
Abstract
There is a growing awareness by consumers and food safety authorities regarding the possible presence of parasites or parasite-related potentially hazardous substances in seafood. Anisakis simplex is among the most frequently occurring parasites in wild-caught marine fish. Except for various visual inspection techniques and PCR-based methods for the detection of more or less intact worms or parasite DNA, respectively, there are at present no validated methods for the quantification of A. simplex proteins in processed fish products. This work describes the development and validation of a sandwich enzyme-linked immunosorbent assay (ELISA) for the quantification and analysis of proteins from A. simplex in seafood products. The ELISA is based on a polyclonal rabbit anti-A. simplex antibody for capture and a biotinylated conjugate of the same antibody for detection. The ELISA is specific for A. simplex and does not cross-react with other species. Recoveries ranged from 72–101% in typical food matrixes, while intra- and inter-assay precisions were <11 and <25%, respectively. With a limit of detection of 1.1 μg A. simplex protein/g of sample, the sensitivity of the A. simplex sandwich ELISA appears to be sufficient to detect even low levels in seafood products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wild-caught marine fish seems to be the only industrially produced food, which is at risk to carry parasites when put on the market [1]. In this respect, the larvae of the parasitic nematode Anisakis simplex, also known as the herring or whale worm, are among the most frequently occurring parasites in virtually all of the commercially exploited fish stocks in temperate seas around the globe.
The apparently most prevalent Anisakis species, A. simplex, has been shown to actually comprise a complex of closely related siblings in many areas of the North Atlantic and adjacent areas [2]. However, in the North Sea and Norwegian Sea, A. simplex sensu stricto appears to be the only species present [2, 3]. The Anisakis life cycle involves various marine mammals as definitive host, planktonic crustaceans—mainly krill—as intermediate host, and fishes and possibly squids as transport host, transferring the larvae from krill to whales. In fish, the majority of the Anisakis larvae occur encapsulated as flat tight spirals in and on the visceral organs and the peritoneum. However, a minor proportion of the larvae usually migrate from the visceral cavity into the flesh, sometimes penetrating deeply into the epaxial musculature of the fish host [4].
Besides the considerable quality-reducing effect of Anisakis larvae with respect to seafood products, they are of direct human health concern, especially regarding the increasing interest in Asian-inspired seafood dishes based on undercooked, brined or marinated, or even raw fish meat (e.g. sushi, sashimi). Thus, anisakiasis, i.e. human infection with live Anisakis larvae, is reported most frequently from Japan, Korea and some southern European countries, especially Spain, where raw or lightly salted or marinated fish is part of the everyday diet. If accidentally ingested by humans, live worms may bore into the intestinal mucosa and elicit an immune reaction which again may cause considerable distress. The clinical manifestations of anisakiasis include epigastric pain, nausea, vomiting, diarrhea, and urticaria. The condition may resemble several other gastrointestinal disorders, such as gastric ulcer, tumors, acute appendicitis or rectal carcinoma [5]. Additionally, the potential of the worm—both dead and alive—to induce hypersensitivity reactions in humans has received increased attention [6–9].
The European Food Safety Authority (EFSA) has recently released a scientific opinion on risk assessment of parasites in fishery products including the elicitation of allergic reactions in consumers [1]. It was assessed that human fishery product-borne parasitic diseases are mainly caused by cestodes, trematodes, and nematodes. Furthermore, it was concluded that only the nematode Anisakis simplex has been found to have the potential to induce IgE-mediated reactions. Patient sensitisation presumably occurs by the ingestion of seafoods infected with live larvae. It has also been demonstrated that occupational exposure of fish processing employees with Anisakis allergens may lead to increased levels of airway hyperactivity and dermatitis [10].
Currently, eleven allergens of A. simplex have been identified, of which most have been expressed as recombinant proteins. Allergic individuals may be exposed either to somatic antigens from dead larvae in food or to excretory-secretory (ES) antigens as a consequence of the expulsion or surgical removal of intact larvae. In some cases, both may apply when the larvae has penetrated the tissue, is killed by the host, and subsequently degenerates inside the host [11]. Several of the identified A. simplex allergens seem to retain their allergenic potential even after freezing and/or heating [12–14] and appear to be pepsin resistant [15]. In a study on the progression of allergens in the food chain, it was suggested that Anisakis-allergic consumers were affected by the intake of meat from chicken that had been fed with a high proportion of Anisakis-containing fishmeal [16]. The somatic antigens are the most abundant, and some of these proteins show cross-reactivity with other ascarid proteins [17]. The muscle protein tropomyosin is mainly responsible for allergenic cross-reactivities in other invertebrates; but, whether Anisakis tropomyosin (Ani s 3) is a clinically relevant allergen is under discussion. In an allergenicity study using sera from Anisakis-sensitive patients and crude Anisakis extract, IgE-binding to Ani s 3 was undistinguishable [18]. However, Guarneri et al. [19] have suggested that Anisakis tropomyosin could play an important role in eliciting food allergy after the ingestion of cooked seafood, since it closely resembles the heat-stable shrimp tropomyosin, which is an important seafood allergen.
The presence of A. simplex in seafood products may present a health risk for consumers. At the same time, it is also regarded as an economic problem because esthetical aspects may lead to a reduced attractiveness of fish consumption. Therefore, food safety authorities and seafood manufacturers are likewise interested in improved methods for the detection of Anisakis in foods. So far, only visual inspection methods of more or less intact worms [20, 21] or PCR methods for the determination of parasite DNA [22, 23] in fish products are available. However, there are at present no validated methods for the quantification of Anisakis protein in processed fish products and seafood. Recently, a sandwich ELISA for the detection of Anisakis in different fish using recombinant A. simplex allergen Ani s 1 has been published [24]. The ELISA has a limit of detection corresponding to about 25 Anisakis larvae per 100 g fish. The applicability of the assay for the quantification of Anisakis proteins in processed foods was not reported. In contrast, several PCR-based methods been developed that are intended for the quantification of parasite residues in food products [22, 23]. PCR- and ELISA-based methods can be regarded as complementary techniques with respect to allergen detection. However, PCR results can only depict the presence of Anisakis-DNA, which may not be correlated to the amount of allergenic protein, especially in processed food [25].
The aim of the current study was therefore to develop and validate a sensitive, specific, and quantitative sandwich ELISA method for the detection of Anisakis proteins in seafood products.
Materials and methods
Anisakis species identification
A. simplex 3rd stage larvae were collected from freshly caught blue whiting (Micromesistius poutassou) during a research cruise in the Northeast Atlantic. The A. simplex larvae were manually harvested from the muscle tissue of the fish, and the capsules were opened and removed. For Anisakis species identification, polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) analysis was used, as described before [26]. DNA from Anisakis larvae (n = 30) from blue whiting was prepared and digested using the restriction enzyme HinfI resulting in typical Anisakis simplex s.s.-specific RFLP patterns in agarose gel analysis. The results of this pre-study will be published separately (by H. Lin and A. Levsen).
Protein extraction from A. simplex
A protein extract was prepared from larvae that were freed from host tissue remains, rinsed in physiological saline, and subsequently homogenized in 0.1 M tris(hydroxymethyl)aminoethane (Tris)/0.5 M glycine buffer (pH 8.7)/1 mM dithioerythriol (DTE) (Sigma-Aldrich) extraction buffer with a rod homogenizer (Braun Vario, Kronberg, Germany). After overnight extraction at 45 °C in a shaking water bath (OLS 200, Grant, Cambridge, UK) and centrifugation for 25 min at 4 °C and 18,000g (J2-MC, Beckham Instruments, Palo Alto, CA), the supernatant was transferred to a glass beaker. Protein was precipitated with ammonium sulfate (Merck, Whitehouse Station, NJ), reaching a saturation of 75%, under continuous stirring at room temperature (RT) followed by centrifugation (25 min, 18,000g, 4 °C). The pellet was resuspended in 50 mM Tris/1 m DTE (pH 8.0) and dialyzed against distilled water in a dialysis tube with 6–8 kDa μm pore size (Spectrapor, Spectrum Medical industries, Los Angeles, CA) and freeze-dried (Heto, Allerød, Denmark) over night. The protein was dissolved in phosphate-buffered saline (PBS) (pH 7.4). Total protein content was determined by Lowry protein assay (BioRad Laboratories, Hercules, CA). Aliquots were stored at −20 °C until use. Additionally, a second extract was prepared using fewer purification steps to prevent potential protein loss. Therefore, A. simplex larvae were added PBS and homogenized by Ultra-Turrax (IKA®-Werke GmbH & Co. KG, Staufen, Germany). The homogenate of the raw extract was centrifuged at 39,200g for 25 min at 4 °C. Total protein content was again determined by Lowry protein assay, and aliquots were stored at −20 °C.
Production, purification, and labeling of anti-A. simplex antibody
A polyclonal antiserum was raised against the (NH4)SO4—precipitated extract from A. simplex larvae in rabbit using the same procedure as previously described [27]. The crude rabbit serum was desalted on PD-10 columns (Amersham Biosciences, Uppsala, Sweden), and purified by using a HiTrap r-Protein A FF-column (Amersham Biosciences), which had previously been washed with 0.1 M sodium citrate buffer pH 4.0 (elution buffer) and reequilibrated with 20 mM sodium phosphate buffer pH 7.0 (binding buffer). After washing with binding buffer, the bound IgG-antibodies were eluted in 0.5 mL fractions from the column with elution buffer, neutralized with 1 M Tris-base pH 8.5, and stored at 4 °C. The fractions were tested for binding activity using indirect ELISA, and total protein was determined by the Lowry method (DC Protein Assay, BioRad, Hercules, CA). The purity of the IgG in the collected fractions was tested on SDS–PAGE under reducing conditions. The purest antibody fractions were pooled prior to buffer exchange to 0.1 M sodium phosphate, 0.15 M NaCl, 0.1% sodium azide (pH 7.4), using a PD-10 column, and finally stored at −80 °C until use. In total, 30 mg of IgG had been obtained from 10 mL of crude rabbit serum.
For covalent conjugation with biotinamidohexanoic acid 3-sulfo-N-hydroxysuccinimide ester sodium salt (Sigma-Aldrich, Steinheim, Germany), the buffer was changed to 0.05 M carbonate-bicarbonate buffer (pH 9), and the antibody concentration was adjusted to 1 mg/mL. The biotin salt was then dissolved in deionized H2O to a concentration of 1 mg/mL and added to the antibody solution at a ratio of 1–6.67. The mixture was vortexed and rotated at RT for 4 h, and subsequently neutralized with 1 M NH4Cl under rotation at RT for 10 min. The total protein concentration of the biotinylated anti-A. simplex antibody was measured after a buffer change to 0.1 M sodium phosphate, 0.15 M NaCl, 0.1% sodium azide, pH 7.4, using PD-10 columns, and the antibody was stored at −20 °C until use.
Protein extraction and sample preparation
All protein extracts and food samples, unless otherwise stated, were prepared as previously described [28]. In brief, homogenized samples (2 g) were extracted with 10 mL of 0.1 M Tris, 0.5 M glycin (pH 8.7) under shaking, overnight at 45 °C and centrifuged at 39,200g for 25 min at 4 °C. Protein extracts were stored at −20 °C, at 4 °C or were used freshly. Prior to analysis using the sandwich ELISA, extracts were diluted at least 1:20 in phosphate-buffered saline (PBS) (Oxoid, Basingstoke, UK) containing 1% bovine serum albumin (BSA).
Evaluation of extraction buffers for seafood samples
The following extraction buffers were tested in the study (Table 1): urea 6 M, pH 8.7; Tris-glycine, 0.1 M Tris, 0.5 M glycine, pH 8.7; HSB, 20 mM NaH2PO4·H2O, 1 M NaCl, pH 7.5; high-salt buffer (HSB), 20 mM NaH2PO4·H2O, 1 M NaCl, pH 4.5; Citrate, 0.5 M citric acid monohydrate, 0.5 M sodium citrate dehydrate, pH 4; sodium carbonate, 50 mM, pH 11; sodium borate, 7 mM disodium tetraborate decahydrate, pH 9.2; PBS, 0.172 M, pH 7.4 (Oxoid). Extractions were performed as described above. Ionic strengths were high (>1 M), medium (0.5–1), and low (<0.5 M).
Western blotting
The NuPage Gel System (Invitrogen, Carlsbad, CA) was used for electrophoretic separation of protein samples by SDS–PAGE, in accordance with the manufacturer’s instructions. All protein samples were applied in equal amounts (10 μg). Samples were prepared with lithium dodecyl sulfate (LDS) sample buffer and dithiothreitol (DDT) reducing agent (all from Invitrogen). Separation was performed under reducing conditions for 40 min at 200 V in 2-(N-morpholino) ethane sulfonic acid (MES) SDS running buffer, using 4–12% Bis-Tris gels and SeeBluePlus2 prestained reference standard with a range of 3–188 kDa.
Proteins were electrophoretically transferred from the gel onto a nitrocellulose membrane (Bio-Rad, Hercules, CA) for 60 min at 30 V with transfer buffer in an XCell II Blot Module (Invitrogen). Tris-buffered saline containing 0.1% Tween 20 (TBS-T, pH 7.6) was used as washing buffer and TBS-T containing 1% BSA was used as blocking and assay buffer for the Western blots. After blocking for 30–60 min, the blot was incubated at 4 °C overnight, with the unlabelled purified anti-A. simplex antibody (3.3 mg/mL) diluted 1:30 000 in assay buffer. The blot was washed (3 × 15 min) and incubated for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (Zymed, San Francisco, CA) diluted 1:5,000 in assay buffer. After washing (3 × 10 min), the membrane was developed with 3, 3′, 5, 5′-tetramethylbenzidine (TMB) substrate solution (Zymed) until bands of satisfactory intensity appeared (2–10 min). All washing and incubation steps were performed under gentle shaking at RT.
Sandwich ELISA procedure
Ninety-six-well flat-bottom polystyrene microtiterplates (Corning Inc., Corning, NY) were coated overnight at 4 °C with 100 μL/well of purified rabbit anti-A. simplex polyclonal antibody diluted in 0.05 M carbonate-bicarbonate buffer (pH 9.6) (Sigma-Aldrich) to 10 μg/mL. Then, the plates were washed with PBS containing 0.05% Tween-20 (PBS-T, pH 7.6) three times, using a programmable automatic plate washer (Skatron Instruments, Lier, Norway). All consecutive washing steps performed between each operation in the tropomyosin sandwich ELISA were the same. PBS containing 1% BSA and 0.05% Tween-20 was used as blocking and assay buffer for the ELISA. All dilutions, except for the initial coating step, were performed in assay buffer, and the plates were sealed with plate-sealing film during the incubations. Unsaturated binding sites on the polystyrene surface of the microtiterplate were blocked by incubation with 250 μL/well of the blocking/assay buffer for 1 h at RT. After washing, a twofold serial dilution of the A. simplex raw extract, prepared as described above, was added in the concentration range 0.98–1,000 ng/mL and served as assay standard. Incubation of standards, buffer blanks, and sample extracts at a minimum dilution of 1:20 was performed in triplicate on each plate, for 1 h at RT using an electronic plate shaker (IKA®-Werke, GmbH & Co. KG, Staufen, Germany) at low speed. After washing, bound A. simplex protein was detected by adding 100 μL/well of biotinylated rabbit anti-A. simplex antibody diluted 1:800 and incubated for 1 h at RT under gentle shaking. Following the next wash, the plates were incubated for 1 h at RT with 100 μL/well of HRP-streptavidin conjugate (Zymed), 1:5,000 dilution. After a final wash, each well was incubated with 75 μL of K-Blue TMB substrate (Neogen, Lexington, KY, USA). After 20 min, the color development was stopped by the addition of 50 μL/well of 2 M H2SO4. Absorbance was read at 450 nm using a 1420 VICTOR2 multilabel plate counter (Wallac, Turku, Finland).
Assay validation: specificity, accuracy, precision, limit of detection, and limit of quantification
The specificity of the assay was evaluated through cross-reactivity studies using extracts of different fish and seafood species, in addition to some potentially food-contaminating arthropods. Extracts from Atlantic salmon (Salmo salar), Atlantic cod (Gardus morhua), bluefin tuna (Thunnus thynnus), mackerel (Scomber scombrus), Atlantic herring (Clupea harengus), haddock (Melanogrammus aeglefinus), blue whiting (Micromesistius poutassou), rainbow trout (Oncorhynchus mykiss), edible frog (Rana esculenta), European squid (Loligo vulgaris), common cockle (Cerastoderma edule), Northern shrimp (Pandulus borealis), blue mussel (Mytilus edulis), edible crab (Cancer pagurus), giant tiger prawn (Panaeus monodon), European brown snail (Helix aspersa aspersa), German cockroach (Blatella germanica), European house dust mite (Dermatophagoides pteronyssinus), and dried fruit mite (Carpoglyphus lactis) were all diluted 1:20 in ELISA assay buffer, which corresponds to a portion of 100% in a typical food matrix. The apparent A. simplex contents obtained from the ELISA assay were compared to the total protein content of the corresponding extract, and recovery rates were calculated.
The accuracy of the method was assessed by performing recovery studies. Two previously measured A. simplex-free food items (fish balls and mackerel in tomato sauce) were homogenized as described above and spiked with A. simplex protein standard at levels of 1, 10, and 100 μg A. simplex/g sample in a total volume of 5 mL extraction buffer. After vortexing and 15-min incubation at RT, an additional portion of 5 mL extraction buffer was added, and the extraction procedure was performed as previously described. For the evaluation of recovery rates, extractions were performed in triplicate, the resulting extracts were analyzed by the Anisakis sandwich ELISA, and the mean values for the recoveries and the standard error of the mean were calculated.
The precision of the method was evaluated as within (intra-assay) and between (inter-assay) assays, by using extracts from cod meat that had been spiked with two, ten, twenty-five, and fifty intact A. simplex larvae, which had been obtained from infected fish caught on research missions. The spiked samples were homogenized together with uninfected cod fish. After extraction, the samples were stored at −20 °C, and each analysis was performed with freshly thawed extract. The intra-assay precision was determined as the mean coefficient of variation (CV) of 7 replicates in one assay. The inter-assay precision was calculated as the mean coefficient of variation (CV) on the basis of triplicate analysis on 7 different days.
The limit of detection (LOD) of the A. simplex sandwich ELISA was calculated from 13 experiments as the mean of the measured content of triplicate buffer blank samples plus three times the standard deviation (SD) of the mean value. The limit of quantification (LOQ) was calculated as the buffer blank mean value plus 10 times the standard deviation (SD) of the mean value in 13 experiments. Additionally, the limit of detection was determined for three food matrixes free from A. simplex protein, i.e. codfish, fish balls, and mackerel with tomato sauce diluted 1:20 in assay buffer, on the basis of 7 experiments with triplicate samples each.
Results
Characterization of the anti-A. simplex antibody
The binding characteristics of the purified anti-A. simplex antibody were evaluated by Western blot analysis, using the raw and purified A. simplex larvae protein extract, and extracts from several fish, seafood, and insects (Fig. 1). The antibody binds specifically to three major A. simplex protein bands at about 41, 67, and 97 kDa, and in addition multiple weaker bands in the entire blot from 14–190 kDa in both extracts from A. simplex. However, there were slight differences in the observable binding patterns of the raw protein extract (lane 2) and the purified extract that had been thermally treated at 45 °C (lane 1). In the raw preparation, the protein band at 97 kDa was less distinct, and fewer protein bands were visible between 14 to 40 kDa, whereas after heating several bands with molecular weight above 67 kDa became visible.

Western blot analysis of various protein extracts of A. simplex larvae and potential cross-reacting species of seafood and insects using purified polyclonal anti-A. simplex antibody. M Molecular weight marker. Protein sizes [kDa] are indicated on the left side of the blot. Lane 1 A. simplex purified extract; lane 2 raw A. simplex extract; lane 3 A. simplex-spiked cod (HSB (pH 4.5) extract); lane 4 A. simplex-spiked cod (PBS extract); lane 5 A. simplex-spiked cod (Tris–glycine extract); lane 6 salmon; lane 7 cod; lane 8 bluefin tuna; lane 9 mackerel; lane 10 herring; lane 11 haddock; lane 12 blue whiting; lane 13 trout; lane 14 shrimp; lane 15 cockroach; lane 16 dried fruit mite; and lane 17 house dust mite
The Western blot analysis (Fig. 1, lanes 6–17) of possible cross-reacting proteins of salmon, cod, tuna, mackerel, herring, and trout revealed only unspecific background signals.
Sandwich ELISA standard curve
The raw A. simplex protein preparation was used as protein standard in the sandwich ELISA in concentrations ranging from 0.98 to 1,000 ng/mL. The working range of the assay was defined as the part of the curve that had a correlation coefficient of r 2 > 0.99. This was achieved both with 2nd degree polynomial regression (r 2 = 0.9998) and linear regression (r 2 = 0.9935) for an eight-point calibration curve ranging from 0.98–125 ng/mL, corresponding to 0.1–12.5 μg A. simplex protein/g food sample (Fig. 2). Therefore, the more user-friendly linear transformation was used for the determination of A. simplex protein concentrations in food. If necessary, serial dilutions of extracts were performed, and those that gave optical density (OD) values closest to the midpoint of the linear part of the standard curve were used to calculate the A. simplex protein concentration.

Representative eight-point calibration curve obtained using the raw A. simplex protein standard in the A. simplex sandwich ELISA. The graph shows the average values of triplicate measurements, the standard deviation in each point, and the 2nd degree polynomial regression curve and coefficient r 2
Influence of buffers on sample extraction
The extraction efficiency was evaluated by extracting homogenized codfish artificially contaminated with 25 A. simplex larvae, with various buffers of different pH and ionic strength (Table 1). Both, the total protein content and A. simplex protein content, were measured in the resulting extracts. The collected data suggested that the extraction buffer used had a vast impact on the extracted amount of total protein and A. simplex protein (Table 1). For total protein extraction, the best efficiencies were achieved by PBS, Tris-glycine, and HSB (pH 4.5), although no correlation could be observed between the yield of total protein and ionic strength or pH. A. simplex proteins appeared to be more extractable under basic than neutral or acidic conditions. It was observed that the most acidic extracts led to precipitation. Furthermore, A. simplex proteins were not detected in the PBS-based extract at all, even if the recovery of total protein was highest in this sample. The three extracts with the highest total protein concentrations were accessed by SDS–PAGE (data not shown) and Western blot (Fig. 1, lanes 3–5). The blot analysis of the Tris-glycine extract (Fig. 1, lane 5) showed two major bands at 41 kDa and about 70 kDa, which resembled the major protein bands in the raw and purified A. simplex extracts in lane 1 and 2. The profiles for the PBS and HSB-4.5 protein extracts were slightly different, with broader, more undefinable protein bands at lower molecular weights. The PBS and HSB-4.5 protein extracts seemed to have the 41-kDa band as well, but less prominent. The HSB extract (lane 3) showed comparable bands at about 65 and 97 kDa, whereas the PBS extract (lane 4) showed multiple weak bands in the range from 30 to 70 kDa. Tris-glycine buffer seemed to be the most effective with respect to Anisakis protein extraction and recovery in the ELISA and was therefore chosen for all subsequent studies.
Specificity
A number of seafoods were analyzed for cross-reactivity in the A. simplex sandwich ELISA. A preliminary screening of extracts from scallop, snail, mussel, squid, salmon, mackerel, herring, bluefin tuna, cod, and sprat in a non-optimized competitive ELISA showed no inhibition with assay concentrations of 10 μg/mL of total protein. The A. simplex protein standard gave total inhibition at the same concentration (data not shown).
Subsequently, extracts from different potentially cross-reacting species of fish, crustaceans, molluscs, and insects were analyzed with the A. simplex sandwich ELISA under real sample conditions (Table 2). The recoveries of A. simplex protein equivalents were calculated and none of the extracts gave rise to ELISA results exceeding 0.002%.
Accuracy
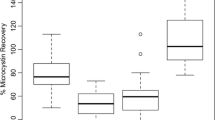
Recovery studies were performed to assess the accuracy of the A. simplex sandwich ELISA. Two blank matrixes were spiked with low, intermediate, and high concentration amounts of A. simplex protein standard before sample extraction and used to determine recovery rates (Table 3). In fish balls, the recoveries ranged from 72 to 88%, and in mackerel in tomato sauce from 84 to 101% with the standard errors of less than 12%.
Precision
The intra- and inter-assay precision for the A. simplex sandwich ELISA was assessed by analyzing codfish spiked with different numbers of A. simplex larvae prior to homogenization and extraction (Table 4). The intra-assay precision, expressed as the coefficient of variation (%CV) ranged from 5 to 11%, and the inter-assay precision ranged from 16 to 25% with the highest uncertainties for the lowest concentrations. The precision studies were performed on relatively moderate concentrations with the highest level corresponding to 30 μg A. simplex protein/g food. Considering, the amount of larvae added to the sample, this can probably be regarded as a quite heavy A. simplex infection and an appropriate level of validation.
Limit of detection and limit of quantification
The LOD of the ELISA corresponded to 0.003 μg/mL measured in assay buffer, equivalent to 0.3 μg A. simplex protein/g food, considering the dilution. The LOQ was 0.01 μg/mL measured in the assay buffer and 1.1 μg A. simplex protein/g food. Additionally, the LOD was assessed in blank samples of codfish, fish balls, and mackerel with tomato sauce resulting in varying LODs in the different matrices. The LOD in codfish and mackerel with tomato sauce was 0.6 μg A. simplex protein/g food and 0.5 μg A. simplex protein/g food in fish balls.
Discussion
The potential contamination of seafood with the fish parasite A. simplex is concerning health authorities, food manufacturers, and consumers. Therefore, survey studies assessing the prevalence of A. simplex in fish and seafood products should be conducted. However, a reliable and sensitive method for the quantitative analysis of A. simplex in foods is not available so far.
ELISA is the most commonly used immunological method for the quantitative detection of food allergens. The principal advantages of these fully quantitative assays are their robustness and sensitivity. Several formats are in use, but all are based on the use of a species-specific antibody. Antibodies may be monoclonal, detecting specifically a single peptide sequence in the target protein, or polyclonal, reacting to many epitopes in the target. Antibodies, in particular polyclonal ones, may also react to similar peptide structures in other related proteins, resulting in cross-reactivity, which generally is limiting the applicability of the assay. However, proteins can be altered by food processing techniques, destroying epitopes, or exposing formerly hidden ones [29, 30]. Therefore, a broader specificity of the antibody seems to be advantageous for the analysis of processed food. In the development of an ELISA method for the detection of A. simplex proteins, we therefore chose to use a polyclonal antibody expecting that proteins and fragments of the dead parasite larvae as well as excretory proteins would be detectable.
In the present study, a sensitive and specific sandwich ELISA for the determination of A. simplex in foods was developed and validated with two different food matrixes. By this method, extractable A. simplex proteins were successfully detected and quantitated in commercially available food products and in fish spiked with A. simplex larvae. A pilot survey on different Norwegian fish products was performed. Several of the tested products contained small amounts of A. simplex protein. These results will be published elsewhere (M.T. Werner et al., 124th AOAC International Meeting and Exposition, Sep. 2010).
Since the A. simplex sandwich ELISA constructed using a purified polyclonal anti-A. simplex antibody, extensive studies on cross-reactivity was performed during the assay validation. The specificity of the anti-A. simplex antibody used in the ELISA was studied by cross-reaction experiments with different fish, molluscs, and insects. The test conditions used in the ELISA to assess the assay specificity were based on the protein concentrations of the raw food ingredients. These conditions would clearly overestimate the real situation in commercial food products, which usually consist of a considerable number of mixed ingredients. None of the species tested showed any cross-reactivity in the A. simplex sandwich ELISA. Furthermore, several species of fish, shrimp, cockroach, and mite [31] were analyzed with Western blot. A number of weak bands in the range of 40–70 kDa were considered to be a result of unspecific binding to the rather high protein amounts applied on the gel and the high anti-A. simplex antibody concentration used. None of these species produced any positive signals in the ELISA.
Additionally, the detection characteristics of the anti-A. simplex antibody were evaluated by Western blot, comparing the binding patterns to two different A. simplex protein extracts. The antibody bound apparently to multiple proteins in the range from 14 to 190 kDa. Three pronounced bands were visible in both the purified and the raw A. simplex extract at about 41, 67 and 97 kDa. The protein bands at 41 and 97 kDa could be identical with the A. simplex allergens Ani s 3 (tropomyosin) [18] and Ani s 2 (paramyosin) [32], respectively, which are the only major somatic allergens described so far [19, 33]. They show extensive homology to tropomyosins and paramyosins from other species causing probably the considerable cross-reactivity of anti-A. simplex-IgE with e.g. German cockroach, chironomids, and dust mites that has been clinically observed [28, 34, 35]. The protein band at 67 kDa might be connected to a thermolabile protein of the same size that had shown antibody binding in mammalian cell cultures [36]. However, to our knowledge, this protein has not been further characterized or regarded as an important immunogen concerning A. simplex allergy. Several other proteins have been identified as allergens from A. simplex. Ani s 1 is a secretory protein of 21 kDa and the only major allergen described with no described function or homology to date [37, 38]. Two allergens, Ani s 4 (9 kDa) and Ani s 6 (7 kDa) [13], are both protease inhibitors [14, 39]. Ani s 5 (15 kDa) [13], Ani s 8 (15 kDa) [40], and Ani s 9 (15 kDa) [41] are all proteins in the SXP/RAL-2 family, whose function is still unclear. Ani s 7 has recently been described as the most important excretory-secretory allergen for diagnosis of infected patients [42].
The key parameters of the A. simplex sandwich ELISA were evaluated and determined in an intra-laboratory validation. The standard curve was linear at concentration levels of 0.98 to 125 ng A. simplex protein/mL, or 0.1 to 12.5 μg A. simplex protein/g food if the sample dilution in the assay is considered. The assay performed well with 1:20 dilution of all tested matrixes. Due to the restricted availability of purified A. simplex protein standard, the method validation had to be modified in some aspects. After determining the optimal ELISA conditions and dilutions of all components in the sandwich ELISA and the initial cross-reactivity testing, the assay performance of the purified A. simplex protein standard and the raw A. simplex protein standard were compared. Since the difference of the observed OD 450 nm absorbance values was approximately 5% at corresponding concentrations and both preparations demonstrated similar antigenicity of the major proteins on Western blot, we decided to switch to the raw A. simplex protein standard in the further validation of the ELISA assay. Regarding the lack of reference materials and standards, and considering errors from sampling and sample preparation, we considered the difference between the two A. simplex protein extracts as insignificant and neglectable. For a further development of the A. simplex sandwich ELISA, the purification of a large amount of A. simplex protein could be reconsidered. Additionally, protein characterization by mass spectrometry should be aspired.
The choice of an appropriate extraction buffer has great impact on the protein composition in the extracts and the recovery from the sample. ELISA performance, however, was not affected by the extraction buffers due to the at least 20-fold sample dilution. Of the eight buffers examined in the present study including a range of different pH and ionic strengths, PBS was shown to be the most effective in terms of total protein yield from the A. simplex-spiked cod samples. However, the amount of total protein extracted was not correlated to the amount of A. simplex protein measured by the ELISA. Extraction by HSB (pH 4.5) and Tris-glycine resulted in similar total protein yield as for PBS. With regard to A. simplex protein, the extractability and ELISA-detectability appeared to be greatly dependent on the pH-values of the buffers. Measurable A. simplex proteins were only obtained under basic conditions, which is congruent to an observation made for other allergens in a previous study [43]. The Tris-glycine extraction buffer was found to be optimal considering the yield of detectable A. simplex proteins from thermally treated samples. This might be connected to the amino acid content in the Tris-glycine buffer, possibly increasing the extraction efficiency of A. simplex proteins. In contrast, PBS was apparently only suited for the extraction of raw samples according to the Western blot results in this study.
The accuracy of the A. simplex sandwich ELISA was determined in recovery experiments, using the raw A. simplex protein standard for two different blank food matrixes. The recovery rates were over 72% for all test concentrations, and variances were low (SEM < 15). The food products analyzed in this study were composed of a variety of ingredients representing authentic seafood products commonly used in Norway. Optimally, more matrixes should have been evaluated in the A. simplex sandwich ELISA to demonstrate the methods general applicability. However, A. simplex protein standard was a limited resource during the method validation and experiments had to be confined to the most relevant ones. In the meantime, we were able to increase the supply of A. simplex protein considerably, so that further studies could be performed. Evaluating the available accuracy data of the A. simplex sandwich ELISA, the method performance was found sufficient for the intended applications. Due to the lack of certified reference materials and standardized allergen protein preparations, the trueness of the method could not be determined.
The repeatability of the A. simplex sandwich ELISA as determined by intra-assay precision was <11% and the reproducibility as measured by inter-assay precision <25% using cod that was artificially infected with A. simplex larvae. The method worked reliable both for low and high numbers of larvae, corresponding to different levels of extracted A. simplex proteins. The numbers of added larvae and the levels of measured protein were not directly correlated reflecting the differences in size and protein content from larva to larva [44]. Because A. simplex larvae can grow in their fish hosts they will vary considerably depending on how many cycles in fish they have passed. However, the method performance considering precision was regarded as satisfactory for the assessed levels, although further investigations including additional food matrixes and concentrations would be desirable.
The sensitivity of the A. simplex sandwich ELISA was evaluated on the basis of buffer blank samples, two blank food matrices, and cod meat without A. simplex protein spike. The assay was sufficiently sensitive to allow quantitative determination of A. simplex protein in seafoods at levels of 1 μg A. simplex protein/g sample. Furthermore, matrix had rather little influence on the measured LODs, ranging from 0.3 μg A. simplex protein/g food sample in blank buffer to 0.6 μg A. simplex protein/g sample in both mackerel in tomato sauce and cod. In terms of A. simplex larvae count, the A. simplex sandwich ELISA was able to detect A. simplex protein originating from only 1 larva in 100 g of fish meat.
For routine analysis, however, the LOD of the A. simplex sandwich ELISA was set to 1 μg A. simplex protein/g sample to reduce the probability of false-positive results and increase the reliability of the assay, which is important for e.g. survey studies commissioned by food authorities. This operative LOD is in agreement with the assay specifications considered generally necessary for the protection of food allergic consumers. Although threshold concentrations and assay requirements are still under discussion [25], there is some kind of consensus that LODs of methods detecting allergens in food matrixes should be somewhere between 1 and 100 μg/g.
In conclusion, a sensitive A. simplex ELISA in sandwich format for the quantitative determination of A. simplex larvae in foods was successfully developed and validated in two different complex food matrixes. This is the first ELISA method for the detection of A. simplex protein that is suited for food analysis. The A. simplex sandwich ELISA could be used for the quality control of raw materials and end products and for food surveys by food safety authorities.
References
EFSA Panel on Biological Hazards (BIOHAZ) (2010) Scientific opinion on risk assessment of parasites in fishery products. EFSA J 8:1543
Mattiucci S, Nascetti G (2006) Molecular systematics, phylogeny and ecology of anisakid nematodes of the genus Anisakis dujardin, 1845: an update. Parasite 13:99–113
Skov J, Kania PW, Olsen MM, Lauridsen JH, Buchmann K (2009) Nematode infections of maricultured and wild fishes in Danish waters: a comparative study. Aquaculture 298:24–28
Levsen A, Lunestad BT (2010) Anisakis simplex 3rd stage larvae in Norwegian spring spawning herring (Clupea harengus L.), with emphasis on larval distribution in the flesh. Vet Parasitol (in press)
Bouree P, Paugam A, Petithory JC (1995) Anisakidosis: report of 25 cases and review of the literature. Comp Immunol Microbiol Infect Dis 18:75–84
Anibarro B, Seoane FJ, Mugica MV (2007) Involvement of hidden allergens in food allergic reactions. J Investig Allergol Clin Immunol 17:168–172
Dominguez-Ortega J, Alonso-Llamazares A, Rodriguez L, Chamorro M, Robledo T, Bartolome JM, Martinez-Cocera C (2001) Anaphylaxis due to hypersensitivity to Anisakis simplex. Int Arch Allergy Immunol 125:86–88
Moreno-Ancillo A, Caballero MT, Cabanas R, Contreras J, Martin-Barroso JA, Barranco P, Lopez-Serrano MC (1997) Allergic reactions to Anisakis simplex parasitizing seafood. Ann Allergy Asthma Immunol 79:246–250
Valls A, Pascual CY, Martin EM (2003) Anisakis and anisakiosis. Allergol Immunopathol (Madr) 31:348–355
Nieuwenhuizen N, Lopata AL, Jeebhay MF, Herbert DR, Robins TG, Brombacher F (2006) Exposure to the fish parasite Anisakis causes allergic airway hyperreactivity and dermatitis. J Allergy Clin Immunol 117:1098–1105
Audicana MT, Kennedy MW (2008) Anisakis simplex: from obscure infectious worm to inducer of immune hypersensitivity. Clin Microbiol Rev 21:360–379
Caballero ML, Moneo I, Gomez-Aguado F, Corcuera MT, Casado I, Rodriguez-Perez R (2008) Isolation of Ani s 5, an excretory-secretory and highly heat-resistant allergen useful for the diagnosis of Anisakis larvae sensitization. Parasitol Res 103:1231–1233
Kobayashi Y, Ishizaki S, Shimakura K, Nagashima Y, Shiomi K (2007) Molecular cloning and expression of two new allergens from Anisakis simplex. Parasitol Res 100:1233–1241
Moneo I, Caballero ML, Gonzalez-Munoz M, Rodriguez-Mahillo AI, Rodriguez-Perez R, Silva A (2005) Isolation of a heat-resistant allergen from the fish parasite Anisakis simplex. Parasitol Res 96:285–289
Caballero ML, Moneo I (2004) Several allergens from Anisakis simplex are highly resistant to heat and pepsin treatments. Parasitol Res 93:248–251
Armentia A, Martin-Gil FJ, Pascual C, Martin-Esteban M, Callejo A, Martinez C (2006) Anisakis simplex allergy after eating chicken meat. J Investig Allergol Clin Immunol 16:258–263
Sugane K, Sun SH, Matsuura T (1992) Radiolabelling of the excretory-secretory and somatic antigens of Anisakis simplex larvae. J Helminthol 66:305–309
Asturias JA, Eraso E, Moneo I, Martinez A (2000) Is tropomyosin an allergen in Anisakis? Allergy 55:898–899
Guarneri F, Guarneri C, Benvenga S (2007) Cross-reactivity of Anisakis simplex: possible role of Ani s 2 and Ani s 3. Int J Dermatol 46:146–150
Levsen A (2007) Detection of some important quality reducing or potentially consumer health hazardous parasites in the flesh of fish—the Norwegian experience. Parasitologia 49:401
Levsen A, Lunestad BT, Berland B (2005) Low detection efficiency of candling as a commonly recommended inspection method for nematode larvae in the flesh of pelagic fish. J Food Prot 68:828–832
Lopez I, Pardo MA (2010) Evaluation of a real-time polymerase chain reaction (PCR) assay for detection of Anisakis simplex parasite as a food-borne allergen source in seafood products. J Agric Food Chem 58:1469–1477
Mossali C, Palermo S, Capra E, Piccolo G, Botti S, Bandi C, D’Amelio S, Giuffra E (2010) Sensitive detection and quantification of Anisakid parasite residues in food products. Foodborne Pathog Dis. 7:391–397
Arilla MC, Ibarrola I, Martinez A, Monteseirin J, Conde J, Asturias JA (2008) An antibody-based ELISA for quantification of Ani s 1, a major allergen from Anisakis simplex. Parasitology 135:735–740
Poms RE, Klein CL, Anklam E (2004) Methods for allergen analysis in food: a review. Food Addit Contam 21:1–31
Farjallah S, Slimane BB, Busi M, Paggi L, Amor N, Blel H, Said K, D’Amelio S (2008) Occurrence and molecular identification of Anisakis spp. from the North African coasts of Mediterranean Sea. Parasitol Res 102:371–379
Faeste CK, Lovik M, Wiker HG, Egaas E (2004) A case of peanut cross-allergy to lupine flour in a hot dog bread. Int Arch Allergy Immunol 135:36–39
Werner MT, Faeste CK, Egaas E (2007) Quantitative sandwich ELISA for the determination of tropomyosin from crustaceans in foods. J Agric Food Chem 55:8025–8032
Besler M, Steinhart H, Paschke A (2001) Stability of food allergens and allergenicity of processed foods. J Chromatogr B Biomed Sci Appl 756:207–228
Maleki SJ (2004) Food processing: effects on allergenicity. Curr Opin Allergy Clin Immunol 4:241–245
Weiler CR (2007) Anisakis simplex and cross-reacting antigens. Int J Dermatol 46:224–225
Perez-Perez J, Fernandez-Caldas E, Maranon F, Sastre J, Bernal ML, Rodriguez J, Bedate CA (2000) Molecular cloning of paramyosin, a new allergen of Anisakis simplex. Int Arch Allergy Immunol 123:120–129
Asturias JA, Eraso E, Martinez A (2000) Cloning and high level expression in Escherichia coli of an Anisakis simplex tropomyosin isoform. Mol Biochem Parasitol 108:263–267
Ferreira F, Hawranek T, Gruber P, Wopfner N, Mari A (2004) Allergic cross-reactivity: from gene to the clinic. Allergy 59:243–267
Johansson E, Aponno M, Lundberg M, Hage-Hamsten M (2001) Allergenic cross-reactivity between the nematode Anisakis simplex and the dust mites Acarus siro, Lepidoglyphus destructor, Tyrophagus putrescentiae, and Dermatophagoides pteronyssinus. Allergy 56:660–666
Raybourne R, Deardorff TL, Bier JW (1986) Anisakis simplex: larval excretory secretory protein production and cytostatic action in mammalian cell cultures. Exp Parasitol 62:92–97
Moneo I, Caballero ML, Gomez F, Ortega E, Alonso MJ (2000) Isolation and characterization of a major allergen from the fish parasite Anisakis simplex. J Allergy Clin Immunol 106:177–182
Shimakura K, Miura H, Ikeda K, Ishizaki S, Nagashima Y, Shirai T, Kasuya S, Shiomi K (2004) Purification and molecular cloning of a major allergen from Anisakis simplex. Mol Biochem Parasitol 135:69–75
Rodriguez-Mahillo AI, Gonzalez-Munoz M, Gomez-Aguado F, Rodriguez-Perez R, Corcuera MT, Caballero ML, Moneo I (2007) Cloning and characterisation of the Anisakis simplex allergen Ani s 4 as a cysteine-protease inhibitor. Int J Parasitol 37:907–917
Kobayashi Y, Shimakura K, Ishizaki S, Nagashima Y, Shiomi K (2007) Purification and cDNA cloning of a new heat-stable allergen from Anisakis simplex. Mol Biochem Parasitol 155:138–145
Rodriguez-Perez R, Moneo I, Rodriguez-Mahillo A, Caballero ML (2008) Cloning and expression of Ani s 9, a new Anisakis simplex allergen. Mol Biochem Parasitol 159:92–97
Anadon AM, Romaris F, Escalante M, Rodriguez E, Garate T, Cuellar C, Ubeira FM (2009) The Anisakis simplex Ani s 7 major allergen as an indicator of true Anisakis infections. Clin Exp Immunol 156:471–478
Poms RE, Capelletti C, Anklam E (2004) Effect of roasting history and buffer composition on peanut protein extraction efficiency. Mol Nutr Food Res 48:459–464
Strømnes E, Andersen K (2003) Growth of whaleworm (Anisakis simplex, Nematodes, Ascaridoidea, Anisakidae) third-stage larvae in paratenic fish hosts. Parasitol Res 89:335–341
Acknowledgments
This study was financially supported by the Orkla Funds. The authors wish to thank Lena Haugland Moen and Christin Plassen at the National Veterinary Institute for technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Werner, M.T., Fæste, C.K., Levsen, A. et al. A quantitative sandwich ELISA for the detection of Anisakis simplex protein in seafood. Eur Food Res Technol 232, 157–166 (2011). https://doi.org/10.1007/s00217-010-1373-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-010-1373-9