
Abstract
Glycosphingolipids (GSL) are a highly heterogeneous class of lipids representing the majority of the sphingolipid category. GSL are fundamental constituents of cellular membranes that have key roles in various biological processes, such as cellular signaling, recognition, and adhesion. Understanding the structural complexity of GSL is pivotal for unraveling their functional significance in a biological context, specifically their crucial role in the pathophysiology of various diseases. Mass spectrometry (MS) has emerged as a versatile and indispensable tool for the structural elucidation of GSL enabling a deeper understanding of their complex molecular structures and their key roles in cellular dynamics and patholophysiology. Here, we provide a thorough overview of MS techniques tailored for the analysis of GSL, emphasizing their utility in probing GSL intricate structures to advance our understanding of the functional relevance of GSL in health and disease. The application of tandem MS using diverse fragmentation techniques, including novel ion activation methodologies, in studying glycan sequences, linkage positions, and fatty acid composition is extensively discussed. Finally, we address current challenges, such as the detection of low-abundance species and the interpretation of complex spectra, and offer insights into potential solutions and future directions by improving MS instrumentation for enhanced sensitivity and resolution, developing novel ionization techniques, or integrating MS with other analytical approaches for comprehensive GSL characterization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
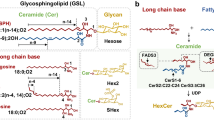
GSL comprise a vast group of remarkably heterogeneous biomolecules that are found in all eukaryotes, as well as some prokaryotes and viruses. GSL are ubiquitous membrane components, which are almost exclusively located on the outer leaflet of cell plasma membranes and in intracellular organelles (Fig. 1) [1, 2]. GSL are also found in body fluids, where they either circulate freely or are transported in lipoproteins [3]. GSL are amphiphilic molecules composed of two distinct parts. The hydrophobic region consists of a ceramide backbone anchored into the plasma membrane, while the hydrophilic region composed of a glycan moiety glycosidically linked to a ceramide backbone faces the extracellular environment [4].

Cross-section and structure of plasma membrane
GSL are classified based on their charges into neutral, acidic, and basic. Neutral GSL (nGSL) include cerebrosides (1 glycan unit) and globosides (≥ 2 glycan units), while acidic GSL (aGSL) are subdivided into sialic acid-containing GSL called gangliosides and sulfated GSL called sulfatides with sulfate group at C3 hydroxyl of Gal. In contrast, basic GSL are rare [2].
The rapid and direct structural elucidation of GSL and other lipids proves to be critical for studying their functional roles in many biological processes as well as the fundamental mechanism of their metabolism and pathogenesis in various diseases. MS-based methods using either atmospheric pressure ionization (API) or matrix-assisted laser desorption/ionization (MALDI) have emerged as powerful techniques for GSL profiling in the lipidomic analysis [5]. Although GSL can be relatively easily ionized and fragmented to product ions, providing information about the head groups (i.e., lipid class) and the type of the ceramide backbone, respectively fatty acyl composition (i.e., lipid species), the precise and in-depth characterization of GSL is quite challenging.
In this review, MS-based techniques tailored for the structural elucidation of GSL are discussed together with various fragmentation and ion activation techniques to study the glycan sequence, linkage positions, and fatty acid composition of GSL.
Mass spectrometry characterization of GSL in biological samples
One of the current major challenges in lipidomics is the difficulty to separate and differentiate isomeric and isobaric species due to the immense structural variability in head groups, acyl chains, number and location of C = C bonds (cis/trans), and regioisomerism (sn-positions), which inhibit the delineation and assignment of their biological roles. The specific functions of the isomer have remained largely unknown due to these challenges [6]. In the case of GSL, the isomer problem is multiplied as many glycans have the same formula (e.g., Glc vs. Gal) as well as there can be distinct linkages in the oligosaccharide chain also with the option for either α- or β-glycosidic bonds, which further complicate GSL analysis [6, 7]. For instance, GlcCer and GalCer have different biological functions with GlcCer required for proper functioning of the epidermis, while GalCer maintains the structure and stability of myelin and the differentiation of oligodendrocytes. GlcCer and GalCer are also accumulated in various lysosomal storage disorders. Accumulation of GlcCer is a typical feature of Gaucher disease, while GalCer is accumulated in Krabbe disease. GlcCer has also been shown to be accumulate in neurodegenerative diseases such as Parkinson’s disease. However, little is still known about their role in tumor progression [8]. In general, a-series gangliosides are known to promote tumor growth while b-series gangliosides may have tumor-suppressive effects. Similarly, antibodies against GD1a and GD1b gangliosides are differentially expressed in various neurological and autoimmune diseases. Differentiation of GSL isomers is therefore crucial because of their distinct biological functions, disease associations, and therapeutic implications [9]. Moreover, the glycosphingolipidome is not only amazingly large, but also expanding with a number of new lipid species. Specifically, GlcCer with less prevalent α linkage rather than β linkage have been recently found [8, 10, 11] together with ceramides either lacking the 1-hydroxyl group [12] or having a fatty acyl attached to the 1-hydroxyl [13] alongside GSL with polyunsaturated very long-chain FA (C26–C36) [14]. Furthermore, humans only synthesize cis (Z) FAs, while trans (E) FAs are not endogenously produced but present in the human body due to dietary intake. They are well known to play an important role in various physiological processes and therefore, the separation of cis/trans isomers is of great interest. The coelution of different lipid subclasses can lead to ion suppression, obscuring the detection of low-abundant lipids [15]. To address these issues, improved separation of lipid subclasses and lipid species together with the ability to distinguish and identify GSL isomers is essential and highly advantageous for the investigation of their physiological role and functions in nature and disease [6, 15].
Several approaches are used to study GSL structures in connection with MS, such as direct infusion (DI), liquid chromatography (LC), supercritical fluid chromatography (SFC), mass spectrometry imaging (MSI), and ion mobility (IM).
General workflows for the analysis of GSL
DI-MS
DI-MS (also termed shotgun lipidomics) is a technique when lipid extracts are directly introduced into the MS instrument without upfront separation [16]. The molecular characterization of lipid species relies either on the accurate m/z determination in the full scan mode or on the detection of specific fragmentation reactions in MS/MS experiments. In that case, the HRMS instruments are preferred due to their ability to differentiate isomeric and isobaric compounds [5, 17, 18]. Multi-dimensional MS-based shotgun lipidomics (MDMS-SL) allows the separation of many lipid (sub)classes through selective ionization of a certain category of lipids in the ion source (i.e., intra-source separation), even if the lipids are minor [16, 19, 20]. Although electrospray ionization (ESI) and MALDI are by far the most widely used ionization techniques in DI-MS, desorption electrospray ionization (DESI) [21], laser ablation electrospray ionization (LAESI) [22], and matrix-free laser desorption ionization (LDI) [23] have also been successfully applied. The major advantage of shotgun analysis is the reproducibility and relative high-throughput capability, allowing rapid acquisition of the full mass spectrum within seconds while providing similar sensitivity to LC/MS approaches, especially in coupling with nano-ESI [24]. On the contrary, the major drawbacks are the possible carry-over effect and susceptibility to ion suppression due to the presence of other major lipids or polar compounds (e.g., phospholipids, polar metabolites, and salts), which limits the ionization capacity and may even completely suppress the signals of minor or poorly ionizable GSL. Thorough sample preparation is required to ensure the removal of these interfering compounds. More detailed reviews on MS-based shotgun lipidomics can be read elsewhere [25, 26].
Sample preparation for shotgun lipidomics is very straightforward and generally involves simple extraction focusing on efficient total lipid extraction and minimal sample cleanup. The total lipid extract can be obtained commonly by chloroform/methanol-based liquid–liquid extraction such as of that Folch (CHCl3/CH3OH; 2:1, v/v) [27] and Bligh-Dyer (CHCl3/CH3OH; 1:2, v/v) [28]. In these protocols, lipids are partitioned into the lower chloroform phase. Polar solvent, such as methanol, ethanol, or ispropylalcohol, is used to increase the solubility of the lipids in the organic phase [29, 30]. These biphasic systems are able to recover wide range of lipids; however, sialylated and sulfated GSL or neutral GSL with at least four glycan residues mostly partition to the methanol-rich layer. On the contrary, neutral GSL with less than four glycan residues and other less polar lipids remain rather in the chloroform-rich layer. Thus, these methods do not provide effective recovery of the amphiphilic and highly polar GSL, as they generally require more aqueous portion [29, 31]. Over the years, several modifications and alternative strategies to these original protocols have been developed. One of them is the method described by Matysh et al. [17], which utilizes the mixture of methyl tert-butyl ether (MTBE) and methanol in ratio 10:3. This method was specifically developed for shotgun lipidomics of samples with excessive amounts of biological matrices. Furthermore, single-phase butanol-methanol (BUME) extraction system firstly described by Löfgren et al. (n-butanol/CH3OH; 3:1, v/v) [32, 33] and further modified by Alshehry et al. (n-butanol/CH3OH; 1:1, v/v) [34] has been reported to provide a similar yield of lipids compared to traditional Folch and Bligh-Dyer methods. Over the past years, monophasic extractions, also termed as protein precipitation methods, have gained popularity and have been applied to the simultaneous analysis of polar and non-polar lipids and other metabolites. The one-phase extraction methods are generally faster, cheaper, and less complex compared to conventional two-phase partition systems, and eliminate the risk of losses during transfer between phases; however, they do not allow the removal of polar and ionic impurities, leading to an increased risk of matrix effects and ion suppression. Thus, the application of one-phase extractions should be limited to polar lipid classes or should be followed by a sample cleanup using liquid–liquid extraction (LLE) and/or solid-phase extraction (SPE) [5, 35, 36]. The one-phase extraction is usually achieved through simultaneous protein precipitation with a variety of organic solvents including methanol, ethanol, acetonitrile, acetone, isopropanol, n-butanol, as well as their mixtures [35,36,37,38].
LC/MS
LC/MS is a key, well-established, and powerful analytical method used in lipidomics, which allows lipid subclass and/or lipid species separation when coupled to MS. The most frequently used ionization technique in LC/MS-based lipidomics is ESI, which is best suited for a wide range of lipids, including GSL due to several significant advantages including high sensitivity, easy coupling with chromatographic techniques, and structural details based on the use of tandem mass spectrometry (MS/MS) with high mass accuracy. In contrast, APCI and APPI are valuable alternatives for less polar lipids [5, 39]. The initial lipid extraction for the lipidomic analysis using LC–MS methods is similar to those described in the “DI-MS” section, i.e., using organic solvents. In contrast to sample preparation used for DI-MS, the sample preparation for LC–MS often requires more rigorous sample cleanup, including SPE to further purify the lipid extract by removing impurities and concentrating the lipids [30]. If needed, depletion of highly abundant lipids such as glycerolipids and phospholipids can be performed using alkaline hydrolysis [40] or special ZrO2/TiO2-based SPE method can be employed for removal of phospholipids to allow the analysis of low-abundant lipid species [41]. In addition, SPE or open column chromatography can be used to fractionate the lipid extract into subfractions [40].
MSI
MSI has become a popular and powerful method perfectly designed for the analysis of solid samples (e.g., tissues) with the ability to simultaneously display both spatial distribution and molecular level information. The most frequently used ionization technique employed in MSI is MALDI, but other ionization techniques could be employed as well, e.g., DESI, LAESI, or secondary ion mass spectrometry (SIMS) [42, 43]. The major advantages of MALDI are minimal sample preparation, high tolerance to salts, and the ability to relatively easily ionize heavily glycosylated GSL, but with lower ionization efficiency compared to ESI [29]. MALDI also has a few limitations, such as the inability to resolve isomers without prior separation and generally high background noise and ion suppression effects due to the formation of matrix clusters [29, 44].
The most widely used MSI technology applied for rapid in situ screening and mapping the spatial distribution of individual lipid species in biological samples is MALDI coupled to time-of-flight analyzers (MALDI-TOF). However, the comprehensive analysis is limited since the MSI is largely based on the qualitative comparison of healthy and diseased samples [45,46,47]. The coupling of MSI with Orbitrap or ion cyclotron resonance has provided deeper insight into the lipidomic complexity of biological samples [48], such as the application of MALDI-Orbitrap using MS/MS spectra to facilitate structural elucidation of even highly complex sulfo-GSL with up to five hexose moieties [49]. MSI techniques generally require minimal sample preparation. In MALDI, the tissue samples are first cryodissected into slices (~ µm), placed on a target surface, co-crystallized and immobilized with a suitable matrix, and then irradiated by laser to produce ions [29]. Common matrices used for GSL analysis include, for example, 2,5-dihydroxybenzoic acid, 1,5-diaminonaphtalene, 4-hydrazinobenzoic acid, 6-aza-2-thiothymine, 6,7-hydroxycoumarin (esculetin), and α-cyano-4-hydroxycinnamic acid [29, 50]. MALDI matrices used in lipidomics were also well-reviewed by Leopold et al. [51].
Ion mobility
In recent years, IM has appealed as a suitable technique for the separation of lipid isomers. However, due to the current resolving power limitations, lipid isomers cannot be fully resolved by IM alone in complex mixtures [6], which led to the interfacing of IM with LC/MS with great potential for the separation of lipid isomers together with increased selectivity and sensitivity [52].
Wojcik et al. [6] utilized ultrahigh-resolution IM separation with traveling waves in a serpentine and extended multipass SLIM platform for selected lipid and glycolipid isomers. They achieved partial separation of GlcSph 18:1;O2 vs. GalSph 18:1;O2 and GlcCer 18:1;O2/18:0 vs. GalCer 18:1;O2/18:0, differing only in the identity of glycan, which was achieved after four passes (~ 60 m path). Moreover, the baseline separation of GD1a and GD1b gangliosides, which only differ in the location of sialic acid residues, has been accomplished even with a minimal possible path of 1.25 m (i.e., without using multipass separation). However, the major issue is the limited number of passes due to increasing peak widths with the increasing number of passes, which reduces the detection window and range of mobilities. May et al. [53] also resolved GD1a and GD1b gangliosides in a standard mixture as doubly sodiated species [M + 2Na]2+ along with two pentasaccharide GSL differing in the location and linkage of fucose. Djambazova et al. [54] have reported a partial separation of GD1a and GD1b isomers with 36:1;O2 and 38:1;O2 ceramide in tissue samples using MALDI-TIMS. Xu et al. [55] have shown an effective resolution of GlcCer and GalCer species from human plasma and cerebrospinal fluids using differential mobility spectrometry coupled to LC/ESI–MS. Sample preparation for IM is similar to DI-MS or LC–MS.
In summary, although the separation and characterization of lipid isomers still remain very challenging and only a limited number of lipidomic studies have been carried out to differentiate GSL isomers using IM technologies, recent advances in chromatography and IM through novel instrumental developments have pushed the popularity of IM forward by enhancing its resolution and sensitivity. The coupling of IM with LC/MS is thus likely to become a very valuable tool capable of efficiently separating and reliably distinguishing various GSL isomers. However, further progress is still needed as potential applications of IS are still being discovered [56].
Structural elucidation of neutral GSL
The MS/MS analysis of GSL relies on various dissociation techniques. Each dissociation technique provides a distinct level of structural information since it cleaves bonds at different locations of the molecule. Their combination can provide complementary structural details of GSL [29].
Biological functions of GSL, as well as other lipids, highly depend on their varying expression levels and structural diversity, including carbon–carbon DB locations, cis/trans isomerism, and the sn-position of the fatty acyl chain(s), which complicate the structural elucidation. The MS/MS analysis is essential in the structural elucidation of GSL. The systematic nomenclature of MS/MS fragments for the carbohydrate part of GSL was proposed by Domon and Costello [57] and includes fragments containing the non-reducing end (i.e., A, B, and C) and the reducing end (i.e., X, Y, and Z). Fragments B, C, Y, and Z correspond to glycosidic cleavages that determine the glycan sequence (Fig. 2A), while A and X fragments are cross-ring cleavages allowing the differentiation of linkage positions (Fig. 2B). Since A, B, and C ions do not include ceramide structure, unlike their X, Y, and Z counterparts, their masses are not affected by the lipid moiety. The nomenclature was later updated by Ann and Adams [58] in order to include more detailed ceramide fragments. The major fragments are shown in Fig. 2C, where the NI and NII fragments are diagnostic ions of the long-chain base of the ceramide moiety. It implies that the fragmentation pathways of GSL can be predicted and structural databases can be constructed in silico to identify GSL by matching MS/MS spectra with the database, which is an indisputable advantage in the structural analysis of GSL.

Neutral GSL are relatively poorly ionized in the negative ion mode due to their basic (i.e., amino glycan-containing) and acidic counterparts [59]; thus, neutral GSL are commonly analyzed in the positive ion mode, where GSL are better ionized and provide abundant Y/Z-pair ion series indicative of sequence information accompanied by less common B/C-type fragments. A/X-type ions involving C–C bond usually require higher energies [60].
Simple neutral GSL (up to 4 monosaccharide units)
Collision-mediated dissociation is a conventional dissociation technique employed for MS/MS experiments. Both low-energy collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) provide structural information for elucidating the glycan sequence in the positive ion mode (i.e., sequential cleavage of a monosaccharide unit represented by a series of Z/Y-type ions) or in the negative ion mode (i.e., B- and C-type ions) and ceramide moiety of GSL. An alternative implemented exclusively for ion trap MS is pulsed Q dissociation (PQD), which deposits higher energies on the ions compared to CID and allows the observation of low m/z fragments that are usually excluded from CID, however at the cost of reduced fragmentation efficiency [29, 61, 62].
Incremental Δm/z indicates the loss of a hexose (Δm/z 162) and N-acetlyhexosamine (Δm/z 204) [29]. In addition, the ceramide composition, respectively, sphingoid bases, can be identified from the specific fragment ions in the positive mode (Table 1) [62].
General fragmentation patterns of simple neutral GSL using CID have previously been extensively studied [62,63,64], as illustrated by examples in Figs. 3 and 4.

Examples of fragmentation behavior of (A) HexCer and (B) Hex2Cer. MS spectra of (C) HexCer and (D) Hex2Cer, and MS2 spectra of (E) HexCer 34:1;O2 and (F) Hex2Cer 34:1;O2 with characteristic fragment ions obtained in human plasma analyzed by HILIC/ESI–MS/MS

Examples of fragmentation behavior of (A) Gb3Cer and (B) Gb4Cer, MS spectra of (C) Gb3Cer and (D) Gb4Cer, MS2 spectra of (E) Gb3Cer 34:1;O2 and (F) Gb4Cer 34:1;O2. MS3 spectra of the respective [Z0]+ ion of (E) Gb3Cer and (F) Gb4Cer together with characteristic fragment ions obtained in human plasma analyzed by HILIC/ESI–MS/MS
To distinguish isomers with subtle structural differences, multistage collisional dissociation has to be usually used, even when coupled with the chromatographic separation. Li et al. [65] showed that MSn of permethylated GSL allowed the differentiation of Gb4Cer (specific ion at m/z 315) and iGb4Cer (specific ion at m/z 357).
Whereas collision-mediated dissociation methods mainly induce glycosidic bond cleavage, ultraviolet photodissociation (UVPD) yields extensive fragmentation patterns of GSL to study neutral and acidic GSL [66], which provides more informative cross-ring fragments of the glycan moieties (A/X-type) and several unique UVPD-specific cleavages at ceramide C–C and C–N bonds that allow differentiation between isomeric glycans and ceramides. In particular, UVPD can reliably locate C = C bonds in the ceramide moieties of GSL. Despite the advantages, UVPD is not widely used due to the lack of automated software tools for spectra annotation; however, there are a few lipidomics applications [61, 66, 67]. For instance, Ryan et al. [68] have reported that 193-nm UV irradiation leads to the extensive fragmentation of both glycan and ceramide parts in neutral GSL and gangliosides along with a certain level of fragmentation close to carbon–carbon double bonds (DB). The analysis of C = C bond location by UVPD is limited due to the low fragmentation efficiency caused by low photon absorption by C = C bond; therefore, a high-power laser or incorporation of more efficient chromophores is needed to improve the sensitivity to locate C = C bonds [69].
In addition, Pham et al. [70] demonstrated the discrimination of GSL epimers, namely, GlcCer/GalCer and GlcSph/GalSph, using a unique fragmentation technique called radical-directed dissociation. The discrimination was based on the inverted abundance of the major fragments caused by the differential ability of GlcCer and GalCer to lose water (i.e., neutral loss of water is easier in Gal than in Glc). However, the application of radical-directed dissociation in the GSL analysis is very limited, mainly due to the complicated sample preparation.
Complex neutral GSL (more than 4 monosaccharide units)
The analysis of complex and heavily glycosylated GSL poses an additional challenge as they suffer from poor ionization efficiency and multistage fragmentation mass spectrometry (MSn) is usually required to achieve step-by-step cleavage of the glycosidic bonds and ceramide backbones. It is known that ESI sensitivity decreases with the increasing length of the glycan chain of GSL due to increased hydrophilicity [59, 71].
Furthermore, a large majority of GSL share Gal–Glc disaccharide core linked to the ceramide moiety, which may complicate the product spectral analysis because the Y0/Z0, Y1/Z1, and Y2/Z2 fragments of different GSL species may have the same mass, despite their intensities vary among GSL species [60]. GSL containing 2-hydroxy fatty acyl groups are also typically characterized by the abundant fragment ion derived from the loss of the hydroxy-acyl group [72].
In some cases, the analysis of underivatized GSL-derived oligosaccharides in the negative ion mode may be advantageous for isomer recognition due to low chemical background noise and low level of cation adduct formation [73, 74]. Negative ion MS/MS spectra of oligosaccharides are generally dominated by a series of B/C-type ions providing information about the glycan sequence. More interestingly, C1 ion can provide additional information about the terminal Gal linked to GlcNAc. The fragmentation of Gal1–4GlcNAc linkage is more facile and thus can be readily cleaved, while Gal1–3GlcNAc linkage is more resistant. However, when Gal is substituted with Fuc, the 1–3 linkage can be cleaved (Table 2) [73].
Generally, cross-ring fragmentation (i.e., A-type ions) is useful for defining the linkage positions between individual monosaccharides [74]. The cross-ring 0,2A-type cleavage is typical for 4-linked GlcNAc or Glc (i.e., type 2 chain), whereas it is not produced for 3-linked GlcNAc (i.e., type 1 chain). It can be used to discriminate between Gb and iGb. In addition, the double glycosidic D-type cleavage (i.e., C–Z double cleavage) is unique to non-substituted 3-linked GlcNAc/Glc or 4-linked GlcNAc/Glc substituted with Fuc (Table 3). Similarly, D-type ions indicate 3-linked GlcNAc substituted with Gal at the 4-position. Taken together, A- and D-type ions are important for the differentiation of type 1/2 chain, and blood group H, Lea, Leb, Lex, and Ley determinants [73].
In case of complex branched GSL-derived oligosaccharides, MS/MS spectra can provide complementary structural information. In MS/MS spectra of [M–H]–, fragment ions derived from 6-linked branches (α) are dominant, while those from 3-linked branches (β) are absent. In contrast, fragment ions from both branches are dominant in MS/MS spectra of [M–2H]2–. Similarly, double cleavage (D-type ion) occurs only at 3-linked branches (Fig. 5) [74].

Scheme of 3- and 6-branched oligosaccharides with typical fragment ions [74]
In-depth description of the MS/MS of oligosaccharides can be found in this review [59]. The differentiation of blood groups A, B, and H, and Lewis blood group Lea, Leb, Lex, and Ley determinants on GSL-derived oligosaccharides has been described in detail [75, 76].
Structural elucidation of acidic GSL
In contrast, acidic GSL (i.e., sulfatides and gangliosides) are broadly analyzed in the negative ion mode since the molecule is readily ionizable due to anionic sulfate groups or sialic acid residues [77]. However, the application of positive ion mode is also possible [60]. GSL with acidic residues usually face in-source or post-source fragmentation, or produce metastable ions, especially when MALDI-MS is used [71, 72].
Sulfatides
Sulfatides provide a prominent diagnostic ion at m/z 97 (i.e., HSO4− ion) [29], which is, however, not observed in ion trap MS because of the low mass cutoff, together with B/C-type fragment ions reflecting the 3-sulfoGal (m/z 259 and 241, for SHexCer) and 3-sulfoGal–Glc (m/z 419 and 403, for SHex2Cer) residues accompanied by the dehydration. Furthermore, the differentiation of non-hydroxylated and hydroxylated sulfatides has been well documented. The α-hydroxylated fatty acid-containing sulfatides are recognized by the unique and prominent ion cluster originated from the primary cleavage of the fatty acyl CO–CH(OH) bond (ion a) accompanied by the direct loss of fatty acyl as a ketene from the precursor ion via the NH–CO bond cleavage (ion b), which further undergoes the water loss (ion c) [77,78,79]. The difference between MS/MS spectra of non-hydroxylated and hydroxylated sulfatides is illustrated in Fig. 6.

Characteristic fragmentation of non-hydroxylated and hydroxylated sulfatides in negative ion MS/MS [78]
Gangliosides
In general, gangliosides may be multiply charged, e.g., [M-H]−, [M-2H]2−, and [M-3H]3−, depending primarily on the number of sialic acids (NeuAc) and on the pH value, which influences the dissociation states [80]. Gangliosides have been considered difficult to analyze as sialic acid is relatively labile and preferentially lost during the ionization process. The neutral loss of sialic acid (m/z 290) [29] or additionally acetylated sialic acid (m/z 332) is favored when two or more sialic acids are present, but the stability can be improved by the formation of metal ions (i.e., [M–2H + Na]−). This is primarily a typical feature of MALDI-MS [72, 81]. The presence of two or more adjacent sialic acids is identified by fragment ions at m/z 581 (i.e., NeuAc–NeuAc), m/z 623 (i.e., NeuAc–NeuAc2), or m/z 914 (i.e., NeuAc–NeuAc–NeuAc2). This predictable fragmentation behavior allows sequencing of the oligosaccharide part of gangliosides and therefore, allows the distinction of the type of branches, i.e., a-, b-, and c-series gangliosides [80]. Generally, sialic acid glycoconjugates can be linked to Gal via 2–3/6 linkages and to GalNAc via 2–6 linkage. There are also α-/β-anomers of sialic acid with α-anomeric form being the most common, while β-anomeric form is typical for free sialic acid. It is also unclear if the β-anomer is present in oligosaccharide chains or ignored/escaped from detection due to low concentration. The linkage positions and anomeric configurations of sialic acid are reflected in the stability of the molecule during the ionization: β2–3 > α2–6 > α2–3 > β2–6 as well as in the characteristic fragmentation patterns in MS/MS spectra (Table 4) [81].
Novel dissociation techniques for structural analysis of GSL
Nowadays, collision-induced dissociation (CID) MS/MS is a well-established dissociation technique providing the head group and ceramide composition. However, the ceramide backbones of GSL as well as other lipids may contain unsaturated FA with one or more carbon–carbon DB (C = C), which cannot be located using collision-mediated techniques alone. Since the structural elucidation of C = C bond location, cis/trans isomers, and sn-position isomers has recently become a hot topic, several novel and selective ion activation technologies have been developed to address this issue [82, 83].
Paternò-Büchi reaction
In 2014, Ma and Xia [84] introduced a novel method of pinpointing C = C bond locations by online coupling of the Paternò-Büchi (PB) reaction with MS/MS. The reaction is fast and highly specific, providing highly abundant diagnostic ions enabling confident localization of C = C bonds [85]. This strategy has also been applied for the large-scale analysis of C = C location isomers of different lipid subclasses in a variety of biological samples; however, it has a potential for the location of C = C bonds in ceramide moieties of GSL as well [86, 87]. Additionally, Bednařík et al. [88] developed an on-tissue PB reaction for the localization of C = C bonds in phospholipids and glycolipids by MALDI-MS using benzaldehyde.
Ozone-induced dissociation
Ozone-induced dissociation (OzID) was firstly implemented in 2008 by Blanksby’s group, who replaced inert collision gas in the mass spectrometer with O3/O2 mixture so that the ozonolysis can occur inside the collision cell [89]. OzID is a gas-phase reaction based on cycloaddition of O3 to unsaturated lipids generating metastable ozonide that spontaneously decays to more stable Criegee and aldehyde diagnostic ions with constant mass differences of 16 m/z. When coupled to soft ionization techniques, the method is highly specific and efficient for assignment not only the position of C = C bonds, but also their stereochemistry in unsaturated lipids [69, 90]. The major drawbacks are the need for modification of commercial instruments and specialized equipment generating ozone together with the longer reaction time (0.2–10 s) needed to accumulate detectable diagnostic ions due to the low ozone density allowed in the ion trap analyzer [85]. This reduces the analysis speed and makes the coupling with HPLC inefficient, which greatly restricts the application of OzID. To circumvent this limitation, OzID has been implemented in a high-pressure IM cell with high O3 density, significantly accelerating ozonolysis and producing abundant C = C fragment ions [91], which also facilitated a coupling with LC, a novel platform for the analysis of isomers [92].
For instance, Barrientos and coworkers have demonstrated the structural analysis of sodiated adduct ions of unsaturated GSL using OzID-MS that yielded more informative cross-ring cleavages compared to protonated ions [93]. Later, they reported that adducts can remarkably influence both the bond cleavage and the fragmentation behavior and illustrated distinct fragmentation patterns of [M + H]+, [M + Na]+, and [M + Li]+ precursor ions of GSL in OzID-MS. Specifically, they reported that [M + H]+ ion primarily undergoes dehydration yielding [M + H-H2O]+ ions followed by the sequential loss of monosaccharide units, while [M + Na]+ and [M + Li]+ adducts dissociate preferably at the DB yielding similar fragmentation patterns, albeit the relative intensities of diagnostic ions were remarkably different [94].
Epoxidation
Recently, several innovative epoxidation reactions have been developed. Although the majority of them have been used for the identification of C = C bonds in other lipids than GSL. These methods can readily be applied for the differentiation of GSL isomers containing unsaturated fatty acyls as well. For example, the epoxidation using metachloroperoxybenzoic acid (m-CPBA) [95] and peracetic acid (PAA) [96] has been used for large-scale identification and spatial mapping of biological C = C isomers. Epoxidation is initiated by the oxidation of unsaturated lipids via m-CPBA or PAA either in solution or on-tissue reaction to generate an epoxide product, which is further subjected to CID-MS/MS analysis generating a pair of diagnostic ions pinpointing the location of C = C bond [96]. Specifically, the m-CPBA epoxidation is completed within minutes and without overoxidized by-products, showing the potential for high-throughput analysis [97] and making the epoxidation a versatile and user-friendly platform with minimal requirements for instrumentation [95]. Recently, Zhang et al. [98] proposed a rapid light-controlled photoepoxidation using benzoin to locate the positions of C = C in various isomers of unsaturated lipids in mouse tissue extracts, in both positive and negative ion modes. Moreover, the rapid switch-on/off electrochemical epoxidation controlled simply by tuning the electrospray voltage was recently developed to locate the C = C bond positions in lipids [99]. Furthermore, chloroauric acid (HAuCl4)-doped solvent introduced into an electrospray has been reported to induce epoxidation [100]. Next, low-temperature plasma (LTP)-induced epoxidation using atmospheric oxygen can also be used for the assignment of C = C bonds. The reaction is performed by blowing the LTP into the mixture of lipids in acetone/water (50/50, v/v) followed by rapid and nearly complete conversion of C = C bonds to the corresponding epoxides [101]. A novel method based on ambient oxidation coupled with air flow-assisted desorption electrospray ionization (AFADESI) was proposed to conveniently and rapidly characterize the spatial distribution of unsaturated lipid isomers using MSI technology [102]. A novel, online, and selective photosensitized oxidation of C = C bonds induced by singlet oxygen (1O2) coupled to CID-MS/MS has been proposed to distinguish positional isomers based on the formation of unique lipid hydroperoxide products (neutral losses) generated promptly after laser irradiation. Characteristic neutral losses arise from cleavage at the location of the hydroperoxide group of the respective lipid hydroperoxide, which was produced by the interaction of unsaturated lipids with 1O2 [103]. Various approaches used for the location of C = C bonds are summarized in Fig. 7.

In summary, CID commonly generates diagnostic ions of the glycan chain, sphingoid base, or N-fatty acyls composition, while more detailed structural information including branching and linkages in the glycan sequence and DB positions in the ceramide backbone can rather be determined using more specialized dissociation techniques (i.e., ECD, EDD, ETD, UVPD, RDD, OzID, 1O2, PB reaction, or epoxidation) [29]. Among the above-mentioned strategies, the PB reaction is the most commonly used method to locate C = C positions in both DI-MS/MS and HPLC/MS/MS workflows, although OzID and epoxidation provide much higher specificity. A more detailed description of the applications of AIMS techniques utilizing novel ion activation methods in the elucidation of unsaturated lipids can be found in the following reviews [69, 82]. Different levels of structural characterization of GSL obtained by different methods are summarized in Fig. 8.

Diagram illustrating the different levels of structural characterization of GSL (modified from [69]). The sn-position level is not used for GSL structures as it is more pronounced in glycolipids and glycerophospholipids. The identified structural levels are named according to Liebisch et al. [124]. Legend: OzID, ozone-induced dissociation; RDD, radical-directed dissociation; MAD, metastable atom-activated dissociation; UVPD, ultraviolet photodissociation; EIEIO, electron impact excitation of ions from organics; CRF, charge-remote fragmentation; CACI, covalent-adduct chemical ionization; IR, infrared spectroscopy
Deciphering GSL structures using chemical derivatization
In addition to MS-based techniques, the chemical derivatization of specific functional groups has a great potential to overcome the analytical barriers that still exist in the differentiation of isomeric and/or isobaric GSL [104]. The derivatization can increase the ionization efficiency of neutral and heavily glycosylated GSL that suffer from low ionization. The derivatization in combination with UHPLC/MS or IM may also allow the separation of isomers by providing highly abundant diagnostic ions alongside a few by-product ions together with the increased sensitivity [105]. Besides decreasing the risk of false identification due to the presence of specific fragments, the derivatization can pose some challenges, e.g., possible contamination or formation of artifacts [36]. Various derivatization approaches for the structural elucidation of GSL isomers are discussed in more detail below.
Permethylation
MS/MS of permethylated GSL, where the active protons in –OH and –NH2 functional groups of GSL react with methyl iodide in alkaline solution (NaOH) of dimethylsulfoxide, is a powerful tool for rigorous glycan sequence and linkage determination, and ceramide composition due to the formation of specific fragments. However, the data analysis of permethylated GSL may be complicated since GSL may have different numbers of reaction sites. Permethylation can be carried out using both natural and isotope-labelled methyl iodide (i.e., 12CH3I, 13CH3I, or 12CD3I) [29]. When combined, the so-called differential isotope labelling technique is very useful in the quantitation of intact GSL, resulting in up to 20-fold signal enhancement compared to their low sensitivity in the presence of total lipid extracts. It has also been observed that alkaline conditions during permethylation significantly reduced ion-suppressing ester-linked lipids, thus encouraging the usefulness of permethylation for the quantitation of low-abundant GSL in complex mixtures [106]. On the contrary, the permethylation in NaOH/DMSO is not suitable for pH-sensitive functional groups of carbohydrates (e.g., O-acetylation) or GSL with polysialic acids, as these may be destroyed under such chemical conditions [31]. The use of permethylation in the structural analysis of GSL in the positive ion mode has already been demonstrated [107]. In addition, Ejsing et al. reported a shogun-based method for quantitative profiling of long-chain sphingoid base metabolites by using 12CD3I, which significantly enhanced the sensitivity [108].
Sialic acid-related derivatization strategies
Acidic GSL, such as gangliosides, include a large number of sialyl-linked glycan isomers with α2,3-, α2,6-, and α2,8-linked polysialic acids typically on the non-reducing ends of glycans [109]. Sialylated GSL are commonly analyzed in the negative ion mode, while they are hard to analyze in the positive ion mode due to their poor ionization efficiency [110]. It should also be stressed that MS cannot fully supply the glycan sequence since it has been well documented that various sialylated oligosaccharides lose sialic acid during in-source and post-source dissociation, decreasing the sensitivity of the molecular ion and dominantly yielding product ions lacking sialic acid, representing a major problem in MALDI-MS analysis of sialylated GSL [111, 112].
The distinct chemical derivatization methods of sialic acids, e.g., esterification [112], amidation [111], permethylation [113], or perbenzoylation [114], have been developed to stabilize the sialylated residues and to improve ionization efficiency of acidic GSL in the positive ion mode. This modification allows highly sensitive and simultaneous identification of both neutral and acidic GSL without ion mode switching, especially for gangliosides with more sialic acids [110]. Hanamatsu et al. [109] developed a method based on ring-opening aminolysis for discrimination between α2,3- and α2,8-linked sialic acid isomers of GSL-glycan by MS. Liu et al. [110] labelled the carboxyl group of sialic acid with an easily ionizable tertiary amine, i.e., N,N-dimethylethylenediamine (DMEN), which enhanced the ionization more than fourfold and provided a diagnostic ion to facilitate rapid structural assignments of gangliosides and discrimination of isomers. This strategy was successfully applied for the simultaneous identification of neutral and acidic GSL in human plasma. Moreover, the labelling approach using 2-(2-pyridilamino)-ethylamine (PAEA) has been developed by Huang’s group. The method has been applied to plasma using MRM-based LC/MS/MS method, which resulted in 15-fold enhancement of ionization compared to underivatized analogs. However, the derivatization efficiency of PAEA approach was not sufficient; thus, only monosialogangliosides (i.e., GM1, GM2, and GM3) were analyzed [115].
Isobaric labelling
Isobaric labelling is a relatively novel technique in glycosphingolipidomics, where samples are labelled with isobaric tags consisting of the same molecule with varying placement of isotopes (e.g., 13C and 15N). To date, two versions of commercially available isobaric tags have been applied to GSL, namely, iTRAQ™ (i.e., isobaric tag for relative and absolute quantitation) reacting with free amines [116] and aminoxyTMT™ (aminoxy tandem mass tag) [117] reacting with aldehyde and ketones.
Other derivatization strategies
There are also other derivatization strategies that can be used for GSL analysis. Peterka et al. [118] introduced highly reproducible derivatization of multiple lipid subclasses, including even simple GSL, using non-hazardous benzoyl chloride. Zheng et al. [119] developed a highly sensitive method for simultaneous analysis of multiple sphingoid bases using 3-(N,N-dimethylamino)propyl isothiocyanate (DMPI) and its isotopically labelled counterpart (d4-DMPI). A few other chemical strategies can be utilized for the elucidation of GSL structures, such as diluted sodium periodide oxidizing an OH group of sialic acid to aldehyde [120], 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) that transforms an OH group of a sphingosine into allyl ketone [121], or method called oxidative release of the neutral glycans (ORNG) by sodium hypochlorite converting a glycosidic bond of a ceramide to nitrile [122].
Conclusions and outlook
GSL represent a diverse class of biomolecules that play crucial roles in biological processes and disease pathogenesis. Their identification and structural elucidation are pivotal for understanding their functions and metabolic pathways. MS has proven to be a powerful tool for GSL analysis due to its sensitivity and ability to provide detailed structural information on glycan sequences, linkage positions, and fatty acid composition. Although current MS approaches provide valuable insights into GSL structures, they often lack sufficient resolution to distinguish isomeric species with subtle structural differences. Moreover, the localization of carbon–carbon DB in the ceramide backbone poses a significant analytical challenge, as conventional dissociation techniques, such as the predominantly used CID, may not provide definitive information on the bond positions and their geometry. To address this issue, novel ion activation techniques, such as the Paternò-Büchi reaction, ozone-induced dissociation, or various types of epoxidation, have been developed and have shown great promise in locating carbon–carbon DB and determining their cis/trans geometry in GSL ceramides, thus overcoming a long-standing challenge in GSL analysis. In addition, other dissociation techniques, such as electron-mediated, photon-mediated, or radical-directed dissociation, may provide complementary insights into GSL structures, despite being less used. Another major limitation is the poor detection and characterization of highly complex and low-abundance GSL, which is hindered by their low ionization efficiency and incomplete or low extraction efficiency by classical extraction methods, as these GSL are more hydrophilic, thus restricting the discovery of novel GSL species. Looking ahead, further advances in MS instrumentation and ion mobility are expected to improve the sensitivity, selectivity, and throughput of GSL analysis. This will enable a comprehensive characterization of GSLs and their isomers in complex biological samples, paving the way for a deeper understanding of their role in health and disease and unlocking the full potential of GSL analysis to advance biomedical research.
References
Merrill AH, Wang MD, Park M, Sullards MC. (Glyco)sphingolipidology: an amazing challenge and opportunity for systems biology. Trends Biochem Sci. 2007;32:457–68. https://doi.org/10.1016/j.tibs.2007.09.004.
Zhang X, Kiechle FL. Review: Glycosphingolipids in Health and Disease. Ann Clin Lab Sci. 2004;34:3–13.
Senn HJ, Orth M, Fitzke E, Wieland H, Gerok W. Gangliosides in normal human serum concentration, pattern and transport by lipoproteins. Eur J Biochem. 1989;181(3):657–62. https://doi.org/10.1111/j.1432-1033.1989.tb14773.x.
Han X. Lipidomics: Comprehensive mass spectrometry of lipids, 1st ed. New Jersey, USA: John Wiley & Sons; 2016.
Holčapek M, Liebisch G, Ekroos K. Lipidomic Analysis. Anal Chem. 2018;90:4249–57. https://doi.org/10.1021/acs.analchem.7b05395.
Wojcik R, Webb IK, Deng L, Garimella SVB, Prost SA, Ibrahim YM, Baker ES, Smith RD. Lipid and glycolipid isomer analyses using ultra-high resolution ion mobility spectrometry separations. Int J Mol Sci. 2017;18:1–12. https://doi.org/10.3390/ijms18010183.
Merrill AH, Sullards MC. Opinion article on lipidomics: Inherent challenges of lipidomic analysis of sphingolipids. Biochim Biophys Acta - Mol Cell Biol Lipids. 2017;1862:774–6. https://doi.org/10.1016/j.bbalip.2017.01.009.
Reza S, Ugorski M, Suchański J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology. 2021;31:1416–34. https://doi.org/10.1093/glycob/cwab046.
Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–625. https://doi.org/10.1093/brain/awf272.
Kain L, Webb B, Anderson BL, Deng S, Holt M, Constanzo A, Zhao M, Self K, Teyton A, Everett C, et al. The Identification of the Endogenous Ligands of Natural Killer T Cells Reveals the Presence of Mammalian α-Linked Glycosylceramides. Immunity. 2014;41:543–54. https://doi.org/10.1016/j.immuni.2014.08.017.
Von Gerichten J, Schlosser K, Lamprecht D, Morace I, Eckhardt M, Wachten D, Jennemann R, Gröne HJ, Mack M, Sandhoff R. Diastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals. J Lipid Res. 2017;58:1247–58. https://doi.org/10.1194/jlr.D076190.
Duan J, Merrill AH. 1-deoxysphingolipids encountered exogenously and made de novo: Dangerous mysteries inside an enigma. J Biol Chem. 2015;290:15380–9. https://doi.org/10.1074/jbc.R115.658823.
Shayman JA, Abe A, Hiraoka M. A turn in the road: How studies on the pharmacology of glucosylceramide synthase inhibitors led to the identification of a lysosomal phospholipase A2 with ceramide transacylase activity. Glycoconj J. 2003;20:25–32. https://doi.org/10.1023/B:GLYC.0000016739.32089.55.
Sandhoff R. Very long chain sphingolipids: Tissue expression, function and synthesis. FEBS Lett. 2010;584:1907–13. https://doi.org/10.1016/j.febslet.2009.12.032.
Damen CWN, Isaac G, Langridge J, Hankemeier T, Vreeken RJ. Enhanced lipid isomer separation in human plasma using reversed-phase UPLC with ion-mobility/high-resolution MS detection. J Lipid Res. 2014;55:1772–83. https://doi.org/10.1194/jlr.D047795.
Hu C, Wang C, He L, Han X. Novel strategies for enhancing shotgun lipidomics for comprehensive analysis of cellular lipidomes. TrAC - Trends Anal Chem 2019;120, https://doi.org/10.1016/j.trac.2018.11.028.
Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl-terf-butyl ether for high-throughput lipidomics. J Lipid Res. 2008;49:1137–46. https://doi.org/10.1194/jlr.D700041-JLR200.
Ståhlman M, Ejsing CS, Tarasov K, Perman J, Borén J, Ekroos K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2009;877:2664–72. https://doi.org/10.1016/j.jchromb.2009.02.037.
Han X. Multi-dimensional mass spectrometry-based shotgun lipidomics and the altered lipids at the mild cognitive impairment stage of Alzheimer’s disease. Biochim Biophys Acta - Mol Cell Biol Lipids. 2010;1801:774–83. https://doi.org/10.1016/j.bbalip.2010.01.010.
Yang K, Zhao Z, Gross RW, Han X. Systematic analysis of choline-containing phospholipids using multi-dimensional mass spectrometry-based shotgun lipidomics. J Chromatogr B Anal Technol Biomed Life Sci. 2009;877:2924–36. https://doi.org/10.1016/j.jchromb.2009.01.016.
Manicke NE, Wiseman JM, Ifa DR, Cooks RG. Desorption Electrospray Ionization (DESI) Mass Spectrometry and Tandem Mass Spectrometry (MS/MS) of Phospholipids and Sphingolipids: Ionization, Adduct Formation, and Fragmentation. J Am Soc Mass Spectrom. 2008;19:531–43. https://doi.org/10.1016/j.jasms.2007.12.003.
Nemes P, Woods AS, Vertes A. Simultaneous imaging of small metabolites and lipids in rat brain tissues at atmospheric pressure by laser ablation electrospray ionization mass spectrometry. Anal Chem. 2010;82:982–8. https://doi.org/10.1021/ac902245p.
Muck A, Stelzner T, Hübner U, Christiansen S, Svatoš A. Lithographically patterned silicon nanowire arrays for matrix free LDI-TOF/MS analysis of lipids. Lab Chip. 2010;10:320–5. https://doi.org/10.1039/b913212k.
Han X, Yang K, Gross RW. Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high-throughput platform f. Rapid Commun Mass Spectrom. 2008;22:2115–24. https://doi.org/10.1002/rcm.3595Microfluidics-based.
Hsu FF. Mass spectrometry-based shotgun lipidomics – a critical review from the technical point of view. Anal Bioanal Chem. 2018;410:6387–409. https://doi.org/10.1007/s00216-018-1252-y.
Wang J, Han X. Analytical challenges of shotgun lipidomics at different resolution of measurements. TrAC - Trends Anal Chem. 2019;121:115697. https://doi.org/10.1016/j.trac.2019.115697.
Folch J, Lees M, Sloane Stanley GHA. simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. https://doi.org/10.1016/s0021-9258(18)64849-5.
Bligh EG, Dyer WJ. A Rapid Method of Total Lipid Extraction and Purification. Can J Biochem Physiol. 1959;37:911–7. https://doi.org/10.1139/o59-099.
Barrientos RC, Zhang Q. Recent advances in the mass spectrometric analysis of glycosphingolipidome – A review. Anal Chim Acta. 2020;1132:134–55. https://doi.org/10.1016/j.aca.2020.05.051.
Saini RK, Prasad P, Shang X, Keum YS. Advances in lipid extraction methods—a review. Int J Mol Sci. 2021;22:1–19. https://doi.org/10.3390/ijms222413643.
Smith DF, Prieto PA. Special Considerations for Glycolipids and Their Purification. Curr Protoc Mol Biol. 1993;22:1–13. https://doi.org/10.1002/0471142727.mb1703s22.
Löfgren L, Ståhlman M, Forsberg GB, Saarinen S, Nilsson R, Hansson GI. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J Lipid Res. 2012;53:1690–700. https://doi.org/10.1194/jlr.D023036.
Löfgren L, Forsberg GB. Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci Rep 2016;6, https://doi.org/10.1038/srep27688.
Alshehry ZH, Barlow CK, Weir JM, Zhou Y, McConville MJ, Meikle PJ. An efficient single phase method for the extraction of plasma lipids. Metabolites. 2015;5:389–403. https://doi.org/10.3390/metabo5020389.
Höring M, Stieglmeier C, Schnabel K, Hallmark T, Ekroos K, Burkhardt R, Liebisch G. Benchmarking One-Phase Lipid Extractions for Plasma Lipidomics. Anal Chem. 2022;94:12292–6. https://doi.org/10.1021/acs.analchem.2c02117.
Jurowski K, Kochan K, Walczak J, Barańska M, Piekoszewski W, Buszewski B. Comprehensive review of trends and analytical strategies applied for biological samples preparation and storage in modern medical lipidomics: State of the art. TrAC - Trends Anal Chem. 2017;86:276–89. https://doi.org/10.1016/j.trac.2016.10.014.
Wong MWK, Braidy N, Pickford R, Sachdev PS, Poljak A. Comparison of single phase and biphasic extraction protocols for lipidomic studies using human plasma. Front Neurol. 2019;10:1–11. https://doi.org/10.3389/fneur.2019.00879.
Sarafian MH, Gaudin M, Lewis MR, Martin FP, Holmes E, Nicholson JK, Dumas ME. Objective set of criteria for optimization of sample preparation procedures for ultra-high throughput untargeted blood plasma lipid profiling by ultra performance liquid chromatography-mass spectrometry. Anal Chem. 2014;86:5766–74. https://doi.org/10.1021/ac500317c.
Wu Z, Bagarolo GI, Thoröe-Boveleth S, Jankowski J. “Lipidomics”: Mass spectrometric and chemometric analyses of lipids. Adv Drug Deliv Rev. 2020;159:294–307. https://doi.org/10.1016/j.addr.2020.06.009.
Karlsson K-A. Preparation of Total Nonacid Glycolipids for Overlay Analysis of Receptors for Bacteria and Viruses and for Other Studies. Methods Enzymol. 1987;138:212–20. https://doi.org/10.1016/0076-6879(87)38018-8.
Song Z, Duan C, Shi M, Li S, Guan Y. One-step preparation of ZrO2/SiO2 microspheres and modification with D-fructose 1,6-bisphosphate as stationary phase for hydrophilic interaction chromatography. J Chromatogr A. 2017;1522:30–7. https://doi.org/10.1016/j.chroma.2017.09.046.
Ge P, Luo Y, Chen H, Liu J, Guo H, Xu C, Qu J, Zhang G, Chen H. Application of Mass Spectrometry in Pancreatic Cancer Translational Research. Front Oncol. 2021;11:1–16. https://doi.org/10.3389/fonc.2021.667427.
Teo CC, Chong WPK, Tan E, Basri NB, Low ZJ, Ho YS. Advances in sample preparation and analytical techniques for lipidomics study of clinical samples. TrAC - Trends Anal Chem. 2015;66:1–18. https://doi.org/10.1016/j.trac.2014.10.010.
Li M, Zhou Z, Nie H, Bai Y, Liu H. Recent advances of chromatography and mass spectrometry in lipidomics. Anal Bioanal Chem. 2011;399:243–9. https://doi.org/10.1007/s00216-010-4327-y.
Torretta E, Fania C, Vasso M, Gelfi C. HPTLC-MALDI MS for (glyco)sphingolipid multiplexing in tissues and blood: A promising strategy for biomarker discovery and clinical applications. Electrophoresis. 2016;37:2036–49. https://doi.org/10.1002/elps.201600094.
Furukawa JI, Sakai S, Yokota I, Okada K, Hanamatsu H, Kobayashi T, Yoshida Y, Higashino K, Tamura T, Igarashi Y, et al. Quantitative GSL-glycome analysis of human whole serum based on an EGCase digestion and glycoblotting method. J Lipid Res. 2015;56:2399–407. https://doi.org/10.1194/jlr.D062083.
Norris JL, Caprioli RM. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem Rev. 2013;113:2309–42. https://doi.org/10.1021/cr3004295.
Ellis SR, Paine MRL, Eijkel GB, Pauling JK, Husen P, Jervelund MW, Hermansson M, Ejsing CS, Heeren RMA. Automated, parallel mass spectrometry imaging and structural identification of lipids. Nat Methods. 2018;15:515–8. https://doi.org/10.1038/s41592-018-0010-6.
Jirásko R, Holčapek M, Khalikova M, Vrána D, Študent V, Prouzová Z, Melichar B. MALDI Orbitrap Mass Spectrometry Profiling of Dysregulated Sulfoglycosphingolipids in Renal Cell Carcinoma Tissues. J Am Soc Mass Spectrom. 2017;28:1562–74. https://doi.org/10.1007/s13361-017-1644-9.
Harvey DJ. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Carbohydrates. Mass Spectrom Rev. 1999;18:349–451. https://doi.org/10.1002/(sici)1098-2787(1999)18:6%3c349::aid-mas1%3e3.0.co;2-h.
Leopold J, Popkova Y, Engel KM, Schiller J. Recent developments of useful MALDI matrices for the mass spectrometric characterization of lipids. Biomolecules. 2018;8:173. https://doi.org/10.3390/biom8040173.
Camunas-Alberca SM, Moran-Garrido M, Sáiz J, Gil-de-la-Fuente A, Barbas C, Gradillas A. Integrating the potential of ion mobility spectrometry-mass spectrometry in the separation and structural characterisation of lipid isomers. Front Mol Biosci. 2023;10:1–21. https://doi.org/10.3389/fmolb.2023.1112521.
May JC, Knochenmuss R, Fjeldsted JC, McLean JA. Resolution of Isomeric Mixtures in Ion Mobility Using a Combined Demultiplexing and Peak Deconvolution Technique. Anal Chem. 2020;92:9482–92. https://doi.org/10.1021/acs.analchem.9b05718.
Djambazova KV, Dufresne M, Migas LG, Kruse ARS, Van de Plas R, Caprioli RM, Spraggins JM. MALDI TIMS IMS of Disialoganglioside Isomers─GD1a and GD1b in Murine Brain Tissue. Anal Chem. 2023;95:1176–83. https://doi.org/10.1021/acs.analchem.2c03939.
Xu H, Boucher FR, Nguyen TT, Taylor GP, Tomlinson JJ, Ortega RA, Simons B, Schlossmacher MG, Saunders-Pullman R, Shaw W, et al. DMS as an orthogonal separation to LC/ESI/MS/MS for quantifying isomeric cerebrosides in plasma and cerebrospinal fluid. J Lipid Res. 2019;60:200–11. https://doi.org/10.1194/jlr.D089797.
Dodds JN, Baker ES. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J Am Soc Mass Spectrom. 2019;30:2185–95. https://doi.org/10.1007/s13361-019-02288-2.
Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5:397–409. https://doi.org/10.1007/BF01049915.
Ann Q, Adams J. Structure determination of ceramides and neutral glycosphingolipids by collisional activation of [M + Li]+ ions. J Am Soc Mass Spectrom. 1992;3:260–3. https://doi.org/10.1016/1044-0305(92)87010-V.
Zaia J. Mass spectrometry of oligosaccharides. Mass Spectrom Rev. 2004;23:161–227. https://doi.org/10.1002/mas.10073.
Guo Z. The structural diversity of natural glycosphingolipids (GSLs). J Carbohydr Chem. 2022;41:63–154.
Bayat P, Lesage D, Cole RB. Tutorial: Ion Activation in Tandem Mass Spectrometry Using Ultra-High Resolution Instrumentation. Mass Spectrom Rev. 2020;39:680–702. https://doi.org/10.1002/mas.21623.
Hořejší K, Jirásko R, Chocholoušková M, Wolrab D, Kahoun D, Holčapek M. Comprehensive identification of glycosphingolipids in human plasma using hydrophilic interaction liquid chromatography—electrospray ionization mass spectrometry. Metabolites. 2021;11:1–24. https://doi.org/10.3390/metabo11030140.
Schweppe CH, Hoffmann P, Nofer JR, Pohlentz G, Mormann M, Karch H, Friedrich AW, Müthing J. Neutral glycosphingolipids in human blood: A precise mass spectrometry analysis with special reference to lipoprotein-associated Shiga toxin receptors. J Lipid Res. 2010;51:2282–94. https://doi.org/10.1194/jlr.M006759.
Karlsson H, Halim A, Teneberg S. Differentiation of glycosphingolipid-derived glycan structural isomers by liquid chromatography/mass spectrometry. Glycobiology. 2010;20:1103–16. https://doi.org/10.1093/glycob/cwq070.
Li Y, Teneberg S, Thapa P, Bendelac A, Levery SB, Zhou D. Sensitive detection of isoglobo and globo series tetraglycosylceramides in human thymus by ion trap mass spectrometry. Glycobiology. 2008;18:158–65. https://doi.org/10.1093/glycob/cwm129.
O’Brien JP, Brodbelt JS. Structural characterization of gangliosides and glycolipids via ultraviolet photodissociation mass spectrometry. Anal Chem. 2013;85:10399–407. https://doi.org/10.1021/ac402379y.
Kirschbaum C, Pagel K. Lipid Analysis by Mass Spectrometry coupled with Laser Light. Anal Sens. 2022;3(6):202200103. https://doi.org/10.1002/anse.202200103.
Ryan E, Nguyen CQN, Shiea C, Reid GE. Detailed Structural Characterization of Sphingolipids via 193 nm Ultraviolet Photodissociation and Ultra High Resolution Tandem Mass Spectrometry. J Am Soc Mass Spectrom. 2017;28:1406–19. https://doi.org/10.1007/s13361-017-1668-1.
Zhang W, Jian R, Zhao J, Liu Y, Xia Y. Deep-lipidotyping by mass spectrometry: recent technical advances and applications. J Lipid Res. 2022;63:100219. https://doi.org/10.1016/j.jlr.2022.100219.
Pham HT, Julian RR. Characterization of glycosphingolipid epimers by radical-directed dissociation mass spectrometry. Analyst. 2016;141:1273–8. https://doi.org/10.1039/c5an02383a.
Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ. Oligosaccharide Analysis By Mass Spectrometry: A Review Of Recent Developments. Anal Chem. 2014;86:196–212. https://doi.org/10.1021/ac403969n.
Hunnam V, Harvey DJ, Priestman DA, Bateman RH, Bordoli RS, Tyldesley R. Ionization and fragmentation of neutral and acidic glycosphingolipids with a Q-TOF mass spectrometer fitted with a MALDI ion source. J Am Soc Mass Spectrom. 2001;12:1220–5. https://doi.org/10.1016/S1044-0305(01)00309-9.
Chai W, Piskarev V, Lawson AM. Negative-ion electrospray mass spectrometry of neutral underivatized oligosaccharides. Anal Chem. 2001;73:651–7. https://doi.org/10.1021/ac0010126.
Chai W, Lawson AM, Piskarev V. Branching Pattern and Sequence Analysis of Underivatized Oligosaccharides by Combined MS/MS of Singly and Doubly Charged Molecular Ions in Negative-Ion Electrospray Mass Spectrometry. J Am Soc Mass Spectrom. 2002;13:670–9.
Zhang H, Zhang S, Tao G, Zhang Y, Mulloy B, Zhan X, Chai W. Typing of blood-group antigens on neutral oligosaccharides by negative-ion electrospray ionization tandem mass spectrometry. Anal Chem. 2013;85:5940–9. https://doi.org/10.1021/ac400700e.
Hořejší K, Jin C, Vaňková Z, Jirásko R, Strouhal O, Melichar B, Teneberg S, Holčapek M. Comprehensive characterization of complex glycosphingolipids in human pancreatic cancer tissues. J Biol Chem. 2023;299:1–22. https://doi.org/10.1016/j.jbc.2023.102923.
Hsu FF, Bohrer A, Turk J. Electrospray ionization tandem mass spectrometric analysis of sulfatide. Determination of fragmentation patterns and characterization of molecular species expressed in brain and in pancreatic islets. Biochim Biophys Acta - Lipids Lipid Metab. 1998;1392:202–16. https://doi.org/10.1016/S0005-2760(98)00034-4.
Yuki D, Sugiura Y, Zaima N, Akatsu H, Hashizume Y, Yamamoto T, Fujiwara M, Sugiyama K, Setou M. Hydroxylated and non-hydroxylated sulfatide are distinctly distributed in the human cerebral cortex. Neuroscience. 2011;193:44–53. https://doi.org/10.1016/j.neuroscience.2011.07.045.
Hsu FF, Turk J. Studies on sulfatides by quadrupole ion-trap mass spectrometry with electrospray ionization: Structural characterization and the fragmentation processes that include an unusual internal galactose residue loss and the classical charge-remote fragmentation. J Am Soc Mass Spectrom. 2004;15:536–46. https://doi.org/10.1016/j.jasms.2003.12.007.
Hájek R, Jirásko R, Lísa M, Cífková E, Holčapek M. Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Characterization of Gangliosides in Biological Samples. Anal Chem. 2017;89:12425–32. https://doi.org/10.1021/acs.analchem.7b03523.
Chai W, Piskarev VE, Mulloy B, Liu V, Evans PG, Osborn HMI, Lawson AM. Analysis of chain and blood group type and branching pattern of sialylated oligosaccharides by negative ion electrospray tandem mass spectrometry. Anal Chem. 2006;78:1581–92. https://doi.org/10.1021/ac051606e.
Lu H, Zhang H, Xu S, Li L. Review of recent advances in lipid analysis of biological samples via ambient ionization mass spectrometry. Metabolites. 2021;11:781. https://doi.org/10.3390/metabo11110781.
Zhao XE, Zhu S, Liu H. Recent progresses of derivatization approaches in the targeted lipidomics analysis by mass spectrometry. J Sep Sci. 2020;43:1838–46. https://doi.org/10.1002/jssc.201901346.
Ma X, Xia Y. Pinpointing double bonds in lipids by paternò-büchi reactions and mass spectrometry. Angew Chemie - Int Ed. 2014;53:2592–6. https://doi.org/10.1002/anie.201310699.
Xia F, Wan JB. Chemical derivatization strategy for mass spectrometry-based lipidomics. Mass Spectrom Rev. 2023;42:432–52. https://doi.org/10.1002/mas.21729.
Ma X, Chong L, Tian R, Shi R, Hu TY, Ouyang Z, Xia Y. Identification and quantitation of lipid C=C location isomers: A shotgun lipidomics approach enabled by photochemical reaction. Proc Natl Acad Sci U S A. 2016;113:2573–8. https://doi.org/10.1073/pnas.1523356113.
Zhang W, Zhang D, Chen Q, Wu J, Ouyang Z, Xia Y. Online photochemical derivatization enables comprehensive mass spectrometric analysis of unsaturated phospholipid isomers. Nat Commun. 2019;10:1–9. https://doi.org/10.1038/s41467-018-07963-8.
Bednařík A, Bölsker S, Soltwisch J, Dreisewerd K. An On-Tissue Paternò-Büchi Reaction for Localization of Carbon-Carbon Double Bonds in Phospholipids and Glycolipids by Matrix-Assisted Laser-Desorption–Ionization Mass-Spectrometry Imaging. Angew Chemie - Int Ed. 2018;57:12092–6. https://doi.org/10.1002/anie.201806635.
Thomas MC, Mitchell TW, Harman DG, Deeley JM, Nealon JR, Blanksby SJ. Ozone-induced dissociation: Elucidation of double bond position within mass-selected lipid ions. Anal Chem. 2008;80:303–11. https://doi.org/10.1021/ac7017684.
Brown SHJ, Mitchell TW, Blanksby SJ. Analysis of unsaturated lipids by ozone-induced dissociation. Biochim Biophys Acta - Mol Cell Biol Lipids. 2011;1811:807–17. https://doi.org/10.1016/j.bbalip.2011.04.015.
Poad BLJ, Green MR, Kirk JM, Tomczyk N, Mitchell TW, Blanksby SJ. High-Pressure Ozone-Induced Dissociation for Lipid Structure Elucidation on Fast Chromatographic Timescales. Anal Chem. 2017;89:4223–9. https://doi.org/10.1021/acs.analchem.7b00268.
Poad BLJ, Zheng X, Mitchell TW, Smith RD, Baker ES, Blanksby SJ. Online Ozonolysis Combined with Ion Mobility-Mass Spectrometry Provides a New Platform for Lipid Isomer Analyses. Anal Chem. 2018;90:1292–300. https://doi.org/10.1021/acs.analchem.7b04091.
Barrientos RC, Vu N, Zhang Q. Structural Analysis of Unsaturated Glycosphingolipids Using Shotgun Ozone-Induced Dissociation Mass Spectrometry. J Am Soc Mass Spectrom. 2017;28:2330–43. https://doi.org/10.1007/s13361-017-1772-2.
Barrientos RC, Zhang Q. Fragmentation Behavior and Gas-Phase Structures of Cationized Glycosphingolipids in Ozone-Induced Dissociation Mass Spectrometry. J Am Soc Mass Spectrom. 2019;30:1609–20. https://doi.org/10.1007/s13361-019-02267-7.
Kuo TH, Chung HH, Chang HY, Lin CW, Wang MY, Shen TL, Hsu CC. Deep Lipidomics and Molecular Imaging of Unsaturated Lipid Isomers: A Universal Strategy Initiated by mCPBA Epoxidation. Anal Chem. 2019;91:11905–15. https://doi.org/10.1021/acs.analchem.9b02667.
Zhang H, Xu M, Shi X, Liu Y, Li Z, Jagodinsky JC, Ma M, Welham NV, Morris ZS, Li L. Quantification and molecular imaging of fatty acid isomers from complex biological samples by mass spectrometry. Chem Sci. 2021;12:8115–22. https://doi.org/10.1039/d1sc01614h.
Feng Y, Chen B, Yu Q, Li L. Identification of Double Bond Position Isomers in Unsaturated Lipids by m-CPBA Epoxidation and Mass Spectrometry Fragmentation. Anal Chem. 2019;91:1791–5. https://doi.org/10.1021/acs.analchem.8b04905.
Zhang J, Zhang Z, Jiang T, Zhang Z, Zhang W, Xu W. Rapidly identifying and quantifying of unsaturated lipids with carbon-carbon double bond isomers by photoepoxidation. Talanta. 2023;260:124575. https://doi.org/10.1016/j.talanta.2023.124575.
Tang S, Cheng H, Yan X. On-Demand Electrochemical Epoxidation in Nano-Electrospray Ionization Mass Spectrometry to Locate Carbon-Carbon Double Bonds. Angew Chemie - Int Ed. 2020;59:209–14. https://doi.org/10.1002/anie.201911070.
Luo K, Chen H, Zare RN. Location of carbon-carbon double bonds in unsaturated lipids using microdroplet mass spectrometry. Analyst. 2021;146:2550–8. https://doi.org/10.1039/d0an02396e.
Zhao Y, Zhao H, Zhao X, Jia J, Ma Q, Zhang S, Zhang X, Chiba H, Hui SP, Ma X. Identification and Quantitation of C=C Location Isomers of Unsaturated Fatty Acids by Epoxidation Reaction and Tandem Mass Spectrometry. Anal Chem. 2017;89:10270–8. https://doi.org/10.1021/acs.analchem.7b01870.
Zhang J, Huo X, Guo C, Ma X, Huang H, He J, Wang X, Tang F. Rapid Imaging of Unsaturated Lipids at an Isomeric Level Achieved by Controllable Oxidation. Anal Chem. 2021;93:2114–24. https://doi.org/10.1021/acs.analchem.0c03888.
Unsihuay D, Su P, Hu H, Qiu J, Kuang S, Li Y, Sun X, Dey SK, Laskin J. Imaging and Analysis of Isomeric Unsaturated Lipids through Online Photochemical Derivatization of Carbon-Carbon Double Bonds**. Angew Chemie - Int Ed. 2021;60:7559–63. https://doi.org/10.1002/anie.202016734.
Lydic TA, Busik JV, Reid GE. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J Lipid Res. 2014;55:1797–809. https://doi.org/10.1194/jlr.D050302.
Lee JC, Byeon SK, Moon MH. Relative Quantification of Phospholipids Based on Isotope-Labeled Methylation by Nanoflow Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry: Enhancement in Cardiolipin Profiling. Anal Chem. 2017;89:4969–77. https://doi.org/10.1021/acs.analchem.7b00297.
Barrientos RC, Zhang Q. Differential Isotope Labeling by Permethylation and Reversed-Phase Liquid Chromatography-Mass Spectrometry for Relative Quantification of Intact Neutral Glycolipids in Mammalian Cells. Anal Chem. 2019;91:9673–81. https://doi.org/10.1021/acs.analchem.9b01206.
Aoki K, Heaps AD, Strauss KA, Tiemeyer M. Mass spectrometric quantification of plasma glycosphingolipids in human GM3 ganglioside deficiency. Clin Mass Spectrom. 2019;14:106–14. https://doi.org/10.1016/j.clinms.2019.03.001.
Ejsing CS, Bilgin M, Fabregat A. Quantitative profiling of long-chain bases by mass tagging and parallel reaction monitoring. PLoS ONE. 2015;10:1–17. https://doi.org/10.1371/journal.pone.0144817.
Hanamatsu H, Nishikaze T, Miura N, Piao J, Okada K, Sekiya S, Iwamoto S, Sakamoto N, Tanaka K, Furukawa JI. Sialic Acid Linkage Specific Derivatization of Glycosphingolipid Glycans by Ring-Opening Aminolysis of Lactones. Anal Chem. 2018;90:13193–9. https://doi.org/10.1021/acs.analchem.8b02775.
Liu Y, Yang L, Li H, Liu J, Tian R. Derivatization strategy for sensitive identification of neutral and acidic glycosphingolipids using RPLC-MS. Int J Mass Spectrom. 2022;482:116937. https://doi.org/10.1016/j.ijms.2022.116937.
Sekiya S, Wada Y, Tanaka K. Derivatization for Stabilizing Sialic Acids in MALDI-MS. Anal Chem. 2005;77:4962–8. https://doi.org/10.1021/ac050287o.
Miura Y, Shinohara Y, Furukawa JI, Nagahori N, Nishimura SI. Rapid and simple solid-phase esterification of sialic acid residues for quantitative glycomics by mass spectrometry. Chem - A Eur J. 2007;13:4797–804. https://doi.org/10.1002/chem.200601872.
Kang P, Mechref Y, Klouckova I, Novotny MV. Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun Mass Spectrom. 2005;19:3421–8. https://doi.org/10.1002/rcm.2210.
Chen P, Werner-Zwansiger U, Wiesler D, Pagel M, Novotny MV. Mass spectrometric analysis of benzoylated sialooligosaccharides and differentiation of terminal α2→3 and α2→6 sialogalactosylated linkages at subpicomole levels. Anal Chem. 1999;71:4969–73. https://doi.org/10.1021/ac990674w.
Huang Q, Liu D, Xin B, Cechner K, Zhou X, Wang H, Zhou A. Quantification of monosialogangliosides in human plasma through chemical derivatization for signal enhancement in LC-ESI-MS. Anal Chim Acta. 2016;929:31–8. https://doi.org/10.1016/j.aca.2016.04.043.
Nabetani T, Makino A, Hullin-Matsuda F, Hirakawa TA, Takeoka S, Okino N, Ito M, Kobayashi T, Hirabayashi Y. Multiplex analysis of sphingolipids using amine-reactive tags (iTRAQ). J Lipid Res. 2011;52:1294–302. https://doi.org/10.1194/jlr.D014621.
Barrientos RC, Zhang Q. Isobaric Labeling of Intact Gangliosides toward Multiplexed LC-MS/MS-Based Quantitative Analysis. Anal Chem. 2018;90:2578–86. https://doi.org/10.1021/acs.analchem.7b04044.
Peterka O, Jirásko R, Vaňková Z, Chocholoušková M, Wolrab D, Kulhánek J, Bureš F, Holčapek M. Simple and Reproducible Derivatization with Benzoyl Chloride: Improvement of Sensitivity for Multiple Lipid Classes in RP-UHPLC/MS. Anal Chem. 2021;93:13835–43. https://doi.org/10.1021/acs.analchem.1c02463.
Zheng SJ, Zheng J, Xiao HM, Wu DM, Feng YQ. Simultaneous quantitative analysis of multiple sphingoid bases by stable isotope labeling assisted liquid chromatography-mass spectrometry. Anal Chim Acta. 2019;1082:106–15. https://doi.org/10.1016/j.aca.2019.07.016.
Hermanson GR. Bioconjugate Techniques, 3rd ed. San Diego, USA: Academic Press; 2013.
Ghidoni R, Sonnino S, Masserini M, Orlando P, Tettamanti G. Specific tritium labeling of gangliosides at the 3-position of sphingosines. J Lipid Res. 1981;22:1286–95. https://doi.org/10.1016/s0022-2275(20)37322-3.
Song X, Ju H, Lasanajak Y, Kudelka MR, Smith DF, Cummings RD. Oxidative release of natural glycans for functional glycomics. Nat Methods. 2016;13:528–34. https://doi.org/10.1038/nmeth.3861.
Domon B, Vath JE, Costello CE. Analysis of derivatized ceramides and neutral glycosphingolipids by high-performance tandem mass spectrometry. Anal Biochem. 1990;184:151–64. https://doi.org/10.1016/0003-2697(90)90028-8.
Liebisch G, Fahy E, Aoki J, Dennis EA, Durand T, Ejsing CS, Fedorova M, Feussner I, Griffiths WJ, Köfeler H, et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J Lipid Res. 2020;61:1539–55. https://doi.org/10.1194/jlr.S120001025.
Funding
Open access publishing supported by the National Technical Library in Prague. This work was financially supported by the Czech Science Foundation (project No. 21-20238S) and by the Ministry of Education, Youth and Sports, Czech Republic (project No. JA344644).
Author information
Authors and Affiliations
Contributions
Karel Hořejší: conceptualization, writing — original draft; review and editing, visualization; Michal Holčapek: writing — review and editing, funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests. Michal Holčapek is a guest editor of ABC but was not involved in the peer review of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection New Trends in Lipidomics with guest editor Michal Holčapek.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hořejší, K., Holčapek, M. Unraveling the complexity of glycosphingolipidome: the key role of mass spectrometry in the structural analysis of glycosphingolipids. Anal Bioanal Chem 416, 5403–5421 (2024). https://doi.org/10.1007/s00216-024-05475-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-024-05475-7