
Abstract
Osteogenesis imperfecta (OI) and other decreased bone density disorders comprise a heterogeneous group of heritable diseases with skeletal fragility. Recently, it was discovered that mutations in SGMS2, encoding sphingomyelin synthetase 2, result in aberrant sphingomyelin metabolism and lead to a novel form of OI termed osteoporosis with calvarial doughnut lesions (OP-CDL) with moderate to severe skeletal fragility and variable cranial hyperostotic lesions. This study describes a Japanese family with the skeletal phenotype of OP-CDL. The affected individuals have moderately severe, childhood-onset skeletal fragility with multiple long-bone fractures, scoliosis and bone deformities. In addition, they exhibit multiple CDLs or calvarial bumps with central radiolucency and peripheral radiopacity. However, SGMS2 sequencing was normal. Instead, whole-exome sequencing identified a novel IFITM5 missense mutation c.143A>G (p.N48S) (classified as a VUS by ACMG). IFITM5 encodes an osteoblast-restricted protein BRIL and a recurrent c.-14C>T mutation in its 5' UTR region results in OI type V, a distinctive subtype of OI associated with hyperplastic callus formation and ossification of the interosseous membranes. The patients described here have a phenotype clearly different from OI type V and with hyperostotic cranial lesions, feature previously unreported in association with IFITM5. Our findings expand the genetic spectrum of OP-CDL, indicate diverse phenotypic consequences of pathogenic IFITM5 variants, and imply an important role for BRIL in cranial skeletogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) and other decreased bone density disorders comprise a phenotypically diverse group of heritable bone dysplasia with increased skeletal fragility, most commonly arising from genetic defects in type I collagen [1]. Rigorous research has, however, broken the narrative of collagen-related OI and unveiled several forms linked to defects in collagen-independent pathways that directly affect the extracellular matrix or bone cell function. In 2019, we expanded this spectrum by reporting novel genetic and phenotypic findings in a rare monogenic form of osteoporosis termed osteoporosis with calvarial doughnut lesions (OP-CDL, OMIM 126550) [2]. The disease is caused by mutations in the SGMS2 gene, resulting in aberrant function of the encoded enzyme sphingomyelin synthetase 2 (SMS2) and sphingolipid metabolism, and consequently abnormal bone matrix mineralization and marked skeletal fragility. Depending on the type of the underlying mutation, affected individuals present with variably severe early-onset osteoporosis with spontaneous fractures and low bone mineral density (BMD) and typically without any of the classical extra-skeletal features of OI. In addition, they portray a distinctive feature of circular sclerotic cranial lesions, also referred to as calvarial doughnut lesions (CDLs). Although several families have been described so far—some prior to the genetic discovery—the disease is still extremely rare and its pathomechanisms incompletely understood [3,4,5].
Here, we describe a family with moderately severe skeletal fragility and multiple sclerotic skull lesions similar to the skeletal phenotype of OP-CDL. However, no pathogenic variant was found in SGMS2. Instead, whole-exome sequencing (WES) revealed a novel heterozygous mutation p.N48S in IFITM5, bringing new knowledge on the genetics behind OP-CDL and the phenotypic spectrum of IFITM5 mutations.
Patients and Methods
Patients
We recruited a mother and her daughter with a monogenic skeletal dysplasia. Before participation in our study, both affected individuals as well as healthy family members signed a written informed consent. All clinical and genetic studies were performed according to ethically approved guidelines. Clinical data from medical records were collected retrospectively for all affected individuals in the family.
Genetic Studies
Genomic DNA was extracted from peripheral blood leukocytes using standard methods or from saliva using Oragene DNA collection kit (DNA Genotek, Ottawa) according to the manufacturer's protocol. All five exons, 5′ UTR and 3′ UTR and a minimum of 30 bases of flanking introns in the SGMS2 gene (NM_002335) were sequenced in two affected and two healthy family members by direct sequencing after polymerase chain reaction (PCR) amplification. Sequencing was done with an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA) and chromatograms analyzed using Sequencher v5.4.6 software.
For WES we included four family members from three generations: the index patient, her affected mother, her healthy father and her healthy brother (Fig. 1). WES was performed at Blueprint Genetics (Espoo, Finland) according to their standard methods (blueprintgenetics.com; Supplementary material). The procedure yielded × 178 median coverage of target bases. Reannotation was done with Annovar [6] and data analyzed with VarAFT 2.16 (http://varaft.eu).

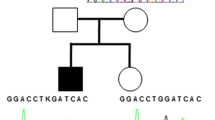
Pedigree of the Japanese family with a novel heterozygous IFITM5 mutation p.N48S. Squares represent males, circles females, black mutation-positive, white mutation-negative, and slash a deceased individual. Asterisks indicate individuals included in WES analysis. Arrows indicate individuals Sanger sequenced for the IFITM5 variant. Generations are shown with Roman numerals
Before exome-wide exploration for causative variants, we first evaluated all variants found in 23 known genes to underlie OI, bone fragility and primary osteoporosis (Figure S1) [7]. Second, we filtered variants in all genes and selected candidate variants with the following criteria: (1) heterozygous variants present in clinically affected subjects (II-3, III-1) and absent in clinically healthy subjects (II-2, III-2); (2) functional variants affecting coding regions or splice junction; and (3) an allele frequency < 0.1% in Genome Aggregation Database (gnomAD) database (v2 and v3) (www.gnomad.broadinstitute.org), the 1000 Genomes Project [8], The Single Nucleotide Polymorphism Database 153 (dsSNP153) (https://ftp.ncbi.nlm.nih.gov/snp/), the Sequencing Initiative Suomi (SISu) database (www.sisuproject.fi), The Human Genomic Variant Search Engine, Varsome [9] and Japanese multiomics panel (jMorp) (https://jmorp.megabank.tohoku.ac.jp/202008/). In silico predictions of the damaging capacity for missense variants were performed using SIFT (www.sift.jcvi.org), PROVEAN (http://provean.jcvi.org/index.php), PolyPhen2 (www.genetics.nwh.harvard.edu/pph2/), UMD Predictor (www.umd.predictor.eu), MutationTaster2 [10]), M-CAP (www.http://bejenaro.stanford.edu/mcap/), and CADD scores (www.cadd.gs.washington.edu). The variants' possible effects on protein conformation were evaluated using HOPE web server (http://www.cmbi.ru.nl/hope/). Segregation and trueness of the selected candidate variants were confirmed by Sanger sequencing (II-3, III-1, II-2, III-2, II-1, II-4) (Fig. 1). Primers and protocols are available upon request from the authors.
Copy number variation (CNV) was analyzed from WES data in all known disease genes by Blueprint Genetics (Espoo, Finland). These genes are supplemented with genes included in The Clinical Genomics Database (> 3350 genes, https://research.nhgri.nih.gov/CGD/) and the Developmental Disorders Genotype–Phenotype Database (DD2GP) (> 1640 genes, https://www.sanger.ac.uk/collaboration/deciphering-developmental-disorders-ddd/). The total number of genes considered as clinically associated in the CNV analysis is at least 3750.
Results
Case Report
The study involves a Japanese family clinically evaluated at Kobe Children’s Hospital, Japan, for an autosomal dominant fragile bone syndrome with CDLs (Fig. 1). The proband is a presently 18-year-old female with a history of multiple long-bone fractures starting at the age of two years. These were all result of low-energy trauma and included three separate left femur fractures, right femur, left tibia, and right tibia; no upper extremity fractures were recorded. The four femoral fractures were treated with recurrent corrective casts or intra-medullary osteosynthesis operations. No vertebral compression fractures were recorded. Her stature is otherwise normal with adult height of 165 cm (+ 1.2 SD). Her mother, presently 47 years, has a similar phenotype of early-onset skeletal fragility. She too has sustained multiple fractures, first at the age of six years. Despite the absence of vertebral compression fractures or exaggerated kyphosis, she has short stature with height of 133 cm (− 4 SD), partially perhaps accounted for by lower-limb deformities and spinal scoliosis. Neither patient exhibits other OI-related extra-skeletal characteristics (blue sclerae, dentinogenesis imperfecta, hearing impairment, or joint laxity), neurological symptoms related to basilar invagination commonly seen in OI, or any of the hallmark features of OI type V features (hyperplastic callus formation, calcification of forearm interosseous membrane, dislocation of the radial head or radiolucent metaphyseal bands) [8, 11]. Neither has received any bisphosphonate or other osteoporosis treatment at any point. The grandmother of the proband was reported to be similarly affected and had short stature but other detailed clinical data were unavailable.
Skeletal radiographs show demineralized bones with moderate deformities (Fig. 2). The proband portrays mild bowing of the femora with diaphyseal cortical thickening, but no deformity of other long bones. Metaphyseal modeling of the tubular bones is normal. Bone mineralization is uneven with alternating areas of densely and poorly mineralized bone. The metaphyses are strikingly osteopenic, while the diaphyses are not evidently demineralized and have normal diaphyseal cortical thickness. It is notable that the metaphyseal radiolucencies of the knee are intermingled with sclerotic striations and cystic, tumor-like lesions. The spine shows only mild scoliosis and no vertebral compression fractures (Fig. 2B, C). In the mother, radiographs of the trunk and legs show more severe skeletal abnormalities with severe bowing of the femora, tibiae, and fibulae, and evident metaphyseal overmodeling of the tubular bones. The metaphyses are severely demineralized, while the bent diaphyses are associated with cortical thickening. Her spine shows mild scoliosis; no lateral spine images were available to assess vertebral height or shape. Lastly, both also exhibit several distinctive changes of the calvaria. Skull radiographs and cranial CT reveal multiple calvarial bumps with central radiolucency and peripheral radiopacity (Fig. 3).

Skeletal radiographs of two patients with a novel heterozygous IFITM5 mutation p.N48S. Images show (A) mild scoliosis, (B, C) normal thoracic and lumbar vertebral height without vertebral compression fractures, (D) mild femoral bowing with diaphyseal cortical thickening, and (E) metaphyseal osteopenia intermingled with sclerotic striations (arrow), cystic (tumor-like) lesions, and preserved cortical thickness of the distal femur and proximal tibia in an 18-year-old female; and (F) severe scoliosis and (G) long-bone deformity with metaphyseal widening (dashed arrow) and diaphyseal bowing (white arrow) in a 47-year-old female

Cranial lesions in two female patients with a novel heterozygous IFITM5 mutation p.N48S. (A) 3D CT showing osseous bumps of the right frontal and left parietal calvaria (black arrows); (B) skull radiograph showing doughnut lesions of the frontal and parietal regions (arrows), corresponding to ones in (A); and (C) Coronal CT showing parietal hyperostosis with central radiolucency in an 18-year-old female. (D) CT showing calvarial thickening with central radiolucency of the left occipital region in a 47-year-old female. (E) Cross-sectional CT image of an adult male patient with a heterozygous SGMS2 mutation p.R50* showing multiple sclerotic lesions
No bone mineral density (BMD) measures are available in either individual. Biochemistry of bone turnover markers, including alkaline phosphatase, have remained constantly normal. For the index, bone histomorphometry, performed on a bone biopsy taken during a hip joint surgery at age 10, portrayed an absence of the birefringent pattern of normal lamellar bone and the presence of fish-scale pattern under polarized light (Fig. 4).

Bone biopsy sample from a female patient with a novel heterozygous IFITM5 mutation p.N48S. Images show an absence of the birefringent pattern of normal lamellar bone and the presence of fish-scale pattern (arrows) under polarized light
Genetic Findings
No pathogenic variants were found in SGMS2 by Sanger sequencing. CNV analysis of known disease genes (> 3650), defined as single exon or larger deletions or duplications, did not reveal any potential variations probable to cause the disease. Screening of the WES data for pathogenic variants in the known OI and primary osteoporosis genes yielded a novel missense mutation c.143A > G (p.N48S) in exon 1 of IFITM5 (Fig. 5). The variant is classified as a variant uncertain significance (VUS) according to the American College of Medical Genetics and Genomics (AMCG). Further filtering of the whole WES data for other single nucleotide variations (SNVs), small insertions and deletions (INDELs), yielded two candidate variants: one missense variant and one splice region variant. These two variants, however, were discarded for their close association with known human genetic diseases with different phenotypes to that of our family (Supplementary Table). Detailed filtering steps and the number of variants after each filtering step for a dominant disorder are shown in supplementary Fig. 1S.

Genetic results in a family with a novel heterozygous IFITM5 missense mutation p.N48S. (A) Sanger sequence image of the heterozygous point mutation in the index patient and a normal sequence in a healthy family member (II-4). (B) The four previously reported mutations in IFITM5 linked to osteogenesis imperfecta. The novel mutation reported in this publication is marked by box
The identified IFITM5 missense variant (p.N48S) is novel with no carriers identified in the aforementioned databases. The variant is predicted to be damaging by all six prediction programs: damaging by SIFT (0.012), and PROVEAN (-4.9), M-CAP (0.310), probably damaging by PolyPhen2 (0.896), pathogenic by UMD Predictor (pathogenicity 90), and disease causing by MutationTaster2 (0.996). It also has a CADD score of 22.7. Automated analysis of the mutant protein by HOPE indicated that the residue is likely located in a transmembrane domain, is smaller than the wild-type residue, and this size difference can affect the protein's contacts with the lipid membrane. According to SwissProt, the 3D structure is different between WT, S40L, S40W, and N48S but unfortunately, the crystal protein structure of IFITM5/BRIL has not been resolved and not enough structural data is available to generate structural models of the protein. In addition, the mutant residue is more hydrophobic than the wild-type residue, which may affect its hydrophobic interactions with the membrane lipids. The mutation also lies in a 100% conserved residue and is likely damaging for the protein. The variant is also only eight amino acids from a previously published IFITM5 locus known to underlie OI [11,12,13]. Lastly, in STRING analysis, we did not find direct connections between IFITM5 and SGMS2.
Discussion
Osteoporosis with cranial lesions (OP-CDL) is a unique form of OI with moderate to severe skeletal fragility and multiple sclerotic cranial lesions. Recently, the genetic cause was identified as mutations in the sphingomyelin synthase 2–encoding SGMS2 and consequent abnormal sphingomyelin metabolism [2]. Here we provide detailed description of two related individuals with the skeletal phenotype of OP-CDL but without a pathogenic variant in SGMS2. Instead, both harbor a novel heterozygous missense mutation p.N48S in IFITM5, a genetic defect previously primarily reported in OI type V and, in a few cases, OI type VI [1, 11,12,13,14,15,16,17,18]. These together expand the genetic background of OP-CDL, underline the importance of differential diagnostics and possibly insinuate a communication between collagen processing and sphingomyelin metabolism in cranial skeletogenesis.
BRIL is a bone-specific modulator of mineralization and mutations in its encoding gene IFITM5 gene have previously been exclusively associated with OI type V or VI [11,12,13,14,15,16,17,18]. To date, only four different IFITM5 mutations have been described [11,12,13,14,15,16,17,18] and the affected patients show great phenotypic variability depending on the contrasting effects of the different mutations. Patients with the recurrent c.-14C>T 5' UTR mutation, affecting the intracellular N terminus of the protein, represent the classical OI type V with interosseous membrane calcification, hyperplastic callus formation, radial head dislocation and characteristic metaphyseal bands [14, 15]. The exonic p.S40L and p.S40W mutations, affecting the coding region of the gene and the transmembrane domain of the protein, portray a phenotype resembling the recessive OI type VI with severe skeletal dysplasia and prenatal bowing of the long bones, broad metaphyses, and coxa vara, and histologically hyperosteoidosis [12, 13]. Recently, a fourth mutation c.-9C>A was described but at a young age of only one month, it is not possible to definitively ascertain the full of extent of the skeletal phenotype [16].
Our patients, though also harboring a mutation in the coding region in close proximity to the S40 mutations, have clinical presentations very different from these. Both exhibit skeletal fragility with postnatal onset of fractures, diaphyseal bowing, and normal metaphyseal modeling and, most distinctively, multiple CDLs. The patients lacked interosseous membrane calcification or hyperplastic callus formation but had relatively thick long-bone cortices [19]. Neither also has blue sclerae, cardiac symptoms, cystic vertebral lesions or other spinal anomalies—clinical features reported in patients with S40 mutations [12, 13]. This may, of course, be a characteristic of this single family and not represent the full clinical phenotype caused by this mutation. Further, prior reports have described delayed and uneven ossification of cranial bones in patients with IFITM5 mutations, such as Wormian bones and granular lytic areas of craniofacial bones [18]. Dagdeviren et al. [20] also described a young female patient with a sclerotic granular bone pattern of the maxillary and mandibular bones with extensive dental problems and a mixture of osteosclerosis and osteolysis of the cranial base. However, she did not have CDL or overt calvarial hyperostosis.
The skeletal manifestation of the present family is classifiable into OP-CDL. However, there are phenotypic differences between the present family and SGMS2-related OP-CDL [2]. Bone fragility, bone deformities, and growth retardation are much more severe in SGMS2-related OP-CDL [2]. Also, vertebral compression fractures were a key finding, neurological complications are common, ALP levels tend to be elevated, and CDLs are more sclerotic. Furthermore, histological examination for bone show impaired mineralization or abnormal bone lamellarity.
Given the phenotypic similarities between SGMS2- and IFITM5-related OP-CDL it may be postulated whether the two syndromes share essential molecular connection or close relation between sphingomyelin metabolism and BRIL. One such hypothetic link may be SP7 (osterix), a transcription factor of osteoblast differentiation, whose expression is under the control of several signaling pathways, endoplasmic reticulum stress and epigenetic factors [21]. SP7 has an activator that acts cooperatively with specificity protein 1 (SP1) and zinc finger protein GLI2 to synergistically induce the BRIL promoter [22], and reports show that complete depletion of SMS2 in osteoblasts by Sp7 promoter-driven Cre-expressing mice (Sp7-Cre; SMS1-CKO; SMS2-KO) induces osteopenia through reduced bone formation [23]. Although our findings would support such interaction, and functional in vitro studies suggest BRIL to similarly partake in matrix mineralization, results from animal studies are unclear and partly conflicting [24,25,26,27]. Also, analysis for possible protein interactions with SwissProt did not find any connections between BRIL and SMS2. Therefore, these suggestions remain on a purely hypothetical basis and warrant further investigations.
We acknowledge certain limitation to our study, mainly concerning the lack of functional assessment of the molecular outcomes of the identified mutation. Also, we acknowledge that having clinical data from only one family provides a limited view on the full spectrum and severity of skeletal and possible extra-skeletal manifestations. Especially, more extensive bone histomorphometry data would have been very informative in determining detailed skeletal outcomes of the IFITM5 mutation. Furthermore, future studies are recommended to address the role of IFITM5 mutations in cranial pathology and its possible links to sphingomyelin metabolism. Lastly, we acknowledge the possibility of random segregation of this variant as only few family members were screened. With the number of tested individuals, the probability of such random segregation would be 1:8. However, taken together, we do consider our findings valuable and important given the novelty of the reported findings.
As conclusion, we report a novel IFITM5 mutation presenting with OP-CDL with moderately severe skeletal fragility and deformities and notable cranial abnormalities, suggesting genetic heterogeneity in OP-CDL. IFITM5-related skeletal fragility should be considered in subjects with OP-CDL like skull lesions. Functional analyses evaluating possible connection between BRIL and sphingomyelin metabolism may shed light on the pathogenetic mechanisms.
Data Availability
Primers and protocols are available upon request from the authors.
References
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, Sillence D, Superti-Furga A, Unger S, Warman ML (2019) Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A 179:2393–2419
Pekkinen M, Terhal PA, Botto LD, Henning P, Mäkitie RE, Roschger P, Jain A, Kol M, Kjellberg MA, Paschalis EP, van Gassen K, Murray M, Bayrak-Toydemir P, Magnusson MK, Jans J, Kausar M, Carey JC, Somerharju P, Lerner UH, Olkkonen VM, Klaushofer K, Holthuis JC, Mäkitie O (2019) Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight. 4:e126180
Jaakkola E, Laine CM, Mäyränpää MK, Falck A, Ignatius J, Mäkitie O (2009) Calvarial doughnut lesions and osteoporosis: a new three-generation family and review. Am J Med Genet A 149A:2371–2377
Couture G, Degboe Y, Baujat G, Cormier-Daire V, Laroche M (2020) Juvenile osteoporosis with calvarial doughnuts: progressive high-turnover bone loss responsive to bisphosphonate therapy. Jt Bone Spine 87:271–272
Baumgartner D, Gassner I, Sperl W, Salzer-Kuntschik M, Judmaier W, Steinmann B (2001) Calvarial, “doughnut lesions”: clinical spectrum of the syndrome, report on a case, and review of the literature. Am J Med Genet 99:238–243
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Robinson ME, Rauch F (2019) Mendelian bone fragility disorders. Bone 126:11–17
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A (2019) VarSome: the human genomic variant search engine. Bioinformatics 35:1978–1980
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362
Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW (2012) A single recurrent mutation in the 5’-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91:343–348
Hoyer-Kuhn H, Semler O, Garbes L, Zimmermann K, Becker J, Wollnik B, Schoenau E, Netzer C (2014) A nonclassical IFITM5 mutation located in the coding region causes severe osteogenesis imperfecta with prenatal onset. J Bone Miner Res 29:1387–1391
Farber CR, Reich A, Barnes AM, Becerra P, Rauch F, Cabral WA, Bae A, Quinlan A, Glorieux FH, Clemens TL, Marini JC (2014) A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Miner Res 29:1402–1411
Lim JY, Bhatia NS, Vasanwala RF, Chay PL, Lim KBL, Khoo PC, Schwarze U, Jamuar SS (2019) A novel Ser40Trp variant in IFITM5 in a family with osteogenesis imperfecta and review of the literature. Clin Dysmorphol 28:120–125
Semler O, Garbes L, Keupp K et al (2012) A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91:349–357
Wu D, Wang Y, Huang H (2020) A novel variant of the IFITM5 gene within the 5’-UTR causes neonatal transverse clavicular fracture: Expanding the genetic spectrum. Mol Genet Genomic Med 8:e1287
Rodriguez Celin M, Moosa S, Fano V (2018) Uncommon IFITM5 mutation associated with severe skeletal deformity in osteogenesis imperfecta. Ann Hum Genet 82:477–481
Guillén-Navarro E, Ballesta-Martínez MJ, Valencia M, Bueno AM, Martinez-Glez V, López-González V, Burnyte B, Utkus A, Lapunzina P, Ruiz-Perez VL (2014) Two mutations in IFITM5 causing distinct forms of osteogenesis imperfecta. Am J Med Genet A 164A(5):1136–1142. https://doi.org/10.1002/ajmg.a.36409 (PMID: 24478195)
Whyte MP, Aronson J, McAlister WH, Weinstein RS, Wenkert D, Clements KL, Gottesman GS, Madson KL, Stolina M, Bijanki VN, Plotkin H, Huskey M, Duan S, Mumm S (2021) Coalescing expansile skeletal disease: delineation of an extraordinary osteopathy involving the IFITM5 mutation of osteogenesis imperfecta type V. Bone 145:115835
Dagdeviren D, Tamimi F, Lee B, Sutton R, Rauch F, Retrouvey JM (2019) Dental and craniofacial characteristics caused by the p.Ser40Leu mutation in IFITM5. Am J Med Genet A 179:65–70
Sinha KM, Zhou X (2013) Genetic and molecular control of osterix in skeletal formation. J Cell Biochem 114:975–984
Kasaai B, Gaumond MH, Moffatt P (2013) Regulation of the bone-restricted IFITM-like (Bril) gene transcription by Sp and Gli family members and CpG methylation. J Biol Chem 288:13278–13294
Matsumoto G, Hashizume C, Watanabe K, Taniguchi M, Okazaki T (2019) Deficiency of sphingomyelin synthase 1 but not sphingomyelin synthase 2 reduces bone formation due to impaired osteoblast differentiation. Mol Med 25:56
Lietman CD, Marom R, Munivez E, Bertin TK, Jiang MM, Chen Y, Dawson B, Weis MA, Eyre D, Lee B (2015) A transgenic mouse model of OI type V supports a neomorphic mechanism of the IFITM5 mutation. J Bone Miner Res 30:489–498
Hanagata N, Li X, Morita H, Takemura T, Li J, Minowa T (2011) Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J Bone Miner Metab 29:279–290
Moffatt P, Gaumond MH, Salois P, Sellin K, Bessette MC, Godin E, de Oliveira PT, Atkins GJ, Nanci A, Thomas G (2008) Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res 23:1497–1508
Patoine A, Husseini A, Kasaai B, Gaumond MH, Moffatt P (2017) The osteogenic cell surface marker BRIL/IFITM5 is dispensable for bone development and homeostasis in mice. PLoS ONE 12:e0184568
Acknowledgements
We thank Mira Aronen and Iuliia Savenko for their help with collecting subject and sample data.
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This study is supported by grants from the Japan Society for the Promotion of Science (No.18H02932) and the Japan Agency For Medical Research and Development (No. 20bm0804006h and 20ek0109486h), Sigrid Jusélius Foundation, the Novo Nordisk Foundation, the Folkhälsan Research Foundation, the Academy of Finland, the Foundation for Pediatric Research, the Helsinki University Research Funds, Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Orion Research Foundation, the Finnish ORL–HNS Foundation, and the Juhani Aho Foundation.
Author information
Authors and Affiliations
Contributions
Study design: MP, SI, OM. Study conduct: all authors. Data collection: RM, MP, DK, NM, YY, SI. Data analysis: all authors. Drafting manuscript: REM and MP. Revising manuscript content: all authors. Approving final version of manuscript: all authors. RM, MP, SI and MO take responsibility for the integrity of the data.
Corresponding author
Ethics declarations
Conflict of interest
Mäkitie RE, Pekkinen M, Morisada N, Kobayashi D, Yonezawa Y, Nishimura G, Ikegawa S, and Mäkitie O states that they have no conflict of interest.
Ethical Approval
All clinical and genetic studies were performed according to ethically approved guidelines.
Consent to Participate
Both affected individuals as well as healthy family members signed a written informed consent prior to participation in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mäkitie, R.E., Pekkinen, M., Morisada, N. et al. A Novel IFITM5 Variant Associated with Phenotype of Osteoporosis with Calvarial Doughnut Lesions: A Case Report. Calcif Tissue Int 109, 626–632 (2021). https://doi.org/10.1007/s00223-021-00878-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-021-00878-5