species conservation status has not been assessed by the IUCN to date (IUCN red list, last accessed 27.07.2023). This cephalopod has a short life cycle (one generation per year; Batista et al. 2021), high fecundity (~ 95,000 eggs; Lima et al. 2017), planktonic paralarvae (Lenz et al. 2015; Lima et al. 2017), and adults display a relatively sedentary life style (Leite et al. 2009a; Lima et al. 2017). O. insularis is targeted by fisheries in Brazil and Mexico, alongside with the sympatric O. americanus, O. maya and O. vulgaris (Lima et al. 2017; Sauer et al. 2021), but no official landing statistics are available. Annual estimates of O. insularis landings in Northeast Brazil are ~ 500 t (Haimovici et al. 2014).
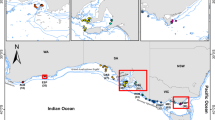
Studies on environmental niche modelling (Lima et al. 2020) found that coastal areas are highly connected by suitable habitat, while most oceanic islands are disconnected from coastal habitats due to high depth. The exception are oceanic archipelagos connected to the coast by shallow seamounts that are hypothesized to work as dispersal corridors between the coast and insular populations, yet this hypothesis remains to be tested. By projecting the current ecological niche to the Last Glacial Maximum (~ 24,000 kya), Lima et al. (2020) suggested that habitat suitability increased strongly along the coast, while no major changes were seen in oceanic islands, but it remains to be tested whether such environmental changes shaped current patterns of genetic diversity in coastal and insular populations. Recent genetic work using a fragment of the mitochondrial COI gene from specimens collected throughout the known species range (Lima et al. 2022) showed four haplotypic groups, two most divergent haplogroups separated by the South Equatorial Current (Fig. 1A), and two less divergent haplogroups separated by high depth between the coast and oceanic islands. These results raise the hypothesis that oceanic currents and habitat availability associated with depth may have driven the diversification of O. insularis. Although valuable, molecular studies relying on mitochondrial markers alone are prone to reflect stochastic processes affecting a single marker, by selective constrains of the mitochondria, and by demographic processes specific to females (Galtier et al. 2009). Understanding how demographic history, in particular temporal changes of effective population size and gene flow, resulted in extant patterns of genetic diversity within populations requires the sampling of independent nuclear markers and the use of coalescent-based methods (Sousa and Hey 2013).

Diversification of Octopus insularis throughout its species range. A Sampling sites in this study. Black arrows depict the major oceanic currents (Sissini et al. 2017): the South Equatorial Current (SEC) runs westwards from the African to the Brazilian coast, splitting into the Brazil Current (BC) running southwards, and the North Brazil Current (NBC) running northwards and continuing as the Caribbean Current (CC). Gray scale represents depth. B Population divergence within O. insularis based on genomic data summarized by an EMU PCA. Single dots represent individuals, colors theirs sampling locations, and placement of individuals in the PCA reflects genomic differentiation based on 99,915 SNPs. Abbreviations of sampling localities: AL Alagoas, AR Rocas Atoll, ASC Ascension Island, BA Bahia, CE Ceará, FN Fernando de Noronha archipelago, OIC Panama, RN Rio Grande do Norte, SPS São Pedro and São Paulo archipelago, STH Saint Helena Island, TM Trindade and Martim Vaz archipelago
Here, using thousands of single nucleotide polymorphisms (SNPs) sampled randomly across the genome of O. insularis, and sampling populations of this species throughout its coastal and disjunct oceanic habitats (Fig. 1A), we perform the first population genomic study in an Octopus species to understand how oceanographic barriers shaped patterns of genetic variability in sedentary species with paralarvae dispersal. First, we identify population structure and associated oceanographic barriers to gene flow along the species range. Second, we infer phylogeographic patterns of island colonization and associated changes in genetic diversity within populations. Lastly, we investigate how temporal changes in effective population size and gene flow are conditioned by changes in habitat suitability and main oceanic currents. Our findings provide general insights into the drivers of diversification in coastal octopods and provide recommendations for conservation and sustainable fisheries of this commercially relevant species.
Material and methods
Sampling of specimens
Seventy-one individuals of Octopus insularis were sampled from 11 localities encompassing large parts of its known species range, including several remote oceanic islands (Table S1, Fig. 1A). Specimens were collected during snorkeling, scuba diving, or were purchased in fish markets, when the exact location of capture was known. Specimens were then stored in 96% ethanol at room temperature and deposited in the mollusks collection of the Federal University of Rio Grande do Norte, Brazil.
Library preparation and sequencing
We extracted DNA from muscle tissue of stored specimens using DNeasy Tissue kits (Qiagen), following the manufacturer’s protocol. We prepared double digest restriction site associated DNA sequencing (ddRADSeq) libraries following the protocol of Peterson et al. (2012), as adapted by Gaither et al. (2015). In short, we used the restriction endonucleases SphI and MluCl (New England Biolabs), following DNA purification with Dynabeads M-270 Streptavidin (Life Technologies), and ligation of the universal P2 adaptors and 24 different P1 adaptors containing individual barcodes of 5 bp, which differed from one another by at least 3 bp. Prior to pooling, we cleaned the DNA libraries using magnetic beads, and pooled individuals in three groups with unique Illumina indices. We then size-selected pooled DNA to recover fragments between 376 and 450 bp using a Pippin Prep (Sage Science). Libraries were quantified using a High Sensitivity DNA Kit on a 2100 Bioanalyzer (Agilent Technologies) and sequenced by QB3 Genomics at UC Berkeley on an Illumina HiSeq 2000, producing 100 bp single end reads. We obtained 260,002,245 raw reads, which after demultiplexing corresponded to an average of 3,298,408 reads per individual (sd 1,900,730 reads/ind; Table S2).
Assembly of RAD-loci and filtering
Since there is no reference genome for Octopus insularis, we assembled de novo RAD-loci using ipyrad (v.0.9.50) (Eaton and Overcast 2020). To assess the robustness of the de novo assembly we considered two sequence similarity thresholds (0.9 and 0.95) that are often used for studies within species (Amor et al. 2020). We only considered reads with length > 70 bp and otherwise kept the default settings of ipyrad. For each assembly we estimated number of all loci assembled across individuals, number of homologous loci after filtering, number of SNPs, percentage of missing data, and number of parsimoniously informative SNPs. Because the two assemblies were similar in their summary statistics (Table S3), we chose a sequence similarity threshold of 0.9 to avoid over splitting of loci. Two samples (SPS3 and FN5) that showed overall low read number (< 200,000) and low number of recovered loci (< 200) were discarded to reduce the amount of missing data (Table S2), leaving 69 individuals from 11 locations for our final assembly.
We exported SNPs in the variant call format (vcf), resulting in 572,012 SNPs and 299,304 homologous loci. We then generated three datasets for downstream analyses (Table S4) with the program vcftools v.0.1.12b (Danecek et al. 2011): (1) the original dataset without further filtering (herein “full”), (2) a more permissive filtered dataset allowing for loci with a maximum of 40% missing data across all 69 individuals (max-missingness 0.6; herein “69inds_40MD”); and (3) a more stringent filtered dataset allowing for a maximum of 20% missing data across individuals after removing the five individuals with the lowest number of loci (max-missingness 0.8 and remove for individuals BA18, RN2A, STH2, OIC1, RN13; herein “64inds_20MD”). All of these data sets contain SNPs linked within the same ddRAD locus, as physical linkage is accounted for by most methods (Table S4). To test if the percentage of missing data reflect the input of reads per individual, rather than a population-specific divergence, we fit a linear regression model to the comparison between the log of raw reads and the log of missing data per individual, using the “69inds_40MD” dataset, and assessed statistical significance.
Population structure
We inferred the most likely number of evolutionarily independent lineages within O. insularis using three complementary methods that rely on different assumptions, using the permissive and stringent datasets (“69inds_40MD”, “64inds_20MD”).
First, we carried out a Principal Component Analysis (PCA) developed for low coverage sequencing data (EMU PCA, Meisner et al. 2021). Conventional PCAs cannot deal with missing entries in the SNP matrix, which are common in ddRAD studies such as the one presented here. Missing data are usually imputed based on the allele frequencies observed either in that specific sampling location or in the total sample (Eaton and Overcast 2020), potentially leading to an under or over-estimation of population structure (Meisner et al. 2021). Alternatively, the imputation method implemented in the EMU PCA does not depend on previously defined groups and thus cannot over-estimate population structure. Missing data from one individual are iteratively replaced by a locus-wide estimate based on the genetic differentiation of the total available data, until the distance between the original matrix and the imputed matrix is minimized. This iterative process considers a fixed number of eigenvalues to summarize genomic information across all loci and individuals. We used a range between one and eleven eigenvalues (the total number of sampling localities) to assess convergence of the results. We converted both datasets to bed format using Plink v1.90b6.22 (Purcell et al. 2007), filtering out alleles with a minor allele frequency < 0.001 (f 0.001).
Second, we inferred the probability of assignment of each individual to a given number of K ancestral clusters, using TESS3R (Caye et al. 2016), also considering the geographic location of the sampled individuals. We converted the vcf-files to the lfmm input format, using the vcf2lfmm function of the R-package LEA (Frichot and François 2015). We conducted 20 independent runs, assuming K between one and eleven and allowing for maximum 1000 iterations.
Lastly, we estimated the co-ancestry matrix between every pair of individuals, using fineRADstructure (Malinsky et al. 2018). In contrast with the previous method, this method uses linkage information within the same ddRAD locus, without any prior assumption based on sampling location. In addition to the permissive and stringent datasets, we also converted vcf-files for the “full” datasets to finerad input files with RADpainter (RADpainter hapsfromVCF), and inferred the co-ancestry matrix with the default settings.
Phylogenetic analysis
We inferred the phylogenetic relationship between individuals, using the permissive and stringent datasets (“69inds_40MD”, “64inds_20MD”).
First, we estimated a maximum likelihood (ML) tree, concatenating all loci. We used fasta files to estimate the most likely evolution model in ModelFinder (Kalyaanamoorthy et al. 2017), and computed a ML tree in IQ-Tree v.2.1.2 (Minh et al. 2020). To evaluate the support values of the inferred topology, we used the ultrafast bootstrap approach (Hoang et al. 2018) with 1000 replicates. We ran the analysis five times, chose the tree with the highest likelihood, and visualized it with ggtree v. 2.4.2. (Yu et al. 2017).
Second, we inferred a coalescent tree that incorporates incomplete lineage sorting, using the SVDquartets (Chifman and Kubatko 2014) and gQMC (Avni et al. 2015) algorithms implemented in tetrad, within ipyrad. This approach uses one randomly chosen SNP per locus for inferring the topology between any combination of four individuals and joins them into a super tree. In addition to the permissive and stringent datasets, we also ran tetrad on the “full” dataset, using 100 non-parametric bootstraps, and constructed a majority rule consensus tree.
Population summary statistics
To assess if coastal and island lineages of O. insularis differ in their levels of genetic diversity, we considered both the permissive and stringent datasets (“69inds_40MD”, “64inds_20MD”).
First, we measured the following individual-level statistics of genetic diversity: (1) expected (He) and observed (Ho) heterozygosity, representing the average and observed number of polymorphic sites in each individual across ddRAD loci; (2) nucleotide diversity per SNP (π*), which represents the average number of nucleotide differences between the two haplotypes from one individual as estimates of effective population size (4Neµ = π); and (3) inbreeding coefficient (FIT), representing the heterozygosity of an individual relative to that observed in the total species range. We did not use the subpopulation inbreeding coefficient FIS, since we had vastly different numbers of samples per subpopulation, possibly biasing the analysis. We converted the datasets into fasta format using PGDSpider (Lischer and Excoffier 2012), calculated He, Ho and FIT in vcftools (Danecek et al. 2011), and π* in DnaSP6 (Rozas et al. 2017). We tested if there was a significant correlation between the percentage of missing data and FIT or π* in each individual, using a linear regression model with R (R Core Team 2017). To test if population-wide π* and FIT were different between lineages, we used a pairwise Student’s t test with Bonferroni–Holm p value correction in R.
Second, to quantify divergence between every population pair, we calculated the pairwise fixation index (FST). We converted the vcf-files to the Arlequin-input format using PGDSpider (Lischer and Excoffier 2012), and used Arlequin ver. 3.5.2.2 (Excoffier and Lischer 2010) to compute pairwise FST, considering the six lineages determined previously. We performed 5000 bootstraps to estimate significance from the null hypothesis of no population structure. We assessed how much genetic variation is explained by four levels of population structure (between the two major regions observed in our Results, within those two major regions, between the six minor regions and between all individuals), by performing a locus-by-locus Analysis of Molecular Variance (AMOVA, Excoffier et al. 1992). We used 10,000 permutations and excluded loci missing in at least one of the lineages.
Lastly, to understand if the genetic diversity observed within each lineage is consistent with changes in effective population size, we computed Tajima’s D using DnaSP (Rozas et al. 2017). We expect lineages that experienced a recent range expansion to have negative values of Tajima’s D, significantly different from zero (p < 0.05).
Demographic analysis
To estimate the past demographic history of O. insularis, we applied demographic modelling with diffusion approximation methods as implemented in the program δaδi (Gutenkunst et al. 2009). First, to assess changes in effective population size within each lineage, we fit one-population models for the lineages containing more than three individuals (i.e. N-Coastal, S-Coastal, S-Oceanic). We compared four demographic models that differ in the number of population size changes (none, one, two) and in the mode of change (none, discrete, exponential) (details in Table S13). Second, to assess gene flow between lineages, we performed two-population models for the two pairs of adjacent lineages with a larger number of individuals (i.e. N-Coastal vs S-Coastal and S-Oceanic vs S-Coastal), which provide some power of estimating demographic parameters. We fitted 15 different demographic models (details in Table S14, following Portik et al. 2017) that incorporate different scenarios of presence and directionality of migration (m), time of a split between lineages (T), and change of effective population size (nu).
For both classes of demographic modelling, to retain a maximum number of segregating sites, we filtered the raw vcf output file (“full”) to only include individuals from the relevant lineages, as in the more stringent dataset (“64ind_20MD dataset”). We excluded loci with more than 40% missing data, and retained the first SNP per ddRAD locus, minimizing physical linkage. We converted the filtered vcf tiles to the δaδi file format, projected down to the number of variants maximizing the number of segregating sites present (Table S15), and calculated the observed one- or two-dimensional folded site-frequency spectra (SFS). We optimized these models using the pipeline developed by Portik et al. (2017) with the following settings (one-population modelling/ two-population modelling): three/four rounds of optimization with 10, 20, 30/15, 10, 5, 5 replicates in each round; with maximum iterations of 5, 10, 50/15, 15, 25, 50 per replicate in each round; and a parameter fold of 3, 2, 1/3, 2, 1, 1 using the default Nelder–Mead method. To guarantee model convergence, we ran each optimization five times. We selected the model with the lowest Akaike information criterion (AIC) score (Akaike 1974), which penalizes the likelihood by the number of parameters, and estimated dAIC and wAIC (Burnham and Anderson 2002). For the two-population models, we carried out a goodness-of-fit test of the most likely demographic model (Barratt et al. 2018) to test general model fit of our inferred parameters. The test is considered passed if the empirical log-likelihood value falls within the distribution of log-likelihood values fitted to simulated SFS.
Results
Assembly of RAD-loci and filtering
Assemblies with alternative similarity thresholds have equivalent summary statistics (Table S3; supplementary Fig. S1), and thus we chose a similarity threshold of 0.9 to avoid over splitting of loci. Assuming this threshold, the “full” dataset consisted of 299,304 homologous loci, with 572,012 SNPs, with 66.34% of missing data (Table S3), a usual value for ddRADseq assemblies (Ivanov et al. 2021); the more permissive “69inds_40MD” dataset consisted of 34,455 loci with 99,915 SNPs, and the more stringent “64inds_20MD” consisted of 25,702 loci with 72,816 SNPs. We found a significant correlation between the number of raw reads and percentage of missing data per individual (adjusted R2: 0.9128, p value 2.2 × 10−16).
Population structure
In the EMU PCA, for the “69inds_40MD” dataset, PC1 and 2 explain 19% and 14% of the genetic variance, respectively. Individuals are clustered into six groups that recapitulate their geographic location (Fig. 1B, Fig. S2A–B): (1) South-coastal (including AL and BA), (2) South-oceanic (the oceanic islands TM), (3) North-SPS (the oceanic islands SPS), (4) North-Caribbean (the coastal OIC), (5) North-Atlantic (the oceanic islands STH, ASC), and (6) North-coastal (including CE, RN, FN, and AR). Results for the more stringent dataset were qualitatively similar (Fig. S2A), and the number of selected eigenvalues did not have an influence on observed results (Fig. S3).
In the TESS3R analysis, using the more permissive dataset (Fig. 2A, B, Fig. S4), the first population split separates samples from the southern coast of Brazil from all the others, with Bahia (BA) and Alagoas (AL) bordering these clusters and containing individuals with admixed ancestry. At K = 3, the northern oceanic islands (São Pedro and São Paulo SPS) form a cluster without showing shared ancestry. At K = 4, the southern populations are divided into a coastal (AL and BA) and an oceanic island cluster (TM), with the individuals from Bahia (BA) showing admixture between these two clusters. At K = 5, the Caribbean individuals are assigned to a new cluster, showing admixture with the northern-coastal cluster. At K = 6, individuals from the most remote oceanic islands (ASC and STH) form their own unmixed cluster. The exception is the individual from Saint Helena, which contains the largest amount of missing data and, therefore, is assigned to every cluster with some probability. Assuming further number of ancestral clusters, we find that previous clusters split equally into two clusters, as thus we refrain from interpreting these biologically. Using the more stringent dataset, we obtained overall concordant results (Fig. S5).

Population structure of Octopus insularis. A Spatial interpolation of ancestral clusters assuming K = 2 using the “69inds_40MD” dataset in TESS3R. B Spatial interpolation of ancestral clusters assuming K = 6 using the “69inds_40MD” dataset in TESS3R. C Co-ancestry matrix of all samples (“full” dataset) inferred in fineRADstructure. Colors on the axis correspond to the individual assignment in the two hierarchical levels shown in A/B
For the fineRADstructure analysis (Fig. 2C) with the “full” dataset, the highest degree of co-ancestry was observed in individuals from São Pedro and São Paulo (N-SPS), followed by individuals from Ascension Island that cluster together with Saint Helena (N-Atlantic), and individuals from the Caribbean (N-Caribbean). These three groups are nested within a northern group that also includes individuals from N-Coastal localities (Ceará, Rio Grande do Norte) and nearby islands (Fernando de Noronha, and Rocas Atoll). All individuals from the archipelago of Trindade and Martim Vaz (S-Oceanic) have a relatively high co-ancestry and are nested in a southern group encompassing individuals from the S-Coastal localities (Alagoas and Bahia), reflecting hierarchical population structure also in the southern group. Results from runs with the more permissive (“69inds_40MD”) and more stringent (“64inds_20MD”) datasets reflected the same relative levels of co-ancestry and hierarchical population structure (Figs. S6, S7).
Phylogenetic analysis
The ML trees estimated with the most likely model (TVM + F + R3) were largely consistent between datasets. Individuals belonging to the island lineages (N-Atlantic, N-SPS, and S-Oceanic) and to the northernmost coastal lineage (N-Caribbean) form well supported (ultrafast bootstrap > 90) monophyletic clades (Fig. 3A). In contrast, individuals from the geographically widespread coastal lineages (N-Costal, and S-Coastal) form paraphyletic clades, with the respective oceanic clades nested within. The N-Caribbean lineage is the sister of the N-Coastal lineage. The clades leading to the two northern oceanic islands (N-Atlantic and N-SPS) show remarkably long branches, reflecting high genetic divergence. While in the most restrictive dataset these two clusters are sister taxa (Figs. S8, S10), this relationship is not supported in the most permissive dataset (Fig. 3A, Fig. S9).

Phylogenetic relationship of the 69 individuals of Octopus insularis. A mid-rooted Maximum Likelihood tree inferred by IQtree from concatenated SNP. B unrooted Coalescent tree inferred by tetrad from resampling unlinked SNPs for 100 non-parametric bootstraps. Individuals are colored according to their lineage identity. Black nodes demarcate UF bootstrap/bootstrap support of > 90, white nodes demarcate bootstrap support of > 50
The topology of the coalescent trees was largely congruent with that from the ML trees. Differences include the monophyly of the S-Coastal lineage with high bootstrap support (> 90), the paraphyly of the N-Caribbean lineage and the monophyly of the N-Coastal lineage with bootstrap support of > 50 (Fig. 3B). Comparing topologies based on the three datasets, we observe that the N-SPS and N-Atlantic lineages form a monophyletic clade in the more permissive dataset (Fig. 3B, Fig. S11), while this monophyly is not recovered in the “full” and more stringent datasets (Figs. S12, S13). Similar to our ML trees, the branch length of lineages from the northern oceanic islands (N-Atlantic and N-SPS) are substantially longer than those of coastal lineages (Fig. S11).
Population summary statistics
Using both datasets, we find that π* and observed (Ho) heterozygosity are lower in the oceanic islands relative to the coastal lineages (Tables S7, S8). Conversely, the inbreeding coefficient FIT is lower in the coastal lineages and highest in all oceanic islands, with N-SPS showing four times higher inbreeding relative to the coast (Fig. 4, Fig. S14, Tables S7, S8). For both π* and FIT, all comparisons between coastal and oceanic lineages yielded significant p values (Tables S5, S6), except the comparisons between S-Oceanic and N-Caribbean (FIT: 0.111, π*: 0.182). We did not find a significant correlation between the percentage of missing data and individual measures of diversity (p values: 0.324 for FIT, 0.145 for π*), confirming that the observed patterns are not driven by missing data.

Genetic diversity and inbreeding coefficients of Octopus insularis within inferred clusters for the 69inds_40MD dataset. A Individual FIT, B π* values. Individuals are grouped by clusters inferred from tess3R at K = 6 and fineRADstrucuture. Boxplots are drawn for each cluster, with the solid black line marking the median, the top and bottom end of the box the 25% and 75% quartile boundaries and the whiskers the 1.5 interquartile range
For both datasets, FST was significant between all six lineages (Tables S9, S10), except for pairwise comparisons where lineages containing three or less individuals (N-SPS, N-Caribbean, N-Atlantic), reflecting the limited sampling. FST values ranged from 0.056 observed between N-Coastal and N-Atlantic, to 0.703 observed between the two oceanic islands (N-SPS and N-Atlantic) of the northern region. The AMOVA of the more permissive dataset (Table S11) showed that variation was highest within individuals (67.88%), followed by variation among the lineages within groups (14.62%), variation explained by the broader regions (North–South, 8.93%), and finally variation within lineages (8.56%). The AMOVA of the more stringent dataset was qualitatively similar (Table S12).
For both datasets, Tajima’s D (Tables S7, S8) was negative for the three coastal lineages (N-Coastal, S-Coastal, N-Caribbean), however, it was only significantly different from demographic stability in the N-Coastal lineage. The three lineages of oceanic islands had Tajima’s D values closer to zero.
Demographic history
For the single population modelling, in all cases the neutral model showed the highest AIC values (Table S16), showing that any model accounting for size changes in effective population size (Ne) explains the observed SFS significantly better. For the S-Coastal and S-Oceanic lineages, the simpler “two_epoch” model showed the lowest AIC score (Fig. S15) and similar residuals to other expansion models (Fig. S16). Assuming this model, parameter estimates indicate an increase in Ne of 1.34 (S-Oceanic) and 4.17 (S-Coastal) times, relative to the Ne of the ancestral population (Na, Table S16, Fig. S17). For the N-Coastal lineage, AIC scores favored the “three_epoch” model incorporating an additional change of Ne. Here, the population first increased to 3.16 times and subsequently to 11.89 times the Na (Table S16, Fig. S17).
For two-population modelling, in both cases we obtained the highest AIC values for the neutral model of no population split, rejecting this scenario (Table S17, Fig. S18). In the case of N-Coastal vs S-Coastal, the model integrating a period without gene flow, followed by secondary contact and asymmetric migration (sec_contact_asym_mig_size) shows the lowest AIC value (Fig. S18). Assuming this model, we find that after the initial population split, the S-Coastal population shrinks to 0.0124 times the Na, while the N-Coastal population increases to 2.33 times of the Na. After secondary contact, both effective population sizes increase to 1.3 and 8.46 times of the Na, respectively (Fig. 5A, Table S17). After secondary contact, migration rates (2 × ancient effective population size × migrants/generation) are highly asymmetric, being 2.6401 from N-Coastal to S-Coastal and 0.0628 in the opposite direction. For S-Coastal vs S-Oceanic, the model incorporating a population split followed by continuous asymmetric migration (asym_mig, Table S14, S17, Fig. S18) showed the lowest AIC values. Assuming this model, S-Coastal increases to an effective population size of 14.1 times the Na after the split, while S-Oceanic increases to 1.19 times the Na. Migration rates are moderately asymmetric at 1.01 from S-Oceanic to S-Coastal and 1.52 in the other direction (Fig. 5B). Residuals of the modelled JSFS are higher in the N-Coastal vs S-Coastal comparison, relative to the S-Coastal vs S-Oceanic comparison (Fig. 5A). Yet, both models passed the goodness-of-fit test (Fig. S19), showing that these idealized models are fair representations of the evolutionary history acting in these populations.

Demographic modelling of two pairs of adjacent lineages of Octopus insularis. The top depicts the best model according to AIC values, with the top block representing the ancestral effective population size (Na), subsequent blocks effective population size (nu1, nu2, nu1a, nu2a, nu1b, nu2b) scaled to Na, arrows between blocks migration (2 × Na × migrants/generation, m12, m21) and height of the blocks represent time since a particular demographic event (2 × Na × generations, T, T1, T2). Bottom shows empirical and modelled SFS as well as per-site residuals and a histogram with the distribution of all residuals. A Best model for N-Coastal vs S-Coastal lineages, B Best model for S-Oceanic vs S-Coastal lineages
Discussion
Oceanic currents and depth cause cryptic diversification in O. insularis
Our genomic methods in Octopus insularis recovered 299,304 ddRAD loci, containing 572,012 linked SNPs, offering the first insights into the genome-wide patterns of genetic differentiation in this species, and into the oceanographic barriers associated with it.
At an older phylogenetic scale, we find that populations of O. insularis are structured into two widely distributed northern and southern groups (Figs. 1B, 2A, C, 3). This deeper division between the northern and southern groups coincides with the South Equatorial Current (SEC) (Fig. 1A), which flows westward from Africa, splitting into two branches at the Brazilian continental shelf (12–14 °S): the Brazil Current (BC) running southwards, and the North Brazil Current (NBC) running northwards and continuing as the Caribbean Current (CC) (Peterson and Stramma 1991). This finding is in agreement with the distribution of the two major haplogroups in mitochondrial DNA (Lima et al. 2022). Given that O. insularis produces up to ~ 95,000 eggs (Lima et al. 2017) and that planktonic paralarvae disperse with oceanic currents (Lima et al. 2017), it is perhaps not surprising to find such a strong role of the SEC in the genetic divergence of this species. This current-mediated North–South division has been reported for other co-distributed species showing a pelagic propagule or larval dispersal, such as corals (Peluso et al. 2018) and mangrove trees (Francisco et al. 2018). Accordingly, a recent review of marine barriers to gene flow, Martins et al. (2021) found the SEC to compose the largest value of phylogeographic concordance among Brazilian coastal organisms, suggesting that this current imposes a major biogeographic barrier across Atlantic species. Studies in other species of octopuses (Octopus vulgaris, Melis et al. 2018; Macroctopus maorum, Higgins et al. 2013) have shown that population structure coincides with oceanic currents in the Mediterranean Sea and the southern Pacific, suggesting that oceanic currents might pose a strong oceanographic barrier for sedentary species with pelagic paralarvae.
At a more recent phylogenetic scale, we find six evolutionarily independent lineages (Fig. 2). The broader northern group is substructured into four lineages: a more widespread coastal lineage encompassing two coastal and two oceanic island localities (N-Coastal), a Caribbean lineage (N-Caribbean), a first oceanic lineage encompassing the archipelago of São Pedro and São Paulo (N-SPS), and a second oceanic lineage encompassing the two islands off the coast of Africa (N-Atlantic), Saint Helena and Ascension. In turn, the broader southern group is substructured into two lineages: a south Brazilian coastal lineage (S-Coastal), and an oceanic lineage representing the archipelago Trindade and Martim Vaz (S-Oceanic). Importantly, these six lineages explain almost double of the genetic variation explained by the two broader groups (AMOVA, Table S11), underscoring their evolutionary significance. This finer population structure is coincident with previous ecological models of habitat suitability for this species that are largely driven by ocean depth (Lima et al. 2020). Oceanic lineages are coincident with volcanic islands that provide suitable habitat highly isolated from the continuous habitat along the coastal shelf of the American continent, with the exception of the S-Oceanic lineage, which is connected to the southern coast of Brazil through a chain of seamounts, the Vitória-Trindade Ridge (Alberoni et al. 2020). Genetic differentiation between populations from oceanic islands and the Brazilian coast have also been reported for dolphins (Oliveira et al. 2019), rockpool blennies (Neves et al. 2016), prosobranch gastropods (Barroso et al. 2016) and corals (Peluso et al. 2018), confirming that such breaks in habitat constitute important drivers of divergence for coastal taxa with very different dispersal rates. Recently, Octopus specimens sighted at several coastal and insular locations in the Caribbean and northern Atlantic have been identified as O. insularis (O’Brien et al. 2021), indicating that the distributional range of this species is still not fully known. Further studies with more extensive sampling may discover new clusters in addition to those presented here.
Island colonization is associated with reduction of genetic diversity
Our phylogenetic analyses shed some light on the evolutionary relationships between the six lineages (Fig. 3). We show that within the southern group, the S-Oceanic clade is nested within the broadly distributed S-Coastal clade, consistent with a colonization of this oceanic archipelago from the southern coast of Brazil. This direction of colonization is concordant with phylogenetic studies in co-distributed fish species (Macieira et al. 2015; Pinheiro et al. 2017; Simon et al. 2021;), which suggest that the colonization of the oceanic archipelago Trindade and Martim Vaz occurred during the LGM (Pinheiro et al. 2017), when seamounts now submerged likely formed an ecological corridor for coastal taxa (Mazzei et al. 2021; Simon et al. 2021). Within the northern group, while the coalescent tree is consistent with a single colonization of the islands from the coast (i.e. N-SPS and N-Atlantic are sister clades; Fig. 3B), the ML tree is consistent with two independent colonization events (i.e. N-SPS and N-Atlantic are not sister clades; Fig. 3A). Yet, the ML analysis using the more stringent dataset is again more consistent with a single colonization event (Figs. S8, S10), corroborating our findings with the coalescent methods, which are more appropriate to study recent radiations due to the expected large amounts of incomplete lineage sorting (Giarla and Esselstyn 2015). Given our finding that these two northern oceanic lineages show the highest genetic differentiation observed in the species (FST: 0.703) and show the longest phylogenetic branches (Fig. 3), more samples would be needed in these islands to establish if colonization occurred once or twice within the northern group. Nevertheless, our results conclusively demonstrate that the island populations represent at least two independent colonization events from the coast—one from the South-Coast and one or two from the North-Coast—allowing us to test evolutionary consequences of island colonization for patterns of genetic diversity.
Regarding genetic diversity segregating within the six lineages of O. insularis, we observe that values of nucleotide diversity (π*) are significantly lower in the three oceanic lineages (N-SPS, N-Atlantic, and S-Oceanic) compared to their respective source of colonization (N-Coastal or S-Coastal; Fig. 4B, Fig. S14, Table S5). Conversely, individual inbreeding coefficients (FIT) are significantly higher in the oceanic lineages (Fig. 4A, Table S6), reflecting the same pattern observed in the co-ancestry matrix (Fig. 2C). These results suggest that island colonization is associated with strong decreases in genetic diversity, probably due to a combination of the founder effect, lower availability of suitable habitat in the islands relative to the coast (Lima et al. 2020, e.g. the available habitat up to 50 m depth in N-SPS is around 1.1 km2 (Ávila et al. 2018)) and to lower recruitment of planktonic paralarvae around islands relative to the coast, as they are “swept out” of their habitat (Johannesson 1988). In accordance with our results, similar decreases of genetic variability in oceanic islands have been reported for several reef fish species (Pinheiro et al. 2017). This suggest that, although marine species of limited dispersal ability such as O. insularis have been able to successfully colonize novel isolated habitats, such as the volcanic islands of the Atlantic, island colonization has led to a significant decrease of genetic variability in the insular range of the species, mirroring what was established in terrestrial systems (White and Searle 2007; Boessenkool et al. 2007).
Demographic history is conditioned on oceanic currents and environmental change
Our demographic models per population detect contrasting demographic histories for coastal and oceanic lineages. For all the three lineages tested, we conclusively reject a scenario of a stable population size in favor of scenarios showing one (for S-Oceanic and S-Coastal lineages) or two (for N-Coastal) instantaneous changes of effective population size. Consistent with relative values of Tajima’s D (Table S7, S8), we estimate that demographic expansions are larger for the two coastal lineages (of 11.96 and 4.17-fold for the N-Coastal and S-Coastal lineages, respectively) than for the oceanic lineage (1.38 fold for the S-Oceanic; Fig. S17). The magnitude of these expansions is consistent with studies of environmental niche modelling that suggested a strong increase of habitat suitability since the LGM along the American coast for this species (Lima et al. 2020), associated with the increase of sea level and the consequent submergence of the continental shelf and increase of sea temperature (Ludt and Rocha 2015). In contrast, in the oceanic islands covered in this study, increases in sea level likely exposed less shallow habitat (Ávila et al. 2018), explaining the more modest expansion of population size.
Our demographic models for two populations reflect similar changes in effective population size, but reveal patterns of genetic connectivity between lineages. For the populations along the coast (N-Coastal vs S-Coastal comparison), the best model reflects a population split without gene flow, followed by a population expansion with gene flow (Fig. 4A). Gene flow is strongly asymmetric, with migration from the N-Coastal to the S-Coastal lineage being 42-fold larger than in the opposite direction. This observation is coincident with the Brazil Current (BC), which moves southwards from the range of the N-Coastal lineage (Fig. 1A). This current has been associated to unidirectional gene flow in rockpool blennies (Neves et al. 2016), suggesting that it may be an important driver of diversification of marine organisms with pelagic dispersal associated to reef habitats.
For the S-Oceanic/S-Coastal comparison (Fig. 4B), the best model consists of a population split with instantaneous size change followed by constant migration. Although migration rates are asymmetric, migration from the coast to the island is only 1.5-fold larger than in the opposite direction. This mildly asymmetric gene flow is consistent with cyclonic eddies that lie north and south of the sea mount chain connecting the coast with Trindade and Martim Vaz (Arruda et al. 2013; Mill et al. 2015), likely facilitating gene flow in both directions (Pinheiro et al. 2015). Together, these findings suggest that oceanic currents not only work as major oceanographic barriers discussed above, but also mediate genetic connectivity between genetically isolated populations.
Implications for conservation biology
As a coastal marine species, O. insularis is part of an ecosystem that is under threat by global change and human exploitation alike. While environmental niche modelling (Lima et al. 2020) shows that suitable habitat for this species has largely increased since the LGM and suggests that O. insularis might further expand its habitat driven by increasing sea surface temperature due to global warming, potential impacts of direct human exploitation such as fisheries have not been investigated in the light of genomic diversity and population connectivity before.
Therefore, our findings on the evolutionary processes shaping the diversification of O. insularis have direct implications for the conservation of this species. As the taxonomic status of the Octopus species mainly targeted by American fisheries has been resolved only recently (Avendaño et al. 2020), little is known about the catch compositions of octopus fisheries in this continent. Annual catches of O. americanus Monfort, 1802 are around 15,000 t in Mexico and 2000 t in Brazil (Jereb et al. 2014). Although O. insularis has a distribution similar to O. americanus, (Avendaño et al. 2020) the reported catch of O. insularis is much lower (around 500 t; Haimovici et al. 2014). Considering that O. insularis was described only in 2008, and since then new studies have identified a greater distribution range than originally thought, the conservation status of its stocks remains unclear, mainly due to the common misidentification between species. With the reduction of pelagic fish stocks due to overexploitation, there is a tendency to target fishing towards cephalopods (Rosa et al. 2019), which was indeed observed by Lopes et al. (2021) on O. insularis fisheries over the last 10 years in Northeast Brazil. Thus, identifying different stocks within this species is crucial to propose management strategies that avoid overexploitation of this important fishery resource.
Our findings on population structure imply that management plans for O. insularis must consider at least six evolutionarily independent units with confined distributions (Fig. 2B).
Moreover, our finding of significantly higher levels of inbreeding and lower genetic diversity in the island lineages relative to the coastal ones (Fig. 4) imply that three island lineages deserve a higher protection status relative to the three coastal ones. Currently, the insular lineages receive some protection status (Marine Protection Atlas, accessed on 24.01.2023): in N-Atlantic, both Rocas Atoll (Rocas Atoll Biological Reserve) and Fernando de Noronha (Fernando de Noronha Marine National Park) are part of protected areas, N-SPS is part of a Brazilian Marine Protected Areas (Environmental Protection Area of São Pedro and São Paulo), S-Oceanic is a protected Brazilian military area, and N-Atlantic is partially (Marine Protection Zone of Saint Helena) or fully protected (MPA of Ascension). Apart from Rocas Atoll, which is a fully protected area, the other archipelagos are only partial no-take areas (Giglio et al. 2018), being exploited mainly by artisanal fishing in part of the territory and potentially exposing the entire stock of the local lineage of O. insularis. In other species, decreased genetic diversity of natural populations has been associated with breakdown of multiple life history traits (Mills 2012; Reed & Frankham 2003; DeWoody et al. 2021), a hypothesis that needs to be evaluated in this system by future studies. Although the relationship between genetic diversity and adaptive potential is discussed in the scientific community (Teixeira and Huber 2021), our finding of low genetic connectivity and high inbreeding in N-SPS and N-Atlantic imply that these populations deserve a higher protection status. We thus advocate that the entirety of Sao Pedro and Sao Paulo archipelago as well as Saint Helena and Ascension Island be classified as areas of no-take as new conservation measures of O. insularis. Restricting fishery activities to the more genetically diverse coastal lineages, educating local stakeholders on the morphological differences between sympatric Octopus species, and establishing set quotas for landings of O. insularis will favor the sustainable management of this economically and ecologically important species.
Data and code availability
Raw reads sequenced for this study were deposited in the NCBI Sequence Read Archive (BioProject Accession PRJNA1024434). All the bioinformatic scripts are available in a public GitHub repository (https://github.com/casparbein/OctopusInsularis).
References
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Control 19(6):716–723. https://doi.org/10.1109/tac.1974.1100705
Alberoni AAL, Jeck IK, Silva CG, Torres LC (2020) The new Digital Terrain Model (DTM) of the Brazilian Continental Margin: detailed morphology and revised undersea feature names. Geo-Mar Lett 40(6):949–964
Amor M, Laptikhovsky V, Norman M, Strugnell J (2017a) Genetic evidence extends the known distribution of Octopus insularis to the mid-Atlantic islands Ascension and St Helena. J Mar Biol Assoc UK 97(4):753–758. https://doi.org/10.1017/S0025315415000958
Amor MD, Norman MD, Roura A, Leite TS, Gleadall IG, Reid A et al (2017b) Morphological assessment of the Octopus vulgaris species complex evaluated in light of molecular-based phylogenetic inferences. Zool Scr 46(3):275–288. https://doi.org/10.1111/zsc.12207
Amor MD, Johnson JC, James EA (2020) Identification of clonemates and genetic lineages using next-generation sequencing (ddRADseq) guides conservation of a rare species, Bossiaea vombata (Fabaceae). Perspect Plant Ecol Evol Syst 45:125544. https://doi.org/10.1016/j.ppees.2020.125544
Arruda WZ, Campos EJ, Zharkov V, Soutelino RG, da Silveira IC (2013) Events of equatorward translation of the Vitoria Eddy. Cont Shelf Res 70:61–73. https://doi.org/10.1016/j.csr.2013.05.004
Avendaño O, Roura Á, Cedillo-Robles CE, González ÁF, Rodríguez-Canul R, Velázquez-Abunader I, Guerra Á (2020) Octopus americanus: a cryptic species of the O. vulgaris species complex redescribed from the Caribbean. Aquat Ecol 54(4):909–925. https://doi.org/10.1007/s10452-020-09778-6
Ávila SP, Cordeiro R, Madeira P, Silva L, Medeiros A, Rebelo AC et al (2018) Global change impacts on large-scale biogeographic patterns of marine organisms on Atlantic oceanic islands. Mar Pollut Bull 126:101–112. https://doi.org/10.1016/j.marpolbul.2017.10.087
Avni E, Cohen R, Snir S (2015) Weighted quartets phylogenetics. Syst Biol 64(2):233–242. https://doi.org/10.1093/sysbio/syu087
Barratt CD, Bwong BA, Jehle R, Liedtke HC, Nagel P, Onstein RE et al (2018) Vanishing refuge? Testing the forest refuge hypothesis in coastal East Africa using genome-wide sequence data for seven amphibians. Mol Ecol 27(21):4289–4308. https://doi.org/10.1111/mec.14862
Barroso CX, Lotufo TMDC, Matthews-Cascon H (2016) Biogeography of Brazilian prosobranch gastropods and their Atlantic relationships. J Biogeogr 43(12):2477–2488. https://doi.org/10.1111/jbi.12821
Barry P, Broquet T, Gagnaire PA (2022) Age-specific survivorship and fecundity shape genetic diversity in marine fishes. Evol Lett 6(1):46–62. https://doi.org/10.1002/evl3.265
Batista AT, Matthews-Cascon H, Marinho RA, Haimovici M (2021) Age growth and seasonal changes in the population structure of an industrial fishery of Octopus insularis in a tropical environment in Northeastern Brazil. J Mar Biol Assoc UK. https://doi.org/10.1017/S0025315421000898
Boessenkool S, Taylor SS, Tepolt CK, Komdeur J, Jamieson IG (2007) Large mainland populations of South Island robins retain greater genetic diversity than offshore island refuges. Conserv Genet 8(3):705–714. https://doi.org/10.1007/s10592-006-9219-5
Bouth HF, Leite TS, Lima FDD, Oliveira JEL (2011) Atol das Rocas: an oasis for Octopus insularis juveniles (Cephalopoda: Octopodidae). Zoologia (curitiba) 28(1):45–52. https://doi.org/10.1590/s1984-46702011000100007
Burkett VR, Fernandez L, Nicholls RJ, Woodroffe CD (2008) Climate change impacts on coastal biodiversity. In: Fenech A, MacIver D, Dallmeier F (eds) Climate change and biodiversity in the Americas. Environment Canada, Ottawa, pp 167–193
Burnham KP, Anderson DR (2002) Model selection and multimodel inference: a practical information-theoretic approach. Springer. https://doi.org/10.1007/978-1-4757-2917-7_3
Caye K, Deist TM, Martins H, Michel O, François O (2016) TESS3: fast inference of spatial population structure and genome scans for selection. Mol Ecol Resour 16(2):540–548. https://doi.org/10.1111/1755-0998.12471
Chifman J, Kubatko L (2014) Quartet inference from SNP data under the coalescent model. Bioinformatics 30(23):3317–3324. https://doi.org/10.1093/bioinformatics/btu530
da Silva EJ, Bezerra LEA, Martins IX (2018) The tropical Octopus insularis (Mollusca, Octopodidae): a natural enemy of the exotic invasive swimming crab Charybdis hellerii (Crustacea, Portunidae). Pan-American J Aquat Sci 13(1):79–83
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. https://doi.org/10.1093/bioinformatics/btr330
DeWoody JA, Harder AM, Mathur S, Willoughby JR (2021) The long-standing significance of genetic diversity in conservation. Mol Ecol 30(17):4147–4154. https://doi.org/10.1111/mec.16051
Duda TF Jr, Kohn AJ, Matheny AM (2009) Cryptic species differentiated in Conus ebraeus, a widespread tropical marine gastropod. Biol Bull 217(3):292–305. https://doi.org/10.1086/bblv217n3p292
Eaton DA, Overcast I (2020) ipyrad: interactive assembly and analysis of RADseq datasets. Bioinformatics 36(8):2592–2594. https://doi.org/10.1093/bioinformatics/btz966
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491. https://doi.org/10.1093/genetics/131.2.479
Filatov DA, Bendif EM, Archontikis OA, Hagino K, Rickaby RE (2021) The mode of speciation during a recent radiation in open-ocean phytoplankton. Curr Biol. https://doi.org/10.1016/j.cub.2021.09.073
Fisher MC, Helser TE, Kang S, Gwak W, Canino MF, Hauser L (2022) Genetic structure and dispersal in peripheral populations of a marine fish (Pacific cod, Gadus macrocephalus) and their importance for adaptation to climate change. Ecol Evol 12(1):e8474. https://doi.org/10.1002/ece3.8474
Flores-Valle A, Pliego-Cárdenas R, Jimenéz-Badillo MDL, Arredondo-Figueroa JL, Barriga-Sosa IDLÁ (2018) First record of Octopus insularis Leite and Haimovici, 2008 in the octopus fishery of a marine protected area in the Gulf of Mexico. J Shellfish Res 37(1):221–227. https://doi.org/10.2983/035.037.0120
Francisco PM, Mori GM, Alves FM, Tambarussi EV, de Souza AP (2018) Population genetic structure, introgression, and hybridization in the genus Rhizophora along the Brazilian coast. Ecol Evol 8(6):3491–3504. https://doi.org/10.1002/ece3.3900
Frankham R (1995) Conservation genetics. Annu Rev Genet 29(1):305–327. https://doi.org/10.1146/annurev.ge.29.120195.001513
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6(8):925–929. https://doi.org/10.1111/2041-210x.12382
Gaither MR, Bernal MA, Fernandez-Silva I, Mwale M, Jones SA, Rocha C, Rocha LA (2015) Two deep evolutionary lineages in the circumtropical glasseye Heteropriacanthus cruentatus (Teleostei, Priacanthidae) with admixture in the south-western Indian Ocean. J Fish Biol 87(3):715–727. https://doi.org/10.1111/jfb.12754
Galtier N, Nabholz B, Glémin S, Hurst GDD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18(22):4541–4550. https://doi.org/10.1111/j.1365-294x.2009.04380.x
Giarla TC, Esselstyn JA (2015) The challenges of resolving a rapid, recent radiation: empirical and simulated phylogenomics of Philippine shrews. Syst Biol 64(5):727–740. https://doi.org/10.1093/sysbio/syv029
Giglio VJ, Pinheiro HT, Bender MG, Bonaldo RM, Costa-Lotufo LV, Ferreira CE et al (2018) Large and remote marine protected areas in the South Atlantic Ocean are flawed and raise concerns: comments on Soares and Lucas (2018). Mar Policy 96:13–17. https://doi.org/10.1016/j.marpol.2018.07.017
Guo B, DeFaveri J, Sotelo G, Nair A, Merilä J (2015) Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks. BMC Biol 13(1):1–18. https://doi.org/10.1186/s12915-015-0130-8
Guo B, Li Z, Merilä J (2016) Population genomic evidence for adaptive differentiation in the Baltic Sea herring. Mol Ecol 25(12):2833–2852. https://doi.org/10.1111/mec.13657
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009) Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5(10):e1000695. https://doi.org/10.1371/journal.pgen.1000695
Haimovici M, Leite TS, Marinho RA, Batista B, Madrid RM, Oliveira JE, Lima LFD, Candice L (2014) As pescarias de polvos do nordeste do Brasil. In Haimovici M, Andriguetto Filho JM, Sunye PS (eds) A pesca marinha e estuarina no Brasil: estudos de caso. FURG, pp 147–160
Higgins KL, Semmens JM, Doubleday ZA, Burridge CP (2013) Comparison of population structuring in sympatric octopus species with and without a pelagic larval stage. Mar Ecol Prog Ser 486:203–212. https://doi.org/10.3354/meps10330
Hoang DT, Chernomor O, Von Haeseler A, Minh BQ, Vinh LS (2018) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35(2):518–522. https://doi.org/10.1093/molbev/msx281
Hoffmann AA, Sgrò CM (2011) Climate change and evolutionary adaptation. Nature 470(7335):479–485. https://doi.org/10.1038/nature09670
Ivanov V, Marusik Y, Pétillon J, Mutanen M (2021) Relevance of ddRADseq method for species and population delimitation of closely related and widely distributed wolf spiders (Araneae, Lycosidae). Sci Rep 11(1):2177
Jereb P, Roper CFE, Norman MD, Finn JK (2014) Cephalopods of the world: an annotated and illustrated catalogue of cephalopod species known to date. Octopods and vampire squids. FAO Species Catalogue for Fisheries Purposes. No 4, Vol 3. FAO, Rome
Johannesson K (1988) The paradox of Rockall: why is a brooding gastropod (Littorina saxatilis) more widespread than one having a planktonic larval dispersal stage (L. littorea)? Mar Biol 99(4):507–513. https://doi.org/10.1007/bf00392558
Johannesson K, Ring AK, Johannesson KB, Renborg E, Jonsson PR, Havenhand JN (2018) Oceanographic barriers to gene flow promote genetic subdivision of the tunicate Ciona intestinalis in a North Sea archipelago. Mar Biol 165(8):1–10. https://doi.org/10.1007/s00227-018-3388-x
Jørgensen HB, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V (2005) Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol Ecol 14(10):3219–3234. https://doi.org/10.1111/j.1365-294x.2005.02658.x
Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14(6):587–589. https://doi.org/10.1038/nmeth.4285
Lande R (1995) Mutation and conservation. Conserv Biol 9(4):782–791. https://doi.org/10.1046/j.1523-1739.1995.09040782.x
Leite TS, Haimovici M, Molina W, Warnke K (2008) Morphological and genetic description of Octopus insularis, a new cryptic species in the Octopus vulgaris complex (Cephalopoda: Octopodidae) from the tropical southwestern Atlantic. J Molluscan Stud 74(1):63–74. https://doi.org/10.1093/mollus/eym050
Leite TS, Haimovici M, Mather J, Oliveira JL (2009a) Habitat, distribution, and abundance of the commercial octopus (Octopus insularis) in a tropical oceanic island, Brazil: Information for management of an artisanal fishery inside a marine protected area. Fish Res 98(1–3):85–91. https://doi.org/10.1016/j.fishres.2009.04.001
Leite TS, Haimovici M, Mather J (2009b) Octopus insularis (Octopodidae), evidences of a specialized predator and a time-minimizing hunter. Mar Biol 156(11):2355–2367. https://doi.org/10.1007/s00227-009-1264-4
Leite TS, Batista AT, Lima FD, Barbosa JC, Mather J (2016) Geographic variability of Octopus insularis diet: from oceanic island to continental populations. Aquat Biol 25:17–27. https://doi.org/10.3354/ab00655
Lenz TM, Elias NH, Leite TS, Vidal EA (2015) First description of the eggs and paralarvae of the tropical octopus, Octopus insularis, under culture conditions. Am Malacol Bull 33(1):101–109
Lima FD, Berbel-Filho WM, Leite TS, Rosas C, Lima SMQ (2017) Occurrence of Octopus insularis Leite and Haimovici, 2008 in the Tropical Northwestern Atlantic and implications of species misidentification to octopus fisheries management. Mar Biodivers 47(3):723–734. https://doi.org/10.1007/s12526-017-0638-y
Lima FD, Ángeles-González LE, Leite TS, Lima SMQ (2020) Global climate changes over time shape the environmental niche distribution of Octopus insularis in the Atlantic Ocean. Mar Ecol Prog Ser 652:111–121. https://doi.org/10.3354/meps13486
Lima FD, Leite TS, Lima SMQ (2022) Seamounts and oceanic currents drive the population structure of Octopus insularis in the Southwest Tropical Atlantic. Aquat Ecol. https://doi.org/10.1007/s10452-022-09955-9
Lima FD, Angeles-Gonzalez LE, Maia H, Leite TS, Cahuich-López M, Mariño-Tapia I, Santana-Cisneros ML, Ardisson P-L, Lima SMQ (2023) Molecular data, ecological niche, and dispersal models reveal a trans-Atlantic shallow-water octopus species. Prog Oceanogr 213:103019
Lischer HE, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28(2):298–299. https://doi.org/10.1093/bioinformatics/btr642
Lopes PFM, Andrade LCA, Pennino MG, Leite TS (2021) The inter-annual fishing variability in Octopus insularis (Leite & Haimovici 2008) as a result of oceanographic factors. Fish Oceanogr 30(5):515–526. https://doi.org/10.1111/fog.12534
Lotze HK, Lenihan HS, Bourque BJ, Bradbury RH, Cooke RG, Kay MC et al (2006) Depletion, degradation, and recovery potential of estuaries and coastal seas. Science 312(5781):1806–1809. https://doi.org/10.1126/science.1128035
Ludt WB, Rocha LA (2015) Shifting seas: The impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J Biogeogr 42(1):25–38. https://doi.org/10.1111/jbi.12416
Macieira RM, Simon T, Pimentel CR, Joyeux JC (2015) Isolation and speciation of tidepool fishes as a consequence of Quaternary sea-level fluctuations. Environ Biol Fishes 98(1):385–393. https://doi.org/10.1007/s10641-014-0269-0
Malinsky M, Trucchi E, Lawson DJ, Falush D (2018) RADpainter and fineRADstructure: population inference from RADseq data. Mol Biol Evol 35(5):1284–1290. https://doi.org/10.1093/molbev/msy023
Martins N, Macagnan L, Cassano V, Gurgel C (2021) Barriers to gene flow along the Brazilian coast: a synthesis and data analysis. Authorea preprint. https://doi.org/10.22541/au.161358135.51187023/v1
Mazzei EF, Pinheiro HT, Simon T, Moura RL, Macieira RM, Pimentel CR et al (2021) Mechanisms of dispersal and establishment drive a stepping stone community assembly on seamounts and oceanic islands. Mar Biol 168(7):1–11. https://doi.org/10.1007/s00227-021-03919-7
Meisner J, Liu S, Huang M, Albrechtsen A (2021) Large-scale inference of population structure in presence of missingness using PCA. Bioinformatics 37(13):1868–1875. https://doi.org/10.1093/bioinformatics/btab027
Melis R, Vacca L, Cuccu D, Mereu M, Cau A, Follesa MC, Cannas R (2018) Genetic population structure and phylogeny of the common octopus Octopus vulgaris Cuvier, 1797 in the western Mediterranean Sea through nuclear and mitochondrial markers. Hydrobiologia 807(1):277–296. https://doi.org/10.1007/s10750-017-3399-5
Mill GN, da Costa VS, Lima ND, Gabioux M, Guerra LAA, Paiva AM (2015) Northward migration of Cape São Tomé rings, Brazil. Cont Shelf Res 106:27–37. https://doi.org/10.1016/j.csr.2015.06.010
Mills LS (2012) Conservation of wildlife populations: demography, genetics, and management. Wiley, Hoboken
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37(5):1530–1534. https://doi.org/10.1093/molbev/msaa015
Moritz C, Agudo R (2013) The future of species under climate change: resilience or decline? Science 341(6145):504–508. https://doi.org/10.1126/science.1237190
Neves JM, Lima SM, Mendes LF, Torres RA, Pereira RJ, Mott T (2016) Population structure of the rockpool blenny Entomacrodus vomerinus shows source-sink dynamics among ecoregions in the tropical Southwestern Atlantic. PLoS ONE 11(6):e0157472. https://doi.org/10.1371/journal.pone.0157472
O’Brien CE, Bennice CO, Leite T (2021) A field guide to distinguishing Octopus insularis and Octopus americanus (Octopoda: Octopodidae). Zootaxa 5060(4):589–594. https://doi.org/10.11646/zootaxa.5060.4.8
Oliveira LRD, Fraga LD, Ott PH, Siciliano S, Lopes F, Almeida R et al (2019) Population structure, phylogeography, and genetic diversity of the common bottlenose dolphin in the tropical and subtropical southwestern Atlantic Ocean. J Mammal 100(2):564–577. https://doi.org/10.1093/jmammal/gyz065
Palomares M-LD, Pauly D (2019) Coastal fisheries: the past, present, and possible futures. Coasts and estuaries. Elsevier, Amsterdam, pp 569–576. https://doi.org/10.1016/b978-0-12-814003-1.00032-0
Pauly D, Zeller D (2016) Catch reconstructions reveal that global marine fisheries catches are higher than reported and declining. Nat Commun 7(1):1–9. https://doi.org/10.1038/ncomms10244
Peijnenburg KTCA, Fauvelot C, Breeuwer JAJ, Menken SBJ (2006) Spatial and temporal genetic structure of the planktonic Sagitta setosa (Chaetognatha) in European seas as revealed by mitochondrial and nuclear DNA markers. Mol Ecol 15(11):3319–3338. https://doi.org/10.1111/j.1365-294x.2006.03002.x
Peluso L, Tascheri V, Nunes FL, Castro CB, Pires DO, Zilberberg C (2018) Contemporary and historical oceanographic processes explain genetic connectivity in a Southwestern Atlantic coral. Sci Rep 8(1):1–12. https://doi.org/10.1038/s41598-018-21010-y
Peterson RG, Stramma L (1991) Upper-level circulation in the South Atlantic Ocean. Prog Oceanogr 26:1–73. https://doi.org/10.1016/0079-6611(91)90006-8
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7(5):e37135. https://doi.org/10.1371/journal.pone.0037135
Pinheiro HT, Mazzei E, Moura RL, Amado-Filho GM, Carvalho-Filho A, Braga AC et al (2015) Fish biodiversity of the Vitória-Trindade Seamount Chain, southwestern Atlantic: an updated database. PLoS ONE 10(3):e0118180. https://doi.org/10.1371/journal.pone.0118180
Pinheiro HT, Bernardi G, Simon T, Joyeux JC, Macieira RM, Gasparini JL et al (2017) Island biogeography of marine organisms. Nature 549(7670):82–85. https://doi.org/10.1038/nature23680
Portik DM, Leaché AD, Rivera D, Barej MF, Burger M, Hirschfeld M et al (2017) Evaluating mechanisms of diversification in a Guineo-Congolian tropical forest frog using demographic model selection. Mol Ecol 26(19):5245–5263. https://doi.org/10.1111/mec.14266
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. https://doi.org/10.1086/519795
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17(1):230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Rosa R, Pissarra V, Borges FO, Xavier J, Gleadall I, Golikov A, Villanueva R et al (2019) Global patterns of species richness in coastal cephalopods. Front Mar Sci 6:1–16. https://doi.org/10.3389/fmars.2019.00469
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Sauer WH, Gleadall IG, Downey-Breedt N, Doubleday Z, Gillespie G, Haimovici M et al (2021) World octopus fisheries. Rev Fisher Sci Aquac 29(3):279–429. https://doi.org/10.1080/23308249.2019.1680603
Selkoe K, D’Aloia C, Crandall E, Iacchei M, Liggins L, Puritz J, von der Heyden S, Toonen R (2016) A decade of seascape genetics: contributions to basic and applied marine connectivity. Mar Ecol Prog Ser 554:1–19. https://doi.org/10.3354/meps11792
Simon T, Pinheiro HT, Santos S, Macieira RM, Ferreira YS, Bernardi G et al (2021) Comparative phylogeography of reef fishes indicates seamounts as stepping stones for dispersal and diversification. Coral Reefs. https://doi.org/10.1007/s00338-021-02178-8
Sissini MN, de Barros Barreto MBB, Széchy MTM, de Lucena MB, Oliveira MC, Gower J et al (2017) The floating Sargassum (Phaeophyceae) of the South Atlantic Ocean–likely scenarios. Phycologia 56(3):321–328. https://doi.org/10.2216/16-92.1
Sodeland M, Jentoft S, Jorde PE, Mattingsdal M, Albretsen J, Kleiven AR et al (2022) Stabilizing selection on Atlantic cod supergenes through a millennium of extensive exploitation. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.2114904119
Sousa V, Hey J (2013) Understanding the origin of species with genome-scale data: modelling gene flow. Nat Rev Genet 14(6):404–414. https://doi.org/10.1038/nrg3446
Teixeira JC, Huber CD (2021) The inflated significance of neutral genetic diversity in conservation genetics. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.2015096118
Volk DR, Konvalina JD, Floeter SR, Ferreira CE, Hoffman EA (2021) Going against the flow: barriers to gene flow impact patterns of connectivity in cryptic coral reef gobies throughout the western Atlantic. J Biogeogr 48(2):427–439. https://doi.org/10.1111/jbi.14010
Watson RA, Tidd A (2018) Mapping nearly a century and a half of global marine fishing: 1869–2015. Mar Policy 93:171–177. https://doi.org/10.1016/j.marpol.2018.04.023
Waycott M, Duarte CM, Carruthers TJB, Orth RJ, Dennison WC, Olyarnik S, Calladine A, Fourqurean JW, Heck KL, Hughes AR, Kendrick GA, Kenworthy WJ, Short FT, Williams SL (2009) Accelerating loss of seagrasses across the globe threatens coastal ecosystems. Proc Natl Acad Sci 106(30):12377–12381. https://doi.org/10.1073/pnas.0905620106
White TA, Searle JB (2007) Factors explaining increased body size in common shrews (Sorex araneus) on Scottish islands. J Biogeogr 34(2):356–363. https://doi.org/10.1111/j.1365-2699.2006.01599.x
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8(1):28–36. https://doi.org/10.1111/2041-210x.12628
Acknowledgements
Sampling and sequencing were supported by the Brazilian National Research Council (CNPq 481492/2013-9) and Coordination for the Improvement of Higher Education Personnel (CAPES, Projeto Ciências do Mar II-23038.004807/2014-01). SMQL is also grateful to the CNPq for the productivity research grant (CNPq 312066/2021-0). We are thankful to the Chico Mendes Institute for Biodiversity Conservation (MMA/ICMBIO) and to the Brazilian Navy for logistics support during the field work.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Brazilian National Research Council (CNPq 481492/2013-9), Coordination for the Improvement of Higher Education Personnel (CAPES, Projeto Ciências do Mar II-23038.004807/2014-01) and Productivity Research Grant (CNPq 312066/2021-0). They provided a grant for FDL and logistic and financial support to TSL, FDL and SMQL.
Author information
Authors and Affiliations
Contributions
FDL, SMQL, and RJP conceived the project. FDL and TSL performed the specimen sampling and HL and LAR produced the genomic data. BB and RJP analyzed the data and interpreted the results. BB and RJP wrote the first version of the manuscript, and FDL, TSL and SMQL contributed to the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethical approval
Government-issued permits were used for collection where applicable, or specimens were purchased on fish markets.
Additional information
Responsible Editor: J. Strugnell.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bein, B., Lima, F.D., Lazzarotto, H. et al. Population genomics of an Octopus species identify oceanographic barriers and inbreeding patterns. Mar Biol 170, 161 (2023). https://doi.org/10.1007/s00227-023-04307-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-023-04307-z