
Abstract
Food fraud is becoming a prominent topic in the food industry. Thus, valid methods for detecting potential adulterations are necessary to identify instances of food fraud in cereal products, a significant component of human diet. In this work, primer–probe systems for real-time PCR and droplet digital PCR (ddPCR) for the detection of these cereal species: bread wheat (together with spelt), durum wheat, rye and barley for real-time PCR and ddPCR were established, optimized and validated. In addition, it was projected to validate a molecular system for differentiation of bread wheat and spelt; however, attempts for molecular differentiation between common wheat and spelt based on the gene GAG56D failed because of the genetic variability of the molecular target. Primer–probe systems were further developed and optimized on the basis of alignments of DNA sequences, as well as already developed PCR systems. The specificity of each system was demonstrated on 10 (spelt), 11 (durum wheat and rye) and 12 (bread wheat) reference samples. Specificity of the barley system was already proved in previous work. The calculated limits of detection (LOD95%) were between 2.43 and 4.07 single genome copies in real-time PCR. Based on the “three droplet rule”, the LOD95% in ddPCR was calculated to be 9.07–13.26 single genome copies. The systems were tested in mixtures of flours (rye and common wheat) and of semolina (durum and common wheat). The methods proved to be robust with regard to the tested conditions in the ddPCR. The developed primer–probe systems for ddPCR proved to be effective in quantitatively detecting the investigated cereal species rye and common wheat in mixtures by taking into account the haploid genome weight and the degree of milling of a flour. This method can correctly detect proportions of 50%, 60% and 90% wholemeal rye flour in a mixture of wholemeal common wheat flour. Quantitative results depend on the DNA content, on ploidy of cereal species and are also influenced by comminution. Hence, the proportion of less processed rye is overestimated in higher processed bread wheat and adulteration of durum wheat by common wheat by 1–5% resulted in underestimation of common wheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cereals and cereal products are a significant source of nutrition. In Germany, the consumption of cereal products averaged 81.7 kg per capita in 2017 and 2018 [1]. Cereal products are among the top 10 products in Europe with the highest risk of food fraud [2]. Food fraud is defined as a mismatch between food claims and actual food properties through deliberate misdeclaration or deliberate omission of claims [3]. Furthermore, it refers to the substitution, manipulation, falsification and adulteration of individual components or of the entire food [2]. In cereal products, the partial or total substitution of value-added components (e.g. substitution of rye flour with cheaper wheat flour) is of particular importance as it represents a financial disadvantage for the consumer. However, in the legal assessment of these products of agricultural origin, adventitious presence as a result of cultivation, harvesting, transport and storage must always be taken into account (e.g. for durum wheat according to Regulation (EU) No. 2016/1238 a permitted proportion of 3%).
The tribus Triticeae evolved about 25 million years ago in the sweet grass family (Poaceae) [4]. Important cereal species in this tribus are common wheat (Triticum aestivum subsp. aestivum), rye (Secale cereale), barley (Hordeum vulgare) and durum wheat (Triticum turgidum subsp. durum) [5]. The tribus consists of the two subtribes: Hordeineae and Triticineae. Hordeineae gave rise to the genus Hordeum with the important representative Hordeum vulgare. The subtribe Triticineae first gave rise to the genus Secale (representative Secale cereale) and finally to the genera Triticum and Aegilops [4]. Starting with the original form Triticum urartu (diploid, genome AA), the tetraploid species Triticum turgidum arose in the genus Triticum through allopolyploidisation with Aegilops speltoides (diploid, genome SS), from which Triticum turgidum subsp. dicoccon (emmer, tetraploid, genome AABB) emerged through cultivation. Further cultivation produced Triticum turgidum subsp. durum (tetraploid, genome AABB) and further Triticum turgidum subsp. turanicum (kamut) [4, 6]. Hybridization of the species Triticum turgidum (genome AABB) with Aegilops tauschii (diploid, genome DD) resulted in the hexaploid Triticum species Triticum aestivum subsp. spelta (hexaploid, genome AABBDD) and Triticum aestivum subsp. aestivum (hexaploid, genome AABBDD) [7]. Another hybridization between Secale cereale (diploid, genome RR) and Triticum aestivum produced Triticale (octoploid, genome AABBDDRR) [8].
Enzyme-linked immunosorbent assay (ELISA) [9], chromatographic methods [9,10,11,12,13], near-infrared spectroscopy (NIRS) [14, 15] and real-time PCR [16,17,18,19,20,21,22] and other molecular techniques [23] were used for the specific detection of cereal species in food. Compared to other analytical parameters, such as proteins and fats, DNA is sufficiently stable, depending on the degree of processing, to be detected even in more processed foods [24]. Therefore, molecular methods based on chromosomal genes can potentially be used for quantification [21]. For the specific detection of rye, the TaqMan® PCR system, based on the single-copy gene for myo-inositol-1-phosphate synthase (MIPS), was developed by Pegels et al. [17]. Casazza et al. were able to detect differences between durum and common wheat in the intron region of the β-tubulin gene on the B genome using the tubulin-based polymorphism method and developed an SYBR Green PCR method for the specific detection of durum wheat [18]. To identify common wheat, Matsuoka et al. used the Starch Synthase II gene (wSSII-D), located on chromosome 7D [19, 25]. The barley system 1, based on the hordein gene and developed by Zeltner et al., showed good specificity not only for Hordeum vulgare, but also for Hordeum murinum [21]. In addition, several systems claimed to be able to distinguish bread wheat from spelt [14, 16, 22].
Compared to real-time PCR, digital PCR (dPCR) allows absolute quantification of copy numbers [26] and is thus used in GMO quantification [27], species detection and quantification [28] and detection of various pathogens in food [26, 29]. A variant of dPCR is droplet digital PCR (ddPCR), in which the reaction mixture is partitioned into about 20,000 smaller reactants (droplets) with an average volume of 0.89 nL [28]. The distribution of DNA in these partitions follows the Poisson distribution. This water-in-oil emulsion is amplified by an endpoint PCR in which each target molecule is amplified in its compartment separately. Hence, in the presence of target DNA in a droplet, amplification and an increase in fluorescence take place. By counting the droplets that show fluorescence and statistical conversion, the absolute copy number of the target DNA can be determined precisely and, in contrast to quantitative real-time PCR, independently from most parameters influencing the amplification efficiency [30]. This allows the determination of the amounts of grain species used. At this time, no method has been published for the quantitative determination of common wheat, durum wheat, rye and barley by ddPCR.
Materials and methods
Sample material
The Institute of Crop Plant Research and Genetics in Gatersleben (IPK Gatersleben, Germany) provided the reference material for the following cereal varieties: common wheat (13), durum wheat (12), spelt (11), barley (12), rye (12), triticale (1), einkorn (1), emmer (1), kamut (1) and buckwheat (1). Sample material for sunflower and pumpkin seeds, whole rye flour, common wheat and durum wheat semolina was obtained from local grocery stores. The other flours were provided by SchapfenMühle GmbH & Co. KG (Ulm, Germany). The complete overview of the sample material is presented in the Electronic Supplementary Material (Supp. Tables 1, 2).
For validation of quantitative analyses, flour blends were prepared from whole rye flour with whole wheat flour (mixture named RW) and rye flour type 997 (i.e. 997 mg ash per 100 g dry mass flour) with wheat flour type 550 (i.e. 550 mg ash per 100 g dry mass flour) (mixture named RoWe) in the mass proportions: 50/50, 60/40 and 90/10, with the first number being the rye proportion and the second number being the common wheat proportion. Semolina blends of durum semolina with common wheat semolina were further produced in mass proportions of 99/1, 98/2, 97/3, and 95/5, with the first number being the durum portion and the second number being the common wheat portion. The flour and semolina were dried to a mass consistency at 70 °C before being homogeneously mixed with a mortar and pestle. Semolina from the different species was finely powdered with mortar and pestle before further used. The semolina mixtures were finally made from ground material by mixing in a Grindomix GM200 (Retsch, Haan, Germany) with repeated scraping.
Primers and probes
Primers and probes (ZEN™ Double-Quenched Probes, reporter: 6-carboxy-fluorescein; quencher: Iowa Black® FQ) were supplied by Integrated DNA Technologies (Coralville, USA) and were designed with the PrimerExpress® software version 2.0 (Thermo Fisher Scientific, Waltham, USA). Table 1 shows the primers and probes for each cereal species-specific PCR system. The complete list of primers and probes used is shown in the Electronic Supplementary Material (Supp. Tables 3, 4).
Sample preparation and DNA extraction
Comminution of the reference material was performed by FastPrep FP120 (Qbiogene, Illkirch, France) using Metal Lysing Tubes, 2 mL with grinding cylinder (MP Biomedicals, Eschwege, Germany). Extraction was performed using a modified CTAB extraction [31]. From the minced material, 200 mg was suspended with 1000 µL of CTAB extraction buffer [c (CTAB) = 20 g/L, c (NaCl) = 1.4 mol/L, c (TRIS) = 0.1 mol/L, c (Na2EDTA) = 0.02 mol/L, pH 8.0], and 100 µL of α-amylase [from Bacillus licheniformis, 880 U/mL] (Sigma, Munich, Germany) was added. Samples were incubated for 45 min at 90 °C with shaking. Then, 10 µL RNase A [c = 30 mg/mL] (Macherey–Nagel, Düren, Germany) was added and incubated again for 45 min at 65 °C with shaking. Further workup was performed as described in [32]. In brief, 10 µL Proteinase K solution (c = 20 mg/L) was added, and the samples were incubated at 60 °C under agitation overnight and centrifuged for 10 min at 12,000×g afterwards. 0.7 mL of chloroform was added to the supernatant, shaken vigorously and centrifuged at 19,000×g for 10 min. The upper phase was transferred into a new vial. Two volumes of CTAB precipitation buffer [c (CTAB) = 5 g/L, c (NaCl) = 0.04 mol/L] were added and incubated 60 min at room temperature. The samples were centrifuged for 5 min at 12,000×g. The pellet was resuspended in 350 μL NaCl solution [c (NaCl) = 1.2 mol/L] and 350 µL of chloroform were added; the samples were vigorously shaken and centrifuged for 10 min at 19,000×g. 0.6 vol of isopropanol was added to the upper phase for nucleic acid precipitation. After 20-min incubation at room temperature, the samples were centrifuged for 10 min at 12,000×g. The resulting pellet was washed with 500 µL ethanol solution (c = 70%) and resolved in 100 µL 0.1 × TE buffer [c (TRIS) = 1 mmol/L, c (Na2EDTA) = 0.1 mmol/L, pH 8.0] and stored as stock solution. Determination of the concentration and purity of dsDNA from the obtained DNA extracts was performed by measuring the optical density at 260 and 280 nm using a ScanDrop photometer (Analytik Jena, Jena, Germany). DNA purity was considered sufficiently if OD260/OD280 was at least 1.8.
DNA-sequencing
Selected samples were sequenced to verify the target regions in the DNA sequence. Three reference samples (R 01/1, R 12/1, R 14/1) for rye were sequenced with primers Rmips01-fw and Rmips01-rv. To screen for a wheat- and spelt-specific system for the gene wSSII-D (AB201447.1), three spelt samples (SP 03/1, SP 13/1, SP 18/1) and three common wheat samples (TRI 03/1, TRI 15/1, TRI 20/2) were sequenced with primers Wss2d01-fw and Wss2d01-rv. Sequencing for the durum wheat-specific β-tubulin gene (MG852148.1) of Triticum durum was performed with primers Dtub05-fw and Dtub03-rv on the reference samples for durum wheat (DU 01/1, DU 08/1, DU 18/1), common wheat (TRI 15/1, TRI 17/1) and kamut (TRI 19,236). Ten reference samples of common wheat (TRI 01/1, TRI 02/1, TRI 03/1, TRI 04/1, TRI 05/1, TRI 16/1, TRI 17/1, TRI 18/2, TRI 19/2, TRI 20/2) and seven reference samples of spelt (SP 05/1, SP 08/1, SP 09/1, SP 13/1, SP 15/1, SP 17/1, SP 18/1) were sequenced with primers Sglia01-fw and Sglia03-rv for alignment with published gene sequences for the γ-gliadin gene (Triticum aestivum—AF144104.1; Triticum spelta—AF120267.1) to test whether or not this gene could distinguish spelt from common wheat, based on a 9-bp insertion in the γ-gliadin gene of common wheat [16].
Conventional PCRs were performed in a reaction mixture of 25 µL total volume consisting of 0.125 µL Hot Star Taq DNA Polymerase [c = 5 U/µL] (Qiagen, Hilden, Germany), 2.5 μL 10 × PCR buffer containing c (MgCl2) = 15 mmol/L (Qiagen, Hilden, Germany), 0.5 μL dNTP mix [c (dATP; dCTP; dGTP; dTTP) = 200 μmol/L each] (Roche Diagnostics, Mannheim, Germany), 500 nmol/L of each primer and 5 µL template DNA [c (DNA) = 100 ng/µL]. Reactions were performed in the GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Darmstadt, Germany) using an initial denaturation step at 95 °C for 15 min followed by 45 cycles of 30 s at 95 °C, 30 s annealing at 5 °C below the melting temperature calculated for the primers and 30 s at 72 °C. A final elongation step was performed for 7 min at 72 °C. Gel electrophoresis was performed in a 2% agarose gel in TAE buffer with 5 µL PCR product using a 50-bp fragment length marker (Gene Ruler™, Thermo Scientific, Waltham, USA). Bands were stained with 1% ethidium bromide solution for visualization.
The PCR products were purified using the QIAquick® PCR Purification Kit (Qiagen, Hilden, Germany). Sequencing of the purified PCR products was performed with the BigDye™ Terminator V 1.1 Cycle Sequencing Kit (Applied Biosystems, Darmstadt, Germany) using the same primers as those in conventional PCR. The reaction conditions of the sequence reaction were chosen analogously to [32]. The annealing temperature of the primers was 5 °C below the melting temperature; see Electronic Supplementary Material (Supp. Table 3). The products were analyzed in the ABI Prism 310 Genetic Analyzer sequencer (Applied Biosystems, Darmstadt, Germany) and evaluated using the Sequence Navigator software version 1.0.1 (Applied Biosystems, Darmstadt, Germany). With Basic Local Alignment Search Tool (BLAST), the sequences were compared with those in the GenBank® database [33].
Real-time PCR
For real-time PCR, a total volume of 25 μL per reaction tube was chosen with a volume of template of 5 µL [c (DNA) = 20 ng/µL], 12.5 µL TaqMan® 2 × Universal PCR Mix (Applied Biosystems, Darmstadt, Germany) and the corresponding volume of forward and reverse primer and probe [c = 10 µM]. A final concentration of 300 nM was chosen for the primers HV-F, HV-R, Rmips02-fw and Rmips02-rv, and a concentration of 600 nM was chosen for Dtub03-fw, Dtub03-rv, Wss2d01-fw and Wss2d01-rv in the PCR assay. For the HV-S probe, the final concentration was 150 nM, and for all other probes, 100 nM were applied.
PCR was performed in 96-well plates using a C1000™ Touch Thermal Cycler coupled with the CFX96™ Real-Time System (Bio-Rad, Munich, Germany) under the following conditions: 95 °C for 10 min; 45 cycles each of 95 °C for 15 s, 58 °C (system HV), 64 °C (system Wss2d01), 60 °C (systems Rmips02, Dtub03) for 1 min each; for the HV system, an elongation step at 72 °C for 10 s was added [21].
Optimization of real-time PCR
The Rmips02, Wss2d01 and Dtub03 systems were optimized with regard to the annealing temperature in temperature ranges of 58–70 °C. The HV system had already been optimized. The optimal primer concentration was determined by combining different primer concentrations (300–900 nM). Tests were performed with forward/reverse primer concentrations of 300 nM/300 nM, 300 nM/600 nM, 300 nM/900 nM, 600 nM/300 nM, 600 nM/600 nM, 600 nM/900 nM, 900 nM/300 nM, 900 nM/600 nM and 900 nM/900 nM in duplicates.
Droplet digital PCR
Reactions were performed in a total volume of 20 µL using a 2.5 µL template [c (DNA) = 10–20 ng/µL]. The master mix consisted of 10 µL ddPCR Supermix for Probes (Bio-Rad, Munich, Germany), 0.5 μmol/L of each primer, 0.2 μmol/L of TaqMan® probe and Aqua bidest. In a twin.tec PCR Plate 96, semi-skirted, blue (Eppendorf, Hamburg, Germany), 39.5 μL of master mix was introduced, 5.64 μL of template was added and the plate was centrifuged at 480 g for 3 min. The mixture was homogenized by repeated pipetting, and was centrifuged again for 3 min at 480 g. Next, two aliquots, each holding 20 µL, were pipetted into the wells of the DG8 Cartridges for the QX100/QX200 Droplet Generator (Bio-Rad, Munich, Germany). Then, 70 μL of Droplet Generation Oil for Probes (Bio-Rad, Munich, Germany) was added to the cartridges. After droplets were generated in the Droplet Generator QX200™ (Bio-Rad, Munich, Germany), the reaction mixtures were pipetted into 96-well reaction plates, and the plates were sealed with Pierceable Foil Heat Seal (Bio-Rad, Munich, Germany) at 180 °C in the PX1 PCR Plate Sealer (Bio-Rad, Munich, Germany). Reaction was performed in the GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Darmstadt, Germany) under the following conditions: initial denaturation step for 10 min at 95 °C followed by 80 cycles of 30 s at 95 °C and 60 s at 64 °C for the system Wss2d01 and 60 °C for the systems Rmips02, Dtub03 and HV. Droplet stabilization was performed at 98 °C for 10 min and subsequently cooled to 4 °C. Droplet reading and copy-number calculation were done using the Droplet Reader QX200 and the QuantaSoft software version 1.7.4 (Bio-Rad, Munich, Germany). Fluorescence was measured in the FAM channel. All thresholds were manually adjusted based on background fluorescence.
Optimization of ddPCR
The systems Rmips02, Dtub03 and Wss2d01 were directly transferred to the ddPCR with regard to the temperature program. For the HV system, an annealing temperature of 60 °C was used for the ddPCR. To optimize the concentrations of primers and probes, 150 nM, 200 nM and 250 nM for the probes and 500 nM, 600 nM and 750 nM for the primers were investigated in duplicates. Data were analyzed using R software version 3.6.3 [34] and an R-script according to Lievens et al. [35]. The evaluation took into account the extent of rain, resolution and the amount of fluorescence.
Validation
The basis for validation in real-time PCR was CEN/TS 17,239–1:2019–06 [36]. The methodology was transferred accordingly for ddPCR. The values from the determination of the copy number by ddPCR were used to set the copy number.
Specificity tests in real-time PCR
The copy number of DNA reference samples was calculated using OD260 according to [36]. To check inclusivity, 11 reference samples were diluted to a calculated copy number of about 40 cp/μL using salmon sperm DNA [c = 20 ng/μL] as background DNA for durum wheat and rye, 10 for spelt, and 12 for common wheat. The determination was performed in duplicates with the respective specific system.
DNA extracts [c = 500 cp/µL] from the following animal and plant species were used in the exclusivity testing: soybean (Glycine max), maize (Zea mays), rice (Oryza sativa), emmer (Triticum dicoccum), triticale (Triticale), oats (Avena sativa), durum wheat (Triticum durum), common wheat (Triticum aestivum), rye (Secale cereale), millet (Sorghum bicolor), flax (Linum usitatissimum), kamut (Triticum turgidum subsp. turanicum), sugar beet (Beta vulgaris), spelt (Triticum spelta), barley (Hordeum vulgare), buckwheat (Fagopyrum esculentum), sunflower (Helianthus annus), pumpkin (Cucurbita pepo), cattle (Bos taurus), pig (Sus scrofa), turkey (Meleagris gallopavo) and chicken (Gallus gallus). The ACT [37] (plant-specific DNA) and myneu [38] (animal-specific DNA) systems were used to confirm the presence of amplifiable DNA.
Determination of the limit of detection and efficiency
DNA extracts of the reference samples were diluted to a final amount of 20 cp/5 μL, 10 cp/5 μL, 5 cp/5 μL, 2 cp/5 μL, 1 cp/5 μL and 0.1 cp/5 μL, respectively, using salmon sperm DNA [c = 20 ng/μL] as background DNA. Twelve PCR replicates were measured in real-time PCR per dilution level. To determine the efficiency of the systems, the concentrations 2500, 500, 100 and 50 copies per preparation were each measured in three PCR replicates. LOD95% was determined using the POD tool [39] in the program R version 3.6.3. By plotting POD (probability of detection) against copy number in logarithmic order, the value for LOD95% can be determined.
To determine the LOD95% in ddPCR, DNA extracts of the reference samples were diluted to a final amount of 20 cp/2.5 µL, 10 cp/2.5 μL, 5 cp/2.5 μL, 2 cp/2.5 μL, 1 cp/2.5 μL and 0.1 cp/2.5 μL, respectively, using DNA from salmon sperm [c = 20 ng/μL] as background DNA. Twelve PCR replicates were measured per dilution level. Following the method of Dobnik et al. [27], the measurements in a ddPCR assay were considered as an overall positive result only when the number of positive droplets was at least three.
Robustness of the systems in ddPCR
The Rmips02, Dtub03, HV and Wss2d01 systems in four replicates per factor combination were examined for robustness. Samples were diluted to 30–40 cp/μL with DNA from salmon sperm [c = 20 ng/μL]. Master mixes for the combinations were prepared and are shown in Table 2. Annealing temperatures from Table 1 were changed up and down by 1 °C. Results were analyzed by a one-factorial analysis of variance and Scheffé test [40] at p = 0.05 each using the IBM® SPSS® Statistics Subscription version 1.0.0.1327 software.
Results
Cereal-specific systems
Rmips02 (rye system)
Sequencing with primers Rmips01-fw and Rmips01-rv on three rye samples reveals two homologous sequences of R 01/1 and R 12/1 with a length of 134 bp. The sequence of R 14/1 shows a deletion at base 110 (see Fig. 1). In preliminary tests, the Rmips01 system showed poor performance with regard to fluorescence maximum in real-time PCR with different rye samples (data not shown). As shown in Electronic Supplementary Material (Supp. Table 4), the calculated melting temperatures of forward and reverse primer differ by 4 K, and between forward primer and probe, the difference is about 14 K. Due to this fact the performance of PCR could be affected. Hence, the Rmips01 system needed to be optimized. Based on the sequence data, the system Rmips02 was developed.

Sequences of samples R 01/1, R 12/1 and R 14/1. Yellow: Rmips01-fw, Rmips01-rv. Turquoise: Rmips02-fw, Rmips02-rv. Magenta: Rmips02-p. Green: Rmips01-p
Dtub03 (durum wheat-specific system)
Sequence analysis of the β-tubulin gene (MG852148.1) from durum wheat showed an insertion of 9 bp, as well as an insertion of 21 bp, when compared to the sequence of common wheat; see alignment in Electronic Supplementary Material (Supp. Fig. 1). These were used for the primer and probe design with specificity for durum wheat. When tested in silico using BLAST [33], the primer–probe system Dtub03 was found to be the most suitable. Primer Dtub03-rv met the criteria for specificity, because there was no match with any other species of genus Triticeae. Sequencing with primers Dtub05-fw and Dtub03-rv resulted in complete sequence alignment of all samples (DU 01/1, DU 08/1, DU 18/1, TRI 15/1, TRI 17/1 and TRI 19,236). The alignment of the sequences is shown in Fig. 2.

Sequencing of β-tubulin gene with the primers Dtub05-fw and Dtub03-rv. Yellow: Dtub03-fw, Dtub03-rv. Green: probe Dtub03-p. Alignment of samples of durum wheat (DU 01/1, DU 08/1, DU 18/1), common wheat (TRI 15/1, TRI 18/1), kamut (TRI 19,236) and Triticum aestivum TUB2_3AL gene (MG852122.1), Triticum aestivum TUB2_3B gene (MG852123.1), Triticum aestivum TUB2_3DL gene (MG852124.1) and Hordeum vulgare beta-tubulin gene (GU017484.1)
Wss2d01 (common wheat- and spelt-specific system)
The Wss2d01 [19] system is also specific for the D genome of spelt, which was confirmed by sequencing (see Fig. 3). Spelt and common wheat could be specifically detected alongside the other cereals using this optimized system.

Sequencing results from wSSII-D gene with primers Wss2d01fw and Wss2d01rv (yellow). Probe Wss2d01p in green. Alignment of the sequenced samples for common wheat (TRI 03/1, TRI 15/1, TRI 20/2), spelt (SP 03/1, SP 13/1, SP 18/1), Triticum aestivum wSSII-B gene (AB201446.1) and Triticum aestivum wSSII-A gene (AB201445.1)
γ-Gliadin gene (GAG56D) to differentiate between common wheat and spelt
The assumed spelt-specific system Sglia01, developed in this work, targets a 9-bp deletion in spelt varieties compared to common wheat varieties. Preliminary tests with the Sglia01 system in real-time PCR showed that the common wheat varieties, TRI 03/1, TRI 04/1, TRI 05/1, TRI 17/1, and TRI 18/2 showed false-positive results for this in the measurement. Furthermore, the spelt variety, SP 17/1, was negative with Sglia01. Results from sequencing seven spelt varieties and ten common wheat varieties confirmed that the aforementioned common wheat varieties do not have a 9-bp insertion in the γ-gliadin gene but that the spelt variety SP 17/1 does indeed have this insertion in this gene, while TRI 03/1, TRI 04/1, TRI 05/1, TRI 17/1, and TRI 18/2 lack this insertion (see Fig. 4). The genetic variability of the 9-bp insertion between spelt and common wheat makes this target unsuitable for differentiation between the two species.

Sequencing results with primers Sglia01-fw and Sglia03-rv (binding sites marked in yellow) and alignment of the gene segment in the γ-gliadin gene of the samples for common wheat (TRI 01/1 to TRI 20/2) and spelt (SP 05/1 to SP 18/1), Triticum aestivum gamma-gliadin (GAG56D) gene (AF144104.1) and Triticum spelta gamma-gliadin (GAG56D) gene (AF120267.1)
Optimization of real-time PCR
For the Rmips02 and Dtub03 system, 60 °C was selected as an optimum annealing and elongation temperature, because maximum fluorescence is reached at almost 60 °C and it is more feasible to work at the same temperature with both systems. Dtub03 amplified kamut and durum wheat with equal specificity. For the system Wss2d01, 64 °C was chosen to avoid nonspecific reactions with durum, kamut, emmer and triticale.
Optimal primer concentrations were 600 nM for Rmips02 and 300 nM for Wss2d01 and Dtub03. All real-time PCR optimization data are presented in the Electronic Supplementary Material (Supp. Table 5–8).
Optimization of ddPCR
For ddPCR, resolution and rain are relevant parameters. In this context, resolution describes the separation between positive and negative droplets. Resolution depends very much on the fluorescence signal of positive droplets and the level of background fluorescence. Rain is the number of droplets between the fluorescence level of positive and the background level. For the systems Rmips02, HV, Dtub03 and Wss2d01, the resolution was above the suggested minimum value of 2.5 [35]; the rain was always below 1%. With higher primer concentrations, the intensities of the fluorescence signals also increased, while the probe concentration did not affect the signal intensity as much. The optimal combination for the Rmips02 system was 500 nM for the primers and 200 nM for the probe (resolution: 9.03; rain: 0.16%; see Fig. 5). For the system Wss2d01, an optimal combination of 600 nM for the primers and 150 nM for the probe was chosen, and for the HV and Dtub03, 600 nM for the primers and 200 nM for the probe were chosen as optimum.

Optimization of the primer and probe concentration in ddPCR for Rmips02. Probe-/primer concentration (in nM): A1&A2 (150/500), B1&B2 (150/600), C1&C2 (150/750), D1&D2 (200/500), E1&E2 (200/600), F1&F2 (200/750), G1&G2 (250/500), H1&H2 (250/600)
Figure 6 presents the results of ddPCR optimization of reference samples at three different dilutions. The droplets were observed in only one fluorescence region in the samples. In the samples with the lowest dilution, the rain appears stronger, because there are significantly more positive droplets, increasing the probability of droplets with intermediate fluorescence. Nevertheless, the relative amount of rain in relation to the number of droplets is not increased. All optimization data can be found in the Supplementary Material (Supp. Tables 9, 10).

Copy-number determination by results of optimized ddPCR systems. a) HV system with samples H 19/1 1:30 (B01, B02), H 19/1 1:150 (D01, D02) and H 19/1 1:750 (F01, F02). b) Dtub03 system with samples DU 06/1 1:50 (B01, B02), DU 06/1 1:250 (D01, D02) and DU 06/1 1:1250 (E01, E02). c) Rmips02 system with samples R 11/1 1:45 (C03, C04), R 11/1 1:225 (E03, E04) and R 11/1 1:1125 (F03, F04). d) Wss2d01 system with samples TRI 19/2 1:20 (A01, A02), TRI 19/2 1:100 (B01, B02), TRI 19/2 1:500 (C01, C02), SP 18/1 1:20 (D01, D02), SP 18/1 1:100 (E01, E02) and SP 18/1 1:500 (F01, F02)
Validation
Specificity tests in real-time PCR
The results of the inclusivity tests showed positive results for the tested systems Rmips02, Dtub03 and Wss2d01, in the reference samples with Cq values ranging from 29.16 to 33.06. For complete results, see the Electronic Supplementary Material (Supp. Tables 11, 12, 13).
The Wss2d01 system gave positive results in the exclusivity tests for spelt and common wheat samples. The Rmips02 system amplified only the rye and triticale samples. Dtub03 also amplified two of 13 common wheat samples (TRI 15/1, TRI 17/1) in addition to durum and kamut samples. Further examination of Schwäbische-Dickkopf Landweizen (TRI 15/1) and LIAO CHUEN NO.5 (TRI 17/1) revealed sequence homology of these common wheat cultivars in the β-tubulin gene. The results of the exclusivity tests are presented in the Electronic Supplementary Material (Supp. Table 14).
Determination of the limit of detection
The LOD95% in real-time PCR (with 95% confidence interval in brackets) in copies per reaction is 4.07 (2.59; 6.35) for Dtub03; 3.17 (1.99; 5.02) for HV; 2.43 (1.49; 3.90) for Rmips02; and 3.08 (1.91; 4.88) for Wss2d01. These data follow the Poisson distribution. Considering that, in case every target sequence is successfully amplified, i.e. copied without any error, the ideal LOD95% is 2.996 copies per reaction, the values obtained are thus close to the best detection limit per reaction achievable [41].
The PCR efficiency of the individual systems could be determined through the calibration series included in real-time PCR. The efficiency can be considered "optimal" if it is 100% ± 10%, and the coefficient of determination (R2) is at least 0.98 [36, 42]. All these criteria could be fulfilled for the systems Dtub03 (E = 100.0%), Rmips02 (E = 93.1%), HV (E = 92.9%) and Wss2d01 (E = 105.8%).
The LOD95% determined in ddPCR (with 95% confidence interval in brackets) in copies per reaction are 9.89 (6.55; 14.84) for Dtub03; 9.48 (6.28; 14.24) for HV; 9.07 (6.00; 13.65) for Rmips02; and 13.26 (8.80; 19.74) for Wss2d01. According to Dobnik et al. [27], test results by ddPCR are considered positive when three droplets give a positive fluorescence signal. Using this minimum value for a positive reaction, a systematic error can be prevented and accidental false-positive results can be excluded; however, this strict requirement leads to higher value for LOD95% (see Fig. 7). The best achievable values for LOD95% are the number of target molecules that are required to have at least three copies per reaction with a probability of 0.05. This number depends on the efficiency of ddPCR assay and on the Poisson statistics as follows:

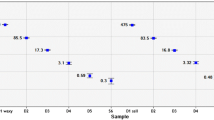
Comparison of LOD95% for Rmips02. a) POD diagram for Rmips02 in real-time PCR. b) POD diagram for Rmips02 in ddPCR. LOD95% shown with 95% confidence interval (CI). (X = nominal DNA concentration; S = number of positive replicates; N = number of tested replicates; green line defines the absolute DNA copy number when POD is 0.95)
The probabilities of positive results in digital PCR results follow the Poisson distribution characterized by the parameter λ.
The probability p for at least three positive droplets [27], i.e., at least three target molecules per reaction, shall be 0.95.
The parameter λ represents in this way the number of target copies per reaction necessary to have at least three copies with the given probability of 0.95.
This means that the probabilities for positive results derived from nominal 0, 1, or 2 copies are 0.05 or less. The reversion of this interrelationship is represented by the following equations.
The solution is found by iteration resulting in λ = 6.2958. This represents the best LOD95% achievable. As a result, the values obtained for LOD95% in ddPCR were higher than those in real-time PCR, where one copy of target molecules shall end up in the reaction with a probability of 0.95. Further data and the figures for the determination of the detection limit are presented in the Electronic Supplementary Material (Supp. Tables 15, 16 and Supp. Figs. 2, 3).
Robustness in ddPCR
Using a factorial analysis of variance (p = 0.05), followed by a Scheffé test (p = 0.05), a significant (p < 0.05) difference was found between combinations 2 and 3 in the Rmips02 system. Combinations 1 and 2 tended to have higher copy numbers (not significant). In combination 2, the highest copy number was determined in all systems, and in combination 3 (except Wss2d01 in combination 4), the lowest copy number was determined. Dtub03 had the smallest variation among combinations. However, positive results were obtained in all combinations and all amplicons. Therefore, all systems were sufficiently robust with respect to the factor combinations tested (primer and probe concentrations of -30%, deviating master mix volumes of 94.3% and 105.7%, temperature differences of ± 1 °C). The statistical calculations are provided in the Electronic Supplementary Material (Supp. Table 17–28).
Analysis of flour and semolina mixtures
From the values calculated for the cereal-specific systems for rye and common wheat, the proportions of flour and semolina of the respective cereal species in the flour and semolina mixtures were calculated according to Eq. 7. For the cereal type G1 or G2, k stands for the measured copy number, and C represents the C-value. The equation establishes a correlation between haploid genome weight and copy number of each cereal species in the mixture.
Equation 7 Calculation of the proportion in % (w/w) of grain type G1 in a binary mix of grain 1 (G1) and grain 2 (G2). The calculation is based on the number of copies (kG1 & kG2) determined for the respective grain type and the corresponding haploid genome weight (CG1 & CG2).
Flour mixtures
For the blends of whole-grain flours (denoted RW x/y, where x and y represent the proportions of rye and wheat), Eq. 7 approximated the ratio of the weighed mass fractions of the two flours. For the 50/50 blend, the mass fraction was 52.8% for rye and 47.2% for common wheat. In the 60/40 and 90/10 blends, the determined flour proportions were 62.6% and 88.8% for rye and 37.4% and 11.2% for common wheat, respectively, which approximately corresponds to the mass proportion of the original initial weight (see Fig. 8a).

a Analysis of the mass fractions in the flour mixes. Blue triangles: proportion of wholemeal rye flour (y1 = 0.8921x1 + 0.086; R2 = 0.9987) – Blue circles: proportion of whole wheat flour (y2 = 0.8921x2 + 0.022; R2 = 0.9987). Red crosses: proportion of rye flour type 997 (y3 = 0.702x3 + 0.3085; R2 = 0.9902) – Red diamonds: proportion of common wheat flour type 550 (y4 = 0.702x4-0.0105; R2 = 0.9902). b Trend analysis of the mass fraction of semolina. Green: proportion of durum wheat semolina (y5 = 0.4776x5 + 0.5183; R2 = 0.9999). Violet: proportion of common wheat semolina (y6 = 0.4776x6 + 0.0041; R2 = 0.9999). Trend values (marked with ×) created with the "TREND" function in Microsoft EXCEL 2019
In contrast to the proportions of flour of the cereal types in the mixture of wholemeal flours, those in the mixtures of flours with different degrees of grinding (RoWe x/y) deviated from the weighed-in proportions. The determined common wheat percentage was too low in absolute terms by 15.1% (in RoWe 50/50), 14.0% (in RoWe 60/40) and 3.8% (in RoWe 90/10). The values determined by ddPCR led to a higher rye content in these samples than was actually weighed in (see Fig. 8a). The copy numbers, shown in Table 3, were determined for the respective cereal species by means of ddPCR. For further data, see Electronic Supplementary Material (Supp. Table 29).
Mixtures of common wheat and durum wheat semolina
With determined proportions of 99.05% durum and 0.95% common wheat semolina, the ratio of this 99/1 mixture was very close to the weighed-in proportion of 99% and 1%, respectively. In the mixture with 5% common wheat, only about half of the weighed-in proportion (2.84%) was determined by this ddPCR method (see Table 4). From the trend analysis, the proportion of durum wheat determined by ddPCR is significantly higher than the actual proportion at each point (see Fig. 8b). The data on the mixtures are given in the Electronic Supplementary Material (Supp. Table 30).
Discussion
Development and optimization of cereal-specific systems
Rye (Secale cereale)
Pegels et al. [17] developed the rye system, which was based on the single-copy MIPS gene and showed good specificity on reference samples. For further optimization, the gene segment was sequenced on three reference samples and was used to develop the Rmips02 system. The Rmips02 system gave positive results in specificity tests for the reference sample of triticale, since triticale is a hybrid of common wheat and rye or durum wheat and rye by allopolyploidization [8].
Common wheat (Triticum aestivum)
Matsuoka et al. [19] developed a system, which targets the Starch synthase II gene (wSSII-D) on chromosome 7D [19, 25] of hexaploid common wheat. Genome D is the target genome, which does not occur in the tetraploid durum wheat genome. Thus, the system Wss2d01 is suitable for distinguishing common wheat from durum wheat as well as the other cereal species tested. Spelt (Triticum spelta), as a closely related species of common wheat, showed a homologous gene sequence in the tested samples. The system Wss2d01 can therefore detect spelt as well as common wheat.
Durum wheat (Triticum durum)
Sequencing demonstrated homology between the sequences of one kamut sample and two common wheat varieties (Schwäbischen Dickkopf-Landweizen and LIAO CHUEN NO.5) in the Dtub03 system. Likewise, homology was expected between kamut and durum wheat given their close relationship [6]. Schwäbischer Dickkopf-Landweizen is a rare regional common wheat variety with a cultivated area of only 1.2 hectares (2012) in the region of Swabia [43]. The common wheat cultivar LIAO CHUEN NO.5 (LIAOCHUN 5) is a Chinese breeding variety [44] and a cross of LIAOCHUN-1 and FRONTANA with a cultivated area of 67,000 ha from 1966 to the late 1970s [45]. To our knowledge, both varieties do not have any economic relevance.
Barley (Hordeum vulgare)
Zeltner et al. [21]. developed the barley system HV for real-time PCR, which provides specific results for barley (Hordeum vulgare) [21]. When transferred to ddPCR, the annealing and elongation temperature was optimized to 60 °C in regards to a potential multiplex ddPCR method.
Differentiation between common wheat and spelt
In previous publications, a 9-bp insertion in the γ-gliadin gene (GAG56D) of common wheat was used to distinguish common wheat from spelt [16, 22]. However, by sequencing the gene segment around the region of the insertion, no insertion was detected in five of ten common wheat reference samples. One of seven spelt reference samples (SP 17/1) exhibited this insertion in the gene sequence, which was considered to be specific for common wheat. This demonstrated that this marker was unsuitable for distinguishing between the two cereal species as no reliable results could be obtained.
As 2nd putative molecular target, a 160-bp insert in the Wx-A1 (HQ625382) gene of spelt was tested by several primer pairs, PCR results however gave false-positive results in common wheat when assessed experimentally, see Electronic Supplementary Material (Supp. Table 4).
Spelt (Triticum spelta or Triticum aestivum subsp. spelta) and common wheat (Triticum aestivum or Triticum aestivum subsp. aestivum) are very closely related in evolutionary biology [7]. Analyses of the growing number of published sequences of Triticum spelta and Triticum aestivum from GenBank® did not provide a molecular basis for differentiating the two species by a PCR system targeting a single genetic sequence. Putative genetic species-specific genetic elements between both species differ remarkably between several breeding lines. Despite several approaches, a distinct molecular system could not be applied to distinguish spelt and common wheat reliably, see Electronic Supplementary Material (Supp. Table 4). In conclusion, it is not possible to distinguish both species reliably by one PCR system for the time being. Thus, additional research is needed. One possible approach to facilitate this could be a multifactorial analysis of single-nucleotide polymorphisms (SNP`s).
Evaluation of the flour mixtures
Equation 7, shown in the results, was used to calculate the proportions in a cereal type. This is achieved by assuming there is a 1:1 mixture of common wheat/rye dsDNA with a dsDNA concentration of 100 ng/μL each. The calculated copy numbers are about 11,364 cp/µL for the rye dsDNA (C-value 8.8 [46]) and about 5780 cp/µL for the common wheat dsDNA (C-value 17.3 [46]). Due to the lower haploid genome weight of rye dsDNA, there is a factor of 1.97 (ratio 17.3:8.8) more copies in the same reaction mixture per μL at the same dsDNA concentration. With this relationship, the flour fraction in the mixtures determined by ddPCR can be calculated using the aforementioned equation. There is a correlation between flour type and DNA content. The flour type is directly related to the degree of milling (proportion of flour/proportion of separated bran). Rye flour type 997 has a degree of milling of about 74–79% and common wheat flour type 550 about 65–72% [47]. A higher degree of milling of a flour in mixtures results in a higher DNA copy number in the extract and thus a higher determined proportion of a cereal species. Accordingly, the flour fractions determined by ddPCR in a flour mixture are only identical to the flour fractions actually weighed if the degrees of grinding of the flours are almost the same. Areas with higher cell density will have a greater amount of DNA similar to the differences between embryo and endosperm in Zea mays [48]. Knowledge of the types of flour is therefore important in making accurate assessments of flour blends in foods. Due to the higher price of rye flour, adulteration by stretching with common wheat flour is more common than doing so with rye.
Evaluation of semolina mixtures
The background for the analysis of these mixtures is Regulation (EU) No. 2016/1238, which stipulates a maximum foreign grain content in durum wheat of 3%. By producing semolina mixtures, a common wheat content of 1%, 2%, 3% and 5% was simulated in the remaining proportion of durum wheat semolina. Thus, the proportion from common wheat semolina is significantly underestimated, and the proportion from durum wheat is overestimated. Since no degree of grinding is usually specified for semolina, a correlation is not applicable.
Applications of ddPCR in analysis of the cereal products
Compared to real-time PCR, results of ddPCR are not subject to the inherent errors of copy-number determination. The assessment of validation parameters for real-time PCR applications requires an exact quantification of the DNA concentration of reference material by photometrical or fluorimetric DNA quantification methods. The same implies to quantitative real-time PCR applications when using standard curves. Digital PCR allows an absolute quantification resulting in a higher degree of reliability. In contrast to real-time PCR, quantification by ddPCR omitted the implementation of a standard curve for the evaluation of the results. Despite the higher complexibility of ddPCR, finally, this saves resources, especially when applied to multiple targets in a single test sample. The differentiation between detection and quantification by several reactions is dispensable.
Droplet digital PCR allows reliable detection and absolute quantification of common wheat, durum wheat, rye and barley. The results of quantification depend on the DNA content of each of the ingredients depend on the DNA content after milling and further processing and are therefore subject to error. This needs to be taking into account by interpreting quantitative data. Within this limitation, quantitative results give indications of adulteration. These analytical results may need to be verified by other methods for legal consequences, this might not necessarily be analytical methods. Our results of flour and semolina mixtures, made of commonly available ingredients, indicate an underestimation of the “less valuable” ingredient common wheat. Based on data presented here, there is no risk of unfair legal consequences. We consider these results of one of several elements to discover less careful production or even fraudulent activities in the food chain.
Conclusion
A molecular method for the differentiation of the cereal species spelt (Triticum spelta) and common wheat (Triticum aestivum) could not be achieved within the scope of this work. The developed systems Wss2d01, Rmips02, Dtub03 and HV were developed for ddPCR and adapted with respect to temperature program and oligo concentrations. In addition, the methods were validated with respect to specificity (inclusivity and exclusivity), detection limit and robustness. Dtub03 also yielded positive results for the common wheat cultivars, Schwäbische-Dickkopf Landweizen and LIAOCHUN 5. Schwäbische-Dickkopf Landweizen is only cultivated in the region of Swabia and therefore has only little economic relevance. LIAO-CHUN 5, as a Chinese variety from the 1970s, is also likely to have little economic relevance in the present.
In the flour and semolina mixtures analyzed, the corresponding cereal species (flour: rye and common wheat; semolina: durum and common wheat) were successfully detected by ddPCR. Application of the barley-specific system in such mixtures is also possible. Quantification of mixtures showed a correlation between the copy number determined by ddPCR and the haploid genome weight of the cereal species was found. Through this correlation, the proportion of cereal species mixtures can be directly calculated from the determined copy number. However, correct assessment of flour mixtures must always take into account the degree of grinding, which has a decisive influence on DNA copy numbers. For further research, more approaches for molecular differentiation of the closely related species spelt and common wheat are needed.
References
Landwirtschaft B (2019) Statistisches Jahrbuch über Ernährung. Landwirtschaft und Forsten der Bundesrepublik Deutschland, Bonn
Committee on the Environment Public Health and Food Safety (2013) Report on the food crisis, fraud in the food chain and the control thereof (2013/2091(INI)). Straßburg
Morin J-F, Lees M (2018) FoodIntegrity Handbook - A guide to food authenticity issues and analytical solutions. Eurofins Analytics France, Nantes
Feldman M, Levy AA (2015) Origin and evolution of wheat and related triticeae species. Alien Introgression Wheat Cytogenet Mol Biol Genomics. https://doi.org/10.1007/978-3-319-23494-6-2
Food and Agriculture Organization of the United Nations (2020) FAOSTAT statistical database. Rome
Khlestkina EK, Röder MS, Grausgruber H, Börner A (2006) A DNA fingerprinting-based taxonomic allocation of Kamut wheat. Plant Genet Resour 4:172–180. https://doi.org/10.1079/pgr2006120
Matsuoka Y (2011) Evolution of polyploid triticum wheats under cultivation: The role of domestication, natural hybridization and allopolyploid speciation in their diversification. Plant Cell Physiol 52:750–764. https://doi.org/10.1093/pcp/pcr018
Wrigley CW, Bushuk W (2010) Rye and triticale: Characteristics and quality requirements. Woodhead Publishing Limited
Koenig A, Konitzer K, Wieser H, Koehler P (2015) Classification of spelt cultivars based on differences in storage protein compositions from wheat. Food Chem 168:176–182. https://doi.org/10.1016/j.foodchem.2014.07.040
Ruibal-Mendieta NL, Dekeyser A, Delacroix DL et al (2004) The oleate/palmitate ratio allows the distinction between wholemeals of spelt (Triticum spelta L.) and winter wheat (T. aestivum L.). J Cereal Sci 39:413–415. https://doi.org/10.1016/j.jcs.2004.02.003
Hong E, Lee SY, Jeong JY et al (2017) Modern analytical methods for the detection of food fraud and adulteration by food category. J Sci Food Agric 97:3877–3896. https://doi.org/10.1002/jsfa.8364
Alves TO, D’Almeida CTS, Scherf KA, Ferreira MSL (2019) Modern approaches in the identification and quantification of immunogenic peptides in cereals by LC-MS/MS. Front Plant Sci 10:1–13. https://doi.org/10.3389/fpls.2019.01470
Bönick J, Huschek G, Rawel HM (2017) Determination of wheat, rye and spelt authenticity in bread by targeted peptide biomarkers. J Food Compos Anal 58:82–91. https://doi.org/10.1016/j.jfca.2017.01.019
Curzon AY, Chandrasekhar K, Nashef YK et al (2019) Distinguishing between Bread Wheat and Spelt Grains Using Molecular Markers and Spectroscopy. J Agric Food Chem 67:3837–3841. https://doi.org/10.1021/acs.jafc.9b00131
Cocchi M, Durante C, Foca G et al (2006) Durum wheat adulteration detection by NIR spectroscopy multivariate calibration. Talanta 68:1505–1511. https://doi.org/10.1016/j.talanta.2005.08.005
Mayer F, Haase I, Graubner A et al (2012) Use of polymorphisms in the γ-gliadin gene of spelt and wheat as a tool for authenticity control. J Agric Food Chem 60:1350–1357. https://doi.org/10.1021/jf203945d
Pegels N, González I, García T, Martín R (2015) Authenticity testing of wheat, barley, rye and oats in food and feed market samples by real-time PCR assays. LWT - Food Sci Technol 60:867–875. https://doi.org/10.1016/j.lwt.2014.10.049
Casazza AP, Morcia C, Ponzoni E et al (2012) A reliable assay for the detection of soft wheat adulteration in Italian pasta is based on the use of new DNA molecular markers capable of discriminating between Triticum aestivum and Triticum durum. J Cereal Sci 56:733–740. https://doi.org/10.1016/j.jcs.2012.08.015
Matsuoka Y, Arami SI, Sato M et al (2012) Development of methods to distinguish between durum/common wheat and common wheat in blended flour using PCR. J Food Hyg Soc Japan 53:195–202. https://doi.org/10.3358/shokueishi.53.195
Carloni E, Amagliani G, Omiccioli E et al (2017) Validation and application of a quantitative real-time PCR assay to detect common wheat adulteration of durum wheat for pasta production. Food Chem 224:86–91. https://doi.org/10.1016/j.foodchem.2016.12.053
Zeltner D, Glomb MA, Maede D (2009) Real-time PCR systems for the detection of the gluten-containing cereals wheat, spelt, kamut, rye, barley and oat. Eur Food Res Technol 228:321–330. https://doi.org/10.1007/s00217-008-0937-4
Von Büren M, Stadler M, Lüthy J (2001) Detection of wheat adulteration of spelt flour and products by PCR. Eur Food Res Technol 212:234–239. https://doi.org/10.1007/s002170000230
García-García A, Madrid R, García T et al (2018) Use of multiplex ligation-dependent probe amplification (MLPA) for screening of wheat, barley, rye and oats in foods. Food Control 84:268–277. https://doi.org/10.1016/j.foodcont.2017.07.037
Busch U (2010) Molekularbiologische Methoden in der Lebensmittelanalytik. Springer, Berlin Heidelberg
Shimbata T, Nakamura T, Vrinten P et al (2005) Mutations in wheat starch synthase II genes and PCR-based selection of a SGP-1 null line. Theor Appl Genet 111:1072–1079. https://doi.org/10.1007/s00122-005-0032-1
Cremonesi P, Cortimiglia C, Picozzi C et al (2016) Development of a droplet digital polymerase chain reaction for rapid and simultaneous identification of common foodborne pathogens in soft cheese. Front Microbiol. https://doi.org/10.3389/fmicb.2016.01725
Dobnik D, Štebih D, Blejec A et al (2016) Multiplex quantification of four DNA targets in one reaction with Bio-Rad droplet digital PCR system for GMO detection. Sci Rep 6:1–9. https://doi.org/10.1038/srep35451
Pinheiro LB, Coleman VA, Hindson CM et al (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 84:1003–1011. https://doi.org/10.1021/ac202578x
Mykytczuk O, Harlow J, Bidawid S et al (2017) Prevalence and Molecular Characterization of the Hepatitis E Virus in Retail Pork Products Marketed in Canada. Food Environ Virol 9:208–218. https://doi.org/10.1007/s12560-017-9281-9
Gürtler P, Gerdes L (2014) Digitale Polymerasekettenreaktion (dPCR). BIOspektrum 20:632–635. https://doi.org/10.1007/s12268-014-0498-y
International Standards Organization (ISO) (2013) Foodstuffs - Methods of analysis for the detection of genetically modified organisms and derived products - Nucleic acid extraction - Amendment 1 (ISO 21571:2005/Amd 1:2013) EN ISO 21571:2013–08
Tetzlaff C, Mäde D (2017) Development of a real-time PCR system for the detection of the potential allergen fish in food. Eur Food Res Technol 243:849–857. https://doi.org/10.1007/s00217-016-2799-5
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
R Core Team (2020) R: a language and environment for statistical computing. R Core Team, Vienna
Lievens A, Jacchia S, Kagkli D et al (2016) Measuring digital PCR quality: performance parameters and their optimization. PLoS ONE 11:e0153317. https://doi.org/10.1371/journal.pone.0153317
Comité Européen de Normalisation (CEN) (2019) Foodstuffs—General guidelines for the validation of qualitative real-time PCR methods - Part 1: Single-laboratory validation; German version CEN/TS 17329–1:2019–06. Brussels
Laube I, Hird H, Brodmann P et al (2010) Development of primer and probe sets for the detection of plant species in honey. Food Chem 118:979–986. https://doi.org/10.1016/j.foodchem.2008.09.063
Laube I, Zagon J, Spiegelberg A et al (2007) Development and design of a “ready-to-use” reaction plate for a PCR-based simultaneous detection of animal species used in foods. Int J Food Sci Technol 42:9–17. https://doi.org/10.1111/j.1365-2621.2006.01154.x
Boenn M (2019) POD: Probability of Detection for Qualitative PCR Methods
Scheffé H (1953) A method for judging all contrasts in the analysis of variance. Biometrika 40:87–104. https://doi.org/10.2307/2333100
Uhlig S, Frost K, Colson B et al (2015) Validation of qualitative PCR methods on the basis of mathematical–statistical modelling of the probability of detection. Accredit Qual Assur 20:75–83. https://doi.org/10.1007/s00769-015-1112-9
Rutledge RG, Côté C (2003) Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res 31:93e–993. https://doi.org/10.1093/nar/gng093
Slow Food Deutschland e. V. Schwäbischer Dickkopf-Landweizen. https://www.slowfood.de/was-wir-tun/arche_des_geschmacks/die_arche_passagiere/schwaebischer_dickkopf_landweizen. Accessed 4 Apr 2020
Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung Genbankinformationssystem des IPK Gatersleben (Akzessionsnummer: TRI 29902). https://gbis.ipk-gatersleben.de/gbis2i/faces/index.jsf. Accessed 3 Apr 2020
He ZH, Rajaram S, Xin ZY, Huang GZ (2001) A History of Wheat Breeding in China. CIMMYT, Mexico, D.F.
Leitch IJ, Johnston E, Pellicer J, Hidalgo O BM (2019) Plant DNA C-values Database. https://cvalues.science.kew.org/. Accessed 5 Dec 2019
Rimbach G, Nagursky J, Erbersdobler HF (2015) Lebensmittel-Warenkunde für Einsteiger, 2. Auflage. Springer Spektrum, Berlin Heidelberg
Papazova N, Malef A, Degrieck I, et al (2005) DNA extractability from the maize embryo and endosperm - relevance to GMO assessment in seed samples. Seed Sci Technol 33:533–542. https://doi.org/10.15258/sst.2005.33.3.01
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Compliance with ethics requirement
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schulze, C., Geuthner, AC. & Mäde, D. Development and validation of a method for quantification of common wheat, durum wheat, rye and barley by droplet digital PCR. Eur Food Res Technol 247, 2267–2283 (2021). https://doi.org/10.1007/s00217-021-03786-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03786-y