
Abstract
The main soluble proteins in bottled wine are pathogenesis-related proteins. Their concentration is influenced by the harvesting technique used, the time of the juice exposition to grape skins and possible microbial infection of grapes. The most typical are namely chitinases and thaumatin-like proteins (TLPs), which accumulate in grapes on ripening. They show a low molecular weight of 20–35 kDa and are resistant to proteolysis as well as the acidic pH of wine. Chitinases are considered the primary cause of heat-induced haze formation because of their irreversible denaturation and aggregation. This process can additionally be affected by the non-protein wine components. We focused on the development of a fast quantification method for wine TLPs using matrix-assisted laser desorption/ionization mass spectrometry. White wine samples were analyzed directly, after dialysis or they were subjected to protein concentrating procedures before the measurements. MS-based quantification was achieved by comparing the peak areas of an internal standard (cytochrome c, thaumatin or myoglobin) and TLPs in the acquired mass spectra. The content of TLPs in commercial wines was determined at milligrams per liter. Chitinases could not be quantified in this way because of a low concentration in the analyzed bentonite-fined wines resulting in missing spectral signals. Anyway, this procedure becomes an alternative to common quantification methods based on liquid chromatography.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteins represent a natural component of wine [1]. They mostly originate from grapes, but can also come from the yeast [2]. Parasitic and contaminating microorganisms such as the fungus Botrytis cinerea can influence the protein composition too [3]. For example, the laccase from B. cinerea has been associated with the phenomenon of sparkling wine gushing [4]. The instability of wine proteins can lead to a haze formation in white wine after bottling [1, 5]. Therefore, bentonite is commonly used to remove excess protein and stabilize wine [6, 7]. The use of peptidases appears to be a promising alternative to the application of bentonite but effective proteolysis under winemaking conditions (pH, temperature) is limited [8]. The main soluble proteins in bottled wine are pathogenesis-related (PR) proteins [9]. They are extracted into the juice during winemaking. The concentration of PR proteins is influenced by the harvesting technique used, the exposure time of the juice to grape skins and possible microbial infection of the grapes [5].
The knowledge of grape proteins and their properties has been stimulated by grapevine genome sequencing projects [10, 11]. Proteomics studies were focused mainly on the diversity of PR proteins and their changes during grape ripening [12, 13]. Interestingly, a decrease in diversity was reported, which is in contrast to a large increase in the total concentration. Plant PR proteins have been classified into many families [14] but only certain representatives occur in grapevine. In particular, class IV chitinases (PR-3 family) and thaumatin-like proteins (TLPs; PR-5 family), which accumulate in grapes during ripening, are the most typical [15]. They show a low molecular weight of 20–35 kDa and are resistant to proteolysis as well as the acidic pH of wine. The PR-3 and PR-5 proteins exist in different molecular forms [13, 16]. Chitinases are considered the primary cause of heat-induced haze formation due to their irreversible denaturation and aggregation [17]. In addition, this process can be affected by the non-protein wine components. The essential factors are, among others, phenolic substances, sulphate anions, and wine pH [2]. Botrytis releases proteases, which degrade PR proteins in wine and their content consequently decreases [18]. Infection of grapes with powdery mildew, on the other hand, leads to increased levels of PR proteins [19].
Juice samples from manually and mechanically harvested Pinot Noir and Sauvignon Blanc grapes were analyzed for the concentration of TLPs and chitinases using reversed-phase (RP) high performance liquid chromatography (HPLC) [20]. The respective peak areas were compared with those of horse heart cytochrome c as a protein standard. Damaged grapes transported for long distances after mechanical harvesting and processed with a time delay were shown to have significantly higher total protein. This increase, for example from 200 (intact Sauvignon Blanc berries) up to 340 mg·L−1 (broken Sauvignon Blanc berries), was found to come from the extraction of unstable proteins from the grape skin [20].
TLPs and chitinases were found applicable as molecular markers for the qualitative characterization of grapevine varietal differences and hierarchical clustering analysis [21]. This employed the varying presence of different isoforms of these proteins not only among varieties but also (in some cases) for the same variety, different year of harvest and/or vineyards. First, concentrated grape juice proteins were separated by HPLC and monitored by UV absorption and electrospray mass spectrometry. Finally, the C8 reversed phase column was replaced by a short protein trap cartridge to increase sensitivity and reduce analysis time [21]. Chitinases and TLPs purified by several chromatographic steps were applied as standards to quantify the same proteins in grape juices and wines by HPLC and ELISA methods [22]. The determined content ranged from a few mg·L−1 in wine up to about 100 mg·L−1 in juice.
The content of TLPs and chitinases in white grape juices was quantified by RP-HPLC and correlated with heat instability tests [23]. Proteins were identified by assigning the respective retention times and quantified using bovine serum albumin. A positive correlation was found for the content of PR proteins (except for one chitinase isoform) and protein instability of the juice. A similar procedure for the relative quantification of PR proteins was described for grape skin and pulp samples [24]. The assigned peaks allowed quantification by comparing their areas with those of a thaumatin standard from Thaumatococcus daniellii yielding relatively high content values of around 580 and 440 mg·L−1, respectively, in the skin extract. These numbers were about twice as high as for pulp. A rapid method for the detection of haze-forming proteins in wine using a fluorescent dye (claimed to bind selectively to TLPs and chitinases) with a detection limit of 2 mg·L−1 has recently been introduced [25].
The aim of this work was to develop a fast and easy method for the determination of TLPs in white wines by means of MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectrometry). Samples of commercial wines and experimental wine made in the laboratory were analyzed directly, after dialysis or they were subjected to ultrafiltration before the measurements. MS-based quantification was achieved by comparing the peak areas of an internal standard such as cytochrome c, myoglobin or thaumatin and TLPs in the acquired mass spectra.
Materials and methods
Wine samples and chemicals
The analyzed wines included: Pálava, Melč 2020, late harvest, Czech Republic; Sauvignon blanc, Brise de France 2019 (SgBF); Sauvignon blanc, Michlovský 2020, Czech Republic (SgM); Chardonnay, Brise de France 2020 (ChBF), and Riesling, Chateau Bzenec 2020, Czech Republic (RR). They were purchased in local alimentary shops and stored at laboratory temperature before opening otherwise kept in a fridge at 4 °C. The unfined white wine (indicated here as experimental wine, EX) was made from a half-fermented juice (“Federweisser”), purchased in September 2020, by completing its fermentation under a stopper with airlock at 23 °C for 1 month. The EX wine was then filtered through a paper filter, clarified by centrifugation at 8,000 × g for 30 min, and stored at 4 °C. Con-A Sepharose 4B and all protein standards were purchased from Merck (Steinheim, Germany). Other chemicals were of analytical purity grade and purchased from Merck. Precision Plus Protein™ Kaleidoscope™ Prestained Protein Standards for electrophoresis were from Bio-Rad (Hercules, CA, USA).
Wine sample concentration and protein assay
Ultrafiltration of wine or dialyzed wine was performed in an Amicon Model 8200 stirred ultrafiltration cell equipped with an Ultracel 10-kDa cutoff membrane filter (Millipore, Billerica, MA, USA). Wine concentrates were stored frozen at – 30 °C. Proteins were determined spectrophotometrically using the bicinchoninic acid assay [26] and bovine serum albumin as a standard.
Wine samples fractionation
Dialysis was carried out with a cellulose acetate dialysis tubing (Sigma–Aldrich, St. Louis, MO, USA) against 0.1% trifluoroacetic acid (TFA) or 25 mM ammonium acetate (the latter solution was used prior to a subsequent treatment with Con-A Sepharose 4B, see further). Mannoproteins (particularly seripauperin 5) were removed by adding 100 µL of wet Con-A Sepharose particles equilibrated in 25 mM ammonium acetate to wine, dialyzed or ultrafiltered wine (300 µL), or in an increased ratio, if necessary, for a complete removal. The resulting slurry was shaken at 1000 RPM and 23 °C for 15 min.
Polyacrylamide gel electrophoresis
Wine proteins were separated by SDS–PAGE according to Laemmli [27] in 12% T-3.3% C resolving and 4% T-3.3% C stacking gels in a Mini-Protean II vertical gel chamber (Bio-Rad). Sample aliquots of wine, dialyzed wine or ultrafiltered wine were evaporated to dryness in a vacuum centrifuge (Eppendorf, Hamburg, Germany). The solid residuum was dissolved in Laemmli sample buffer, heated at 100 °C for 5 min and cooled down on ice. Electrophoresis was run at a constant voltage of 120 V until the marker dye reached the bottom of the gel. The gels were then incubated in 1% trichloroacetic acid for fixation, repeatedly washed in deionized water and the separated proteins finally visualized by a colloidal Coomassie Brilliant Blue G-250 staining [28]. Gel images from a calibrated ImageScanner (Amersham Biosciences, Uppsala, Sweden) were processed using GelAnalyzer 19.1 (http://www.gelanalyzer.com/).
Sample preparation for MALDI
Wine samples were deposited on an MSP BigAnchor 96 BC microScout Target (Bruker Daltonik, Bremen, Germany) by a two-layer preparation technique. First, 1 µL of sinapinic acid (SA, 10 mg·mL−1 in acetone) was deposited at a chosen position and left to dry for making a thin layer of matrix crystals. Then 1 µL of wine (as such, dialyzed or ultrafiltered) was pipetted at the matrix crystals layer, overlaid with the same amount of another SA solution, 10 mg·mL−1 in 0.1% TFA:acetonitrile (ACN), 1:1, v/v, and left to dry for a final crystallization. For the quantification of TLPs, the latter matrix solution contained a selected protein internal standard (5 – 50 ng·µL−1). Stock solutions of the internal standards were made in 0.1% TFA at 5 mg·mL−1 and then diluted into the matrix solution as needed.
MALDI measurements and protein identifications
Intact protein mass spectra of wine samples were acquired on a Microflex LRF20 MALDI-TOF mass spectrometer (Bruker Daltonik) equipped with a 60-Hz nitrogen laser (λmax = 337 nm) and operating in the reflector positive ion mode. Parameters of the instrument in the reflector mode were as follows: IS1 voltage (accelerating): 19.0 kV; IS2 voltage (extraction): 15.5 kV; lens voltage: 9.0 kV; reflector voltage: 20.0 kV; detector voltage: 1670 V; and pulsed ion extraction delay time of 500 ns. The examined mass range was between m/z 1000 and 30,000. All mass spectra were accumulated from 2000 laser shots and averaged from randomly chosen positions at the sample spots (“partial random walk”). Protein Calibration Standard I (Bruker Daltonik) was employed for external calibrations. The acquisition and evaluation software used were flexControl 3.4 and flexAnalysis 3.4, respectively (Bruker Daltonik). Quantitative data from the MALDI spectra were obtained using mMass 5.5.0 [29] by summing peak areas for [M + H]+, [M + 2H]2+, or, if applicable, [2M + H/Na]+ ions.
Protein identifications from SDS–PAGE gels after in-gel digestion were performed on a nanoflow liquid chromatography system coupled to MALDI-TOF/TOF MS/MS via a spotter device as described previously [3]. The highly glycosylated yeast seripauperin 5 (PAU5) was identified after its isolation from the dialyzed and ultrafiltered SgM wine using a microgradient separation device [30, 31]. An aliquot containing 5 μg protein in 30 μl of 0.1% TFA was loaded onto an in-house made microcolumn (250 μm i.d. × 20 mm) packed with 5.0-μm C4 particles (Dr. Maisch, Ammerbuch-Entringen, Germany). Elution was performed with 16 μl of 35% and 8 μl of 30% ACN in 0.1% TFA, loaded into the syringe in the order given. The other wine components were washed off from the column before eluting PAU5 using a stepwise gradient (24 µl) comprising of 10% (8 μl), 20% (8 μl), and 30% (8 μl) ACN in 0.1% TFA. In-solution digestions of the seripauperin fraction (reduced by dithiothreitol) using SOLu trypsin, GluC or chymotrypsin (all Sigma–Aldrich) in a ratio of enzyme-to-substrate of 1:20 then followed in 100 mM NH4HCO3 for 16 h. The process was stopped by adding 1% TFA, 1:9, v/v. The digest was loaded onto a microcolumn (250 μm i.d. × 30 mm) packed with 5.0-μm C18 particles (Dr. Maisch) and equilibrated in 0.1% TFA. Peptide elution was achieved in 96 0.5-μl fractions using a 48-μl gradient from 8 to 48% ACN in 0.1% TFA directly onto a MALDI AnchorChip™ 384 target (Bruker Daltonik). Each eluted drop was immediately covered by 0.5 μl of CHCA matrix (5 mg ml−1 in ACN/0.1% TFA, 1:1, v/v) and left to crystallize on air. PAU5 identification by MS/MS was run on an ultrafleXtreme MALDI-TOF/TOF instrument as described [3]. Serine-bound O-glycans comprising 1–9 hexose units [32] were included as variable modifications for searches in Peaks Studio X (Bioinformatics Solutions, Waterloo, ON, Canada). Tolerance intervals of 50 ppm and 0.5 Da were applied for peptide and fragment masses, respectively. The combined data from the digestions with different proteases were analyzed with an unspecific enzyme digestion search parameter. Protein database was limited to 24 seripauperin sequences from the UniprotKB database and a set of common contaminants (https://www.thegpm.org/crap/).
Results
MALDI measurements with wine samples
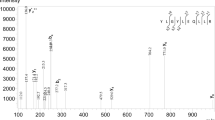
The wine proteins of interest with a molecular mass of 5–30 kDa were detectable by MALDI-TOF mass spectra using SA matrix (Fig. 1). Well-visible peaks appeared in the region of low protein masses between m/z 5000–10,000. But the most characteristic were a high peak at m/z 10,615, a peak cluster centered at m/z 17,000 and namely a group of peaks in the m/z region of 20,000 – 25,000. An additional high peak at m/z 13,016 was observable in the experimental wine made in our laboratory (Fig. 1). Wine samples containing a low amount of proteins, which thus provided less intense spectral peaks when measured directly, were concentrated via ultrafiltration (e.g. 50-fold). We also tested adding of dry Sephadex G-25 Medium beads (25% w/v) to absorb water in 1-ml aliquots within 10 min as an instant step to increase protein concentration. The top liquid layer upon the Sephadex swelling was collected by brief centrifugation in a tabletop mini-centrifuge. This treatment resulted in a volume reduction by sevenfold. This number corresponded well with an increase in protein concentration as determined using BCA protein assay method. For example, a wine containing 0.13 ± 0.02 mg·mL−1 protein was concentrated to 0.94 ± 0.05 mg·mL−1.

MALDI-TOF mass spectra of wine proteins. Samples were deposited on the target using the SA matrix and two-layer matrix deposition technique. The top and bottom spectrum (EX and SgBF, respectively) refer to an ultrafiltered wine; the middle spectrum was acquired directly with bottled wine. The spectra were acquired in the reflector positive ion mode
PR-5 and PAU5 protein signals
The group of peaks observed in the m/z region of 20,000 – 25,000 was attributed to TLPs (PR-5 family) by an obvious analogy with previous literature data [33, 34]. Class IV chitinases, which belong to the PR-3 family and show molecular masses above 25 kDa [33, 34], could not be detected in MALDI-TOF mass spectra when using bottled white wine, dialyzed or even concentrated (ultrafiltered) wine. Measurements in the reflector mode yielded at least six different signals (Fig. 1). The highest TLP peak appeared at m/z 21,228 accompanied by a neighbor peak at m/z 21,430. Accordingly, the peak at m/z 10,615 was attributed to a doubly charged TLP-derived ion. Other TLP signals were e.g. m/z 22,237 and 23,426. The peak cluster centered at m/z of 17,000 was assigned to the highly glycosylated yeast protein seripauperin 5 (PAU5) by molecular mass and literature data [4, 35]. The mass range of this signal group spanned the m/z interval of 15,000 – 18,500 indicating the presence of differently glycosylated proteoforms (with a constant mass difference of 162 Da) containing numerous mannose units in the molecule. The highest peak was slightly different for individual wine samples, usually either m/z 16,789 or 16,952. A similar cluster observed around m/z 8,500 was assigned to doubly charged PAU5 ions. The PAU5 peak clusters could be eliminated from the mass spectra by a treatment of wine samples with Con-A Sepharose equilibrated in 25 mM ammonium acetate prior to the measurements (Fig. 2).

The use of Con-A-Sepharose 4B to remove seripauperin 5 (PAU5). MALDI-TOF mass spectra of wine proteins were measured using the SA matrix and two-layer matrix deposition technique. Top panel, dialyzed SgM; bottom panel, the same wine after PAU5 removal by affinity chromatography on Con-A Sepharose 4B. The spectra were acquired in the reflector positive ion mode
Electrophoresis and identification of wine proteins
Figure 3 shows a result of SDS–PAGE performed with EX, SgBF and SgM samples. The visualized protein bands were analyzed for their retention factors and the corresponding molecular mass by GelAnalyzer 19.1. This software provides an option to read the intensity volume of staining (the gel images were obtained with a calibrated scanner), which was employed to estimate the relative proportion of TLPs. Protein identification was then achieved by MALDI-TOF/TOF tandem mass spectrometry (MS/MS) and NCBI Protein database search after in-gel tryptic digestion (Supplementary file). In the EX sample, Vitis vinifera TLPs were found in multiple bands at molecular masses between 20 and 25 kDa (accession numbers: gi|7406716, gi|520729528, gi|605603680, gi|7406714, and gi|89242714) together with osmotin (gi|1839046), and some yeast proteins (e.g. a cell wall mannoprotein CIS3, gi|151944971, and another cell wall protein CCW14, gi|323353722). All these stained proteins represented 28% of the total intensity volume of the resolved EX sample proteins. V. vinifera class IV chitinase was detected at 27 kDa (gi|2306811), whereas the band at 34 kDa displayed a yeast exo-beta-1,3-glucanase (gi|323304773). The thick protein band at around 60 kDa was found to contain V. vinifera glycosyl hydrolase (invertase, gi|296084197), pectinesterase (gi|359479995) and laccase 2 from Botrytis cinerea (gi|15022489). SgM sample was shown to contain many Botrytis proteins, particularly in the molecular mass region of 30–60 kDa, otherwise the protein content in the most intense protein bands was similar for the other wine samples. Also, similar intensity volume percentages of TLPs were found for SgBF and SgM (39 and 36%, respectively). RR and ChBF wines showed lower numbers of 28 and 15%, respectively.

SDS–PAGE of wine samples. Electrophoresis was carried out in a 12% resolving gel and the separated proteins were visualized by Coomassie Brilliant Blue G-250. Top panel shows (from the left) protein standards with the indicated molecular mass values, wine protein samples (as indicated by the top labels). The calculated amount of the loaded protein was 50 µg (EX), 75 µg (SgBF) and 37 μg (SgM). Bottom panel shows an example of reading the intensity volumes of the stained protein bands (EX separation lane, see the top panel) using GelAnalyzer software
PAU5 was confirmed only after the separation of seripauperin protein fraction using microgradient chromatographic device. It is a highly glycosylated yeast membrane protein secreted under low oxygen and cold stress conditions [36]. A non-glycosylated PAU5 signal observed at m/z 12,905 Da corresponds well with the theoretical molecular mass and the presence of an N-terminal acetylation and methionine oxidation (Supplementary file).
The presence of the peaks corresponding to PAU5 with at least 13 attached hexose units agrees with the yeast glycosylation mechanism starting in the endoplasmic reticulum and continuing in Golgi apparatus, where a minimal sugar count is required for the successful protein processing and secretion [37]. The highest recognizable number of sugars bound to PAU5 protein in our experiments was 36. Database searches with combined MALDI-TOF/TOF MS/MS data for PAU5 digested by different proteases allowed to confirm 32 hexoses at five O-glycosylation sites (Supplementary file). Interestingly, no peptides or glycopeptides from the C-terminus of PAU5 were detected although Pegg et al. [32] previously determined 3/4 hexoses at Ser109 and Ser110 in this region. But it has been reported that the variability of yeast strains and fermentation conditions strongly affects the glycosylation state of seripauperins [38]. The upper mass range limit at m/z 4000 for effective precursors of the MALDI-TOF/TOF instrument precluded analysis of large PAU5 glycopeptides.
Choice of protein standards for quantification
The protein standards evaluated in this study for the quantification of wine TLPs included horse cytochrome c (CYT), chicken lysozyme, horse myoglobin (MYO) and thaumatin (THA) from Thaumatococcus daniellii. These proteins were chosen because of their molecular mass values of 12.4, 14.4, 17.0 and 22.2 kDa, respectively, which fall in the mass region of interest in MALDI mass spectra. The standards were dissolved in the SA matrix solution at a desired concentration and added to a wine sample deposited on the target prior to crystallization (see Materials and methods). CYT, MYO and THA were finally selected because of the quality of their well-developed and symmetric [M + H]+ intact protein peaks reflecting a desirable purity.
CYT could be applied directly to all wine samples. As expected, MYO interfered with the PAU5 signal cluster and its application was possible only in the absence of PAU5 or after its removal using the affinity binding to Con-A Sepharose 4B, which did not influence the signals of TLPs (Fig. 2). On the other hand, THA was applicable only indirectly because of its mass interference with TLPs in wine. The dialyzed Pálava white wine, which showed no signals of TLPs in MALDI-TOF mass spectra but provided a measurable peak cluster of PAU5 isoforms, was applied as a background sample to determine the peak area ratios of CYT versus THA when added individually as internal standards. This numerical factor then allowed conversions of CYT peak areas measured with real samples into THA peak areas (considered a good substitute for unavailable standards of TLPs) by a simple recalculation. The experimental determination was initiated by measurements with a series of 5–50 ng of each of the two internal standards. When mutually compared, the determined peak area ratios of CYT/PAU5 and THA/PAU5 provided a linear dependence: Area ratio (CYT/PAU5) = 6.561 × Area ratio (THA/PAU5) + 9.058 (R2 = 0.98).
The obtained peak area ratios CYT/PAU5 were divided by the corresponding THA/PAU5 ratios, which resulted in an average CYT/THA peak area conversion factor of 9.66 for the relative laser energy setting of 64% (Table 1). As expected, this factor was influenced by laser intensity: increasing the relative intensity value to 72% resulted in its reduced value of 5.12. It was, therefore, necessary to use the same laser setting for the factor determination as well as measurements with real samples. Measuring with CYT and subsequent recalculations to the corresponding THA peak areas were applied to quantify TLPs in white wines. Control quantifications with MYO after the removal of PAU5 were performed in parallel. MYO plus THA binary mixtures were first analyzed using different weight ratios of the proteins in the range of 5 to 50 ng in MALDI probes. Then the corresponding peak areas were compared to find another average factor, in this case applicable for recalculation (conversion) of the MYO content to that of THA, as illustrated in Fig. 4. A MYO/THA conversion factor value of 1.30 was determined by averaging multiple experiments.

MALDI-TOF spectra of model mixtures containing horse MYO and T. daniellii THA used as standards in this work. Panel (A) MYO and THA (5 ng of each), B MYO (10 ng) and THA (5 ng). The spectra were acquired with SA matrix. MYO provided the following peaks: m/z 16,952 ([M + H]+ and 8477 ([M + 2H]2+). The corresponding THA peaks were at m/z 22,162 ([M + H]+) and 11,081 ([M + 2H]2+)
Quantification of TLPs in real wine samples
The five white wine samples EX, ChBF, RR, SgBF and SgM with the known total protein contents and electrophoretic protein patterns were used to quantify TLPs by MALDI-TOF MS employing CYT and MYO as internal standards. Direct measurements without any previous wine treatment indicated a large difference in their TLP contents. Except for SgM, where the TLP protein peaks were well pronounced in the mass spectrum and which was only dialyzed to achieve a better crystallization with matrix on the target for quantification, the other samples were concentrated by ultrafiltration to make the TLP peaks more visible (Fig. 1). RR and ChBF wines were additionally dialyzed prior to ultrafiltration. The concentration factors were the following: SgM – 0.91, EX – 80, ChBF – 50, RR – 50, SgBF – 50.
Quantification experiments were performed using increasing amounts of the internal standard CYT or MYO (10–50 ng) applied in the matrix solution. The ultrafiltered wine samples were measured as such or properly diluted. The determined peak area ratios were then converted to THA/TLP ratios from which the TLP content was calculated as an average value. This approach was chosen to minimize possible systematic errors from pipetting and an inhomogeneous distribution of sample-matrix cocrystals influencing the ionization process at different places of sample spots. Figure 5 depicts measurements with EX wine and CYT internal standard. An example of results is provided in Table 2.

MALDI-TOF mass spectrometric quantification of TLPs in EX wine using CYT as an internal standard. The spectra were acquired with SA matrix, 1 μl of wine ultrafiltrate was deposited on the target. From the top, 10 ng CYT (A), 20 ng CYT (B) and 30 ng CYT (C). CYT provided the following peaks: m/z 12,361 ([M + H]+) and 6181 ([M + 2H]2+). The highest peak of TLPs (m/z 21,229) was attributed to VVTL1 according to the literature and protein identification results
The MYO standard was used is in parallel with CYT. The only difference for this approach resided in the need to remove Pau5 from wine samples by Con-A Sepharose 4B prior to their pipetting on MALDI target. Figure 6 provides representative mass spectra documenting the analysis of RR wine measured as a dialyzed and ultrafiltered sample (Table 3).

MALDI-TOF mass spectrometric quantification of TLPs in RR wine using MYO as an internal standard. The spectra were acquired with SA matrix, 1 μl of ultrafiltered dialyzed wine was deposited on the target. From the top, 5 ng MYO (A), 10 ng MYO (B) and 20 ng MYO (C). MYO provided the following peaks: m/z 16,952 ([M + H]+) and 8477 ([M + 2H]2+). TLPs were registered as a series of peaks between m/z 21,230 and 25,601. As can be seen, no Pau5 signals are visible after the treatment with Con-A Sepharose
Table 4 summarizes the experimental data obtained for all wine samples analyzed in this study. It also shows an obvious correlation of the three applied approaches to find the content of TLPs: reading staining intensity volume from calibrated image scans of SDS–PAGE gels and two MALDI-TOF MS quantifications with either CYT or MYO as internal standards. The highest content of TLPs appeared in the commercial SgM wine, followed by EX and ChBF wines, both comparable in this parameter. The lowest content of TLPs was in SgBF and RR wines, roughly tenfold less than in SgM.
Discussion
Proteins in wine show a large technological and economical importance as they influence its stability. TLPs and chitinases were previously determined using quantifications based on HPLC and comparisons of the corresponding peak areas with those of protein standards [20, 22, 23]. The aim of this work resided in using MALDI-TOF MS as an alternative to HPLC to achieve shorter analysis times with microliter sample amounts. The possibility of measuring proteins in wine by MALDI-TOF MS without any sample pretreatment has been long known [39]. Quantitative MALDI measurements are primarily hampered by the inhomogeneous distribution of matrix and analyte across the target surface resulting in a poor shot‐to‐shot and sample‐to-sample reproducibility [40]. A common solution how to overcome this inherent irreproducibility resides in using internal standards. They do not necessarily need to be isotopically labelled as the use of a structural analog can also provide reliable results [41]. There are many beneficial features of MALDI-based quantification analyses: speed, ease of performance, possible automation and high throughput, sensitivity, versatility and low cost [42]. Peak heights of different analytes measured in an equimolar ratio may largely vary but relative intensities for a repeated sample run are constant [41]. Absolute quantification with an internal standard is based on establishing the constant of proportionality by measurements with a fixed amount of one compound and varying amounts of the other one. Peak intensity or area ratios are determined.
Although a thaumatin protein was available (i.e. THA from Thaumatococcus daniellii) as a possible internal standard, it could not be used directly because of the mass interference with TLPs in real wine samples. We, therefore, chose two commonly available proteins CYT and MYO as internal standards because their molecular masses were relevant for this purpose and not interfering. THA was expected to have its ionization properties equal or at least largely similar to those of wine TLPs. The sequence identity and similarity of THA (gi|1482417275) and V. vinifera osmotin (gi|89242714) or V. vinifera thaumatin-like protein 1 (gi|520729528) is 55 and 65%, respectively, in a pairwise alignment. Our initial experiments were successfully aimed at obtaining peak are ratios CYT/THA and MYO/THA applicable for calculations of the TLPs content of wines from MALDI-TOF MS experimental data. The application of CYT was straightforward but this was not the case for MYO. Most of the wines analyzed in this work displayed peaks of various glycosylated forms of PAU5 in the mass spectra centered at m/z 17,000. It is almost exactly the same mass as that of MYO. It was, therefore, necessary to remove the interfering PAU5 before applying the sample on the target (Fig. 2). The highly glycosylated PAU5 from Saccharomyces cerevisiae has been associated with the gushing phenomenon of sparkling wine upon bottle opening. This excessive over-foaming is largely correlated with the absence of PAU5 in wine samples as the protein has a foam-stabilizing role [35]. The PAU5 level in wine is affected by bentonite treatment. In addition, different yeast strains produce differing PAU5 protein concentrations in the culture supernatant and the production is influenced by many factors such as inoculum amount, temperature, pH, light intensity, and agitation rate [38]. The sequence of S. cerevisiae PAU5 (UniProtKB access. no. P43575) comprises 11 Ser and 12 Thr residues considered as potential O-mannosylation sites. Proteomic analyses of white sparkling wines demonstrated that seripauperins were the most abundant yeast proteins [32]. Con-A Sepharose, which has been established for affinity purifications of mannose-containing glycoproteins [43], was found efficient for the PAU5 removal but this represented an additional step when working with MYO contrary to the use of CYT.
The collection of white wines available for quantification comprised three common products (ChBF, RR, SgBF), one wine with an attribute (SgM) and one experimental wine made in the laboratory (EX). Their protein composition was resolved by SDS–PAGE and the resulting gel patterns were documented as images by a calibrated scanner. The subsequent protein identification analysis by nLC-MALDI-TOF/TOF MSMS after in-gel digestion confirmed the presence of TLPs in a region corresponding to molecular masses of 20–25 kDa. Chitinase bands at around 30 kDa were absent or very weak for most samples with the exception of the EX wine (Fig. 3). No peaks of chitinases were registered in the MALDI-TOF mass spectra and thus only TLPs could be quantified in this way although we originally planned to quantify the PR-3 and PR-5 proteins together. The literature shows enough chitinases in juice samples, but their amount is largely reduced by vinification [22] and this was obviously our case. The intensity volumes in the scanned images allowed estimating the proportional content of TLPs, which differed among the studied wines and appeared between 15% (ChBF) and 39% (SgBF). These estimates, however, should be taken with caution: the mentioned gel separation areas were found to contain some yeast proteins in addition to TLPs (e.g. CIS3 mannoprotein). But the yeast proteins were only a minor part as deduced from the number of their sequenced peptides compared to those derived from TLPs. Similar proportions of TLPs have been reported for Sauvignon blanc and Chardonnay grape juices [23, 33].
The highest protein level was found in SgM (37 mg·L−1) and this wine also contained the highest amount of TLPs (21 mg·L−1 as determined using the CYT standard; Table 4). The difference in TLPs was by almost 20-fold compared to SgBF and doubled compared to EX wine. Then it is not surprising that SgM could be measured by MALDI-TOF MS without any concentrating pretreatment providing well-developed peaks of TLPs (Fig. 1). The other analyzed wines were first concentrated by ultrafiltration to make these protein peaks more pronounced for a better reading of peak areas. We used to process 100-ml wine aliquots but the same effect in a reasonable time is possible with milliliter or lower volumes using centrifugal ultrafiltration cartridges. Dialysis was included to all samples to avoid on-target crystallization difficulties observed with the late harvest Pálava wine lacking measurable TLPs but containing more sugar. The concentration values of TLPs from MALDI-TOF MS were largely comparable with the estimates based on the intensity volumes of the corresponding protein bands in polyacrylamide gels (Table 4). Previous HPLC-based studies have reported TLP content values of 0.5–36.5 mg·L−1 in white wines and 85–105 mg·L−1 in grape juices [22], or 5.2–67.0 mg·L−1 in grape juices [23].
The use of the CYT standard was quick and easy in connection with MALDI-TOF MS, whereas applying MYO required one additional chromatographic step in the sample preparation procedure to remove PAU5. The TLPs content values determined using MYO were comparable to those obtained in the parallel measurements with the CYS standard but, in some cases, they appeared significantly lower (e.g. for SgM and ChBF). This result may reflect unpredictable retention effects of the affinity chromatography, which could reduce the intensity of the inspected protein signals in the m/z range of 20,000–25,000. Both the CIS3 and CCW14 cell wall proteins from S. cerevisiae are O- as well as N-glycosylated according to the Uniprot database and it is expectable they bind to Con-A Sepharose. Although the observed signal maxima were clearly assigned to TLPs, the respective peak areas could include a yeast proteins contribution. The use of MYO standard after the Con-A Sepharose chromatography removing some yeast proteins would, therefore, provide more accurate quantification results. The internal standards CYT and MYO were optimized in amounts of 10–50 ng per sample spot on the target. Adding them into the matrix solution became very convenient when measuring more samples at once. No premixing of sample aliquots with different amounts of the standards was necessary. A few spiked matrix solutions were applicable to all samples in the analyzed series. The calculated relative standard deviation (RSD) values, which show the precision of the obtained averages, appeared at 15 – 30%. Previous standard deviation data from HPLC-based quantifications of TLPs and chitinases allow to calculate most of the RSDs in the range of 10–20% [22] or even higher with some extreme values of more than 80% [23]. It seems that the precision of our MALDI-based method is thus quite comparable in this regard. The recent report on the quantification of TLPs and chitinases using a fluorescent dye determined the minimal concentration of proteins to form haze of 12 mg L−1 [25]. Our procedure goes below this limit and the lowest standard concentration (10 ng per µL of the matrix solution) appears just at this concentration level.
Conclusion
We have shown the applicability of MALDI-TOF MS for quantifications of TLPs in white wine samples with CYT or MYO as internal standards. Although we could not quantify chitinase in the same way because of the missing spectral signals, this approach appears very useful for a fast reading of the TLP content. Not all wine samples are suitable for a direct measurement. A dialysis step (optionally diafiltration as its faster alternative) and/or ultrafiltration are useful for wines, which cannot be measured directly because of the presence of disturbing low-molecular-weight compounds hampering crystallization with the matrix or a low overall protein concentration.
Data availability
The data that support the findings of this study are available from the corresponding author, MŠ, upon reasonable request.
References
Ferreira RB, Piçarra-Pereira MA, Monteiro S, Loureiro VB, Teixeira AR (2001) The wine proteins. Trends Food Sci Tech 12:230–239. https://doi.org/10.1016/S0924-2244(01)00080-2
Tian B, Harrison R, (2021) In: Cosme F, Nunes FM, Filipe-Ribeiro L (eds) Chemistry and biochemistry of winemaking, wine stabilization and aging, IntechOpen, London. https://doi.org/10.5772/intechopen.92445
Perutka Z, Šufeisl M, Strnad M, Šebela M (2019) High-proline proteins in experimental hazy white wine produced from partially botrytized grapes. Biotechnol Appl Biochem 66:398–411. https://doi.org/10.1002/bab.1736
Kupfer VM, Vogt EI, Ziegler T, Vogel RF, Niessen L (2017) Comparative protein profile analysis of wines made from Botrytis cinerea infected and healthy grapes reveals a novel biomarker for gushing of sparkling wines. Food Res Int 99:501–509. https://doi.org/10.1016/j.foodres.2017.06.004
Waters EJ, Alexander G, Muhlack R, Pocock KF, Colby C, O’Neill BK, Høj PB, Jones P (2005) Preventing protein haze in bottled white wine. Aust J Grape Wine R 11:215–225. https://doi.org/10.1111/j.1755-0238.2005.tb00289.x
Salazar FN, Zamora F, Canals JM, Lopez F (2010) Protein stabilization in sparkling base wine using zirconia and bentonite: influence on the foam parameters and protein fractions. J Int Sci Vigne Vin 44:51–58
Sauvage FX, Bach B, Moutounet M, Vernhet A (2010) Proteins in white wines: Thermo-sensitivity and differential adsorbtion by bentonite. Food Chem 118:26–34. https://doi.org/10.1016/j.foodchem.2009.02.080
Albuquerque W, Seidel L, Zorn H, Will F, Gand M (2021) Haze formation and the challenges for peptidases in wine protein fining. J Agric Food Chem 69:14402–14414. https://doi.org/10.1021/acs.jafc.1c05427
Waters EJ, Shirley NJ, Williams PJ (1996) Nuisance proteins of wine are grape pathogenesis-related proteins. J Agric Food Chem 44:3–5. https://doi.org/10.1021/jf9505584
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pè ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quétier F, Wincker P (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467. https://doi.org/10.1038/nature06148
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Demattè L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Van de Peer Y, Salamini F, Viola R (2007) A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2:e1326. https://doi.org/10.1371/journal.pone.0001326
Monteiro S, Piçarra-Pereira MA, Loureiro VB, Teixeira AR, Ferreira RB (2007) The diversity of pathogenesis related proteins decreases during grape maturation. Phytochemistry 68:416–425. https://doi.org/10.1016/j.phytochem.2006.11.014
Marangon M, Van Sluyter SC, Haynes PA, Waters EJ (2009) Grape and wine proteins: their fractionation by hydrophobic interaction chromatography and identification by chromatographic and proteomic analysis. J Agric Food Chem 57:4415–4425. https://doi.org/10.1021/jf9000742
Van Loon LC, Van Strien EA (1999) The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol Mol Plant Pathol 55:85–97. https://doi.org/10.1006/pmpp.1999.0213
Deytieux C, Geny L, Lapaillerie D, Claverol S, Bonneu M, Donéche B (2007) Proteome analysis of grape skins during ripening. J Exp Bot 58:1851–1862. https://doi.org/10.1093/jxb/erm049
Gazzola D, Pasini G, Tolin S, Curioni A, Vincenzi S (2017) Characterization of chitinase isoforms from grape juice. Ital J Food Sci 29:50–62. https://doi.org/10.14674/1120-1770/ijfs.v551
Marangon M, Van Sluyter SC, Neilson KA, Chan C, Haynes PA, Waters EJ, Falconer RJ (2011) Roles of grape thaumatin-like protein and chitinase in white wine haze formation. J Agric Food Chem 59:733–740. https://doi.org/10.1021/jf1038234
Van Sluyter SC, McRae JM, Falconer RJ, Smith PA, Bacic A, Waters EJ, Marangon M (2015) Wine protein haze: mechanisms of formation and advances in prevention. J Agric Food Chem 63:4020–4030. https://doi.org/10.1021/acs.jafc.5b00047
Tian B, Harrison R, Jaspers M, Morton J (2015) Influence of ultraviolet exclusion and of powdery mildew infection on Sauvignon Blanc grape composition and on extraction of pathogenesis-related proteins into juice. Aust J Grape Wine R 21:417–424. https://doi.org/10.1111/ajgw.12135
Pocock KF, Hayasaka Y, Peng Z, Williams PJ, Waters EJ (1998) The effect of mechanical harvesting and long-distance transport on the concentration of haze-forming proteins in grape juice. Aust J Grape Wine R 4:23–29. https://doi.org/10.1111/j.1755-0238.1998.tb00131.x
Hayasaka Y, Adams KS, Pocock KF, Baldock GA, Waters EJ, Høj PB (2001) Use of electrospray mass spectrometry for mass determination of grape (Vitis vinifera) juice pathogenesis-related proteins: a potential tool for varietal differentiation. J Agric Food Chem 49:1830–1839. https://doi.org/10.1021/jf001163+
Le Bourse D, Conreux A, Villaume S, Lameiras P, Nuzillard JM, Jeandet P (2011) Quantification of chitinase and thaumatin-like proteins in grape juices and wines. Anal Bioanal Chem 401:1541–1549. https://doi.org/10.1007/s00216-011-4912-8
Salazar FN, López F, Chiffelle I, López R, Peña-Neira A (2012) Evaluation of pathogenesis-related protein content and protein instability of seven white grape (Vitis vinifera L.) clones from Casablanca Valley, Chile. Eur Food Res Technol 234:509–515. https://doi.org/10.1007/s00217-012-1659-1
Tian B, Harrison R, Morton J, Deb-Choudhury S (2015) Proteomic analysis of sauvignon blanc grape skin, pulp and seed and relative quantification of pathogenesis-related proteins. PLoS ONE 10:e0130132. https://doi.org/10.1371/journal.pone.0130132
Mierczynska-Vasilev A, Vasilev A, Reilly T, Bindon K, Vasilev K (2022) Fluorescence sensing technology for the rapid detection of haze-forming proteins in white wine. Food Chem 374:131770. https://doi.org/10.1016/j.foodchem.2021.131770
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. https://doi.org/10.1016/0003-2697(85)90442-7
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. https://doi.org/10.1038/227680a0
Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25:1327–1333. https://doi.org/10.1002/elps.200305844
Strohalm M, Kavan D, Novák P, Volný M, Havlíček V (2010) mMass 3: A cross-platform software environment for precise analysis of mass spectrometric data. Anal Chem 82:4648–4651. https://doi.org/10.1021/ac100818g
Franc V, Řehulka P, Medda R, Padiglia A, Floris G, Šebela M (2013) Analysis of the glycosylation pattern of plant copper amine oxidases by MALDI-TOF/TOF MS coupled to a manual chromatographic separation of glycans and glycopeptides. Electrophoresis 34:2357–2367. https://doi.org/10.1002/elps.201200622
Franc V, Řehulka P, Raus M, Stulík J, Novak J, Renfrow MB, Šebela M (2013) Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: Role of cysteine alkylation during sample processing. J Proteomics 92:299–312. https://doi.org/10.1016/j.jprot.2013.07.013
Pegg CL, Phung TK, Caboche CH, Niamsuphap S, Bern M, Howell K, Schulz BL (2021) Quantitative data-idependent acquisition glycoproteomics of sparkling wine. Mol Cell Proteomics 20:100020. https://doi.org/10.1074/mcp.RA120.002181
Pocock KF, Hayasaka Y, McCarthy MG, Waters EJ (2000) Thaumatin-like proteins and chitinases, the haze forming proteins of wine, accumulate during ripening of grape (Vitis vinifera) berries and drought stress does not affect the final levels per berry at maturity. J Agric Food Chem 48:1637–1643. https://doi.org/10.1021/jf9905626
Van Sluyter SC, Marangon M, Stranks SD, Neilson KA, Hayasaka Y, Haynes PA, Menz RI, Waters EJ (2009) Two-step purification of pathogenesis-related proteins from grape juice and crystallization of thaumatin-like proteins. J Agric Food Chem 57:11376–11382. https://doi.org/10.1021/jf902365r
Kupfer VM, Vogt EI, Siebert AK, Meyer ML, Vogel RF, Niessen L (2017) Foam-stabilizing properties of the yeast protein PAU5 and evaluation of factors that can influence its concentration in must and wine. Food Res Int 102:111–118. https://doi.org/10.1016/j.foodres.2017.09.060
Luo Z, Van Vuuren HJJ (2008) Stress-induced production, processing and stability of a seripauperin protein, Pau5p, in Saccharomyces cerevisiae. FEMS Yeast Res 8:374–385. https://doi.org/10.1111/j.1567-1364.2008.00355.x
De Pourcq K, De Schutter K, Callewaert N (2010) Engineering of glycosylation in yeast and other fungi: current state and perspectives. Appl Microbiol Biotechnol 87:1617–1631. https://doi.org/10.1007/s00253-010-2721-1
Mann MA, Frisch LM, Vogel RF, Niessen L (2021) Influence of fermentation conditions on the secretion of seripauperin 5 (PAU5) by industrial sparkling wine strains of Saccharomyces cerevisiae. Food Res Int 139:109912. https://doi.org/10.1016/j.foodres.2020.109912
Szilágyi Z, Vas G, Mády G, Vékey K (1996) Investigation of macromolecules in wines by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 10:1141–1143. https://doi.org/10.1002/(SICI)1097-0231(19960715)10:9%3c1141::AID-RCM607%3e3.0.CO;2-4
Duncan MW, Roder H, Hunsucker SW (2008) Quantitative matrix-assisted laser desorption/ionization mass spectrometry. Brief Funct Genomic Proteomic 7:355–370. https://doi.org/10.1093/bfgp/eln041
Kang MJ, Tholey A, Heinzle E (2001) Application of automated matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for the measurement of enzyme activities. Rapid Commun Mass Spectrom 15:1327–1333. https://doi.org/10.1002/rcm.376
Šebela M (2023) The use of matrix-assisted laser desorption/ionization mass spectrometry in enzyme activity assays and its position in the context of other available methods. Mass Spectrom Rev 42:1008–1031. https://doi.org/10.1002/mas.21733
Campbell KP, MacLennan DH (1981) Purification and characterization of the 53,000-dalton glycoprotein from the sarcoplasmic reticulum. J Biol Chem 256:4626–4632. https://doi.org/10.1016/S0021-9258(19)69481-0
Acknowledgements
This work was supported by the ERDF project “Plants as a tool for sustainable global development” (No. CZ.02.1.01/0.0/0.0/16_019/0000827) and Czech-Austrian Mobility project 8J20AT015 provided by the Ministry of Education, Youth and Sports, Czech Republic and Agency for Education and Internationalization, Austria.
Funding
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Contributions
Conceptualization: MŠ, MMD; Methodology: MŠ, ZP; Formal analysis and investigation: MR, MŠ, ZP; Writing—original draft preparation: MŠ, MR, ZP; Writing—review and editing: MŠ, MMD; Funding acquisition: MŠ, MMD.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Compliance with ethical standards
This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rašková, M., Perutka, Z., Marchetti-Deschmann, M. et al. Quantification of thaumatin-like proteins in white wine using MALDI-TOF mass spectrometry. Eur Food Res Technol 250, 69–81 (2024). https://doi.org/10.1007/s00217-023-04366-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-023-04366-y