
Abstract
A validated method for the simultaneous determination of prominent volatile cleavage products (CPs) of β-carotene in cell culture media has been developed. Target CPs comprised β-ionone (β-IO), cyclocitral (CC), dihydroactinidiolide (DHA), and 1,1,6-trimethyltetraline (TMT). CPs were extracted by solid-phase extraction applying a phenyl adsorbent, eluted with 10% (v/v) tetrahydrofuran in n-hexane, and identified and quantified by gas chromatography-mass spectrometry with electron impact ionization. Method validation addressed linearity confirmation over two application ranges and homoscedasticity testing. Recoveries from culture media were between 71.7% and 95.7% at 1.0 μg/ml. Precision of recoveries determined in intra-day (N = 5) and inter-day (N = 15) assays were <2.0% and <4.8%, respectively. Limit of detection and limit of quantification of the analysis method were <18.0 and <53.0 ng/ml for β-IO, CC, and TMT, whereas 156 and 474 ng/ml were determined for DHA, respectively. Although extractions of blank matrix proved the absence of interfering peaks, statistical comparison between slopes determined for instrumental and total method linearity revealed significant differences. The method was successfully applied in selecting an appropriate solvent for the fortification of culture media with volatile CPs, including the determination of their availability over the incubation period. For the first time, quantification of volatile CPs in treatment solutions and culture media for primary cells becomes accessible by this validated method.

Cultured primary rat hepatocytes in phase contrast after nuclea staining with DAPI including a chromatogram (GC-MS) of volatile cleavage products of b-carotene, which are presumed to exert genotoxic effects on hepatocytes and pneumocytes
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nutritional supplementation with vitamins is considered an important issue in health provision thus gaining increasing relevance in food industry [1, 2]. Within the administered vitamin portfolio, β-carotene (BC), a precursor of vitamin A (retinol), has been ascribed a central role in cancer prevention and therapy which is related to the antioxidant property of carotenoids with their conjugated polyene structure predestined for free radical and singlet oxygen scavenging [3]. Additionally, BC reduces the risk of cardiovascular diseases, cataract development, and macula degeneration. The outstanding quencher function of BC advocates dietary supplementation [4–6]. BC accumulates in target organs, with highest concentrations in human liver and lung tissue [7] and enrichment in mitochondria and nucleus which are also considered primary sites of effect [3, 8]. Causal relation between BC supplementation and cancer protective effects has been investigated recently in two comprehensive human intervention trials, the ATBC [9] and the CARET study [10, 11], both providing a daily oral administration of 20–30 mg BC [6]. Remarkably, BC-treated probationers with long smoking history and/or asbestos exposition showed a 16–28% increased lung cancer incidence entailing an increased mortality risk in comparison with placebo controls. This required a premature abandoning of both studies [9–12]. Concomitant chronic alcohol abuse enhanced toxic effects on hepatic level causing carcinogenesis [4, 9]. This apparent paradox has been related to pro-oxidant and pro-carcinogenic properties of BC under particular conditions, such as pronounced daily BC supplementation in combination with high oxygen partial pressure, oxidative stress, and high levels of reactive oxygen species (ROS) all prevailing in lung tissues of affected smokers [13–16].
Apparently, BC degradation products generated by excentric (non-)enzymatic cleavage in the radical-rich lung environment of smokers are responsible effectors, and these cleavage products (CPs) may promote carcinogenesis in manifold ways. In inflamed lung tissue, an increased expression of marker proteins for cancer progression [17], downregulation of tumor suppressors, and localized proliferation of alveolar macrophages (orneutrophils) have been observed [18]. Activated neutrophils release H2O2, O −2 , and myeloperoxidase (MPO), an enzyme which activates (tobacco) pro-carcinogens [19, 20] but is also involved in the formation of protein-, polysaccharide-, and DNA-radicals leading to protein fragmentation, oxidation of cell membranes, and DNA strand breaks, respectively [21–24]. Moreover, MPO catalyzes the formation of HClO from H2O2 and Cl− [21, 25], which induces BC degradation towards non-volatile long chain CPs, e.g., aldehydes (so-called apo-carotenals), epoxides, and carbonyls, but also further to volatile short-chain CPs. These CPs can influence the O −2 release of neutrophils and promote their apoptosis, thus reducing clearance of cancer cells [3, 26]. CPs can also induce enzymes of the cytochrome P450 family (CYP) which activate pro-carcinogenic smoke constituents with their successive binding to DNA [6, 13]. Subsequent changes in cell growth and cell cycle induce a progression of malignant effects [17, 18]. CYPs also induce increased levels of ROS mainly in liver and lung [6, 13]. Consequently, CPs seem to represent key triggers in the development of latent tumors [6, 15].
Particularly, inflammatory effects may be highly relevant for the generation of CPs and have been simulated in vitro by degradation of BC in HClO solutions of concentrations expected also under in vivo conditions and the application of resulting CP mixture to cell cultures. Such studies demonstrated not only an impairment of mitochondria [3, 27] but also provided clear evidence for genotoxic effects of CPs, i.e., treatment of primary hepatocytes with CPs caused significant increases of micronucleated cells, chromosomal aberrations, and sister chromatid exchanges [28–30]. Although profiles of resulting CPs were derived, concentrations of the volatile CP species were not addressed, and their quantification is missing [31].
Selected volatile target CPs have mostly been addressed by gas chromatography-mass spectrometry (GC-MS) and GC-ion trap-MS in the analysis of volatile odor causing and aroma compounds in plants [32–34] and river water [35]. Applied clean-up and pre-concentration strategies comprised headspace solid-phase microextraction (HS-SPME) [33, 34, 36], headspace co-distillation [32], simultaneous distillation-extraction (SDE) [32, 37], stir bar sorptive extraction [37], and purge-and-trap analysis [33] but also liquid–liquid extraction (LLE) [31]. Recoveries and related precision for selected CPs, if determined, were highly variable depending on the analyte species and matrix [33]. GC-MS with off-line extraction was mainly applied for compound profiling or semi-quantitative determination [31, 34, 36]. Some methods, such as SDE, are time and solvent consuming and promote sample loss. Others, such as HS-SPME or purge-and-trap, are highly efficient in extracting volatiles but require equilibrium times between 60 and 120 min and increased temperatures of 35–50 °C. Extended exposure times in combination with continuous heating and open-air conditions increased the risk for artificial product formation especially in the presence of complex mixtures of reactive analytes and matrix compounds and might promote oxidation of volatile organic compounds [32]. To the knowledge of the authors, no validated methods for volatile CPs of BC are available, especially for their determination in cell cultures.
The presented work aims to optimize a GC-MS method with previous off-line solid-phase extraction (SPE) for clean-up applicable for the identification and quantification of volatile CPs recently postulated as relevant degradation products of BC, i.e., β-ionone (β-IO), cyclocitral (CC), dihydroactinidiolide (DHA), and 1,1,6-trimethyltetraline (TMT; Fig. 1) [31]. Due to the lack of some commercially available CP standards, validated methods for these volatiles are missing, consequently excluding standardization of CP application solutions and quantification of CPs in cell cultures. Based on recent results, cultures of primary rat hepatocytes and pneumocytes have been selected as key cellular models in the investigation of possible adverse effects of CPs on the DNA level and thus their genotoxicity [28–30]. Therefore, method optimization and validation has to address equally application solutions and cell culture media containing CPs. Although the current focus is on volatile CPs, a parallel analysis of samples for volatile and non-volatile CPs by GC-MS and LC-ESI-MS is a mid-term objective. This approach will allow for both a true standardization of CP application mixtures derived from HClO degradation and quantification of volatile CPs in culture media after incubation of cell cultures with treatment solutions.

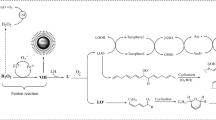
Postulated degradation pathways of β-carotene by treatment with HClO/ClO− resulting in the formation of volatile target cleavage products according to [31] including their relevant physicochemical properties
Materials and methods
Reagents and chemicals
CC (purity >90%) was obtained from SAFC (St. Louis, MO, USA). TMT (purity 95%) was synthesized by VeZerf Laborsynthesen GmbH (Idar-Oberstein, Germany). β-IO; (purity >96%) was obtained from Alfa Aesar (Ward Hill, MA, USA). DHA (purity >98%) was synthesized by International Laboratory (San Bruno, CA, USA). Linalool (Lin; purity >97%) and methylisoeugenol (Mie; purity >98%) were purchased from SAFC and used as internal standards (IS) due to the absence of commercially available deuterated analytes. Tetrahydrofuran (THF; water-free and without stabilizator) was from Merck (Darmstadt, Germany). Methanol, n-hexane (in unisolv quality), minimum essential medium Eagle (MEM), which was applied in cell cultures, and Tween 20 were all purchased from Sigma Aldrich (St. Louis, MO, USA). Ultrapure water was prepared in a quality higher than 18.2 MΩ cm by a Milli-Q Plus 185 system (Millipore S.A., Molsheim, France).
Gas chromatography-mass spectrometry
Analyses were performed on an Agilent HP-G1800A GCD System coupled to an MS GDC Electron Ionization Detector operating in electron impact mode applying 70 eV. The system was equipped with a HP Automatic Liquid Sampler G1512 (all from Agilent Technologies, Palo Alto, CA, USA). Automatic MS tuning was performed with perfluorotributylamine (PFTBA) addressing 69, 219, and 502 m/z. Analytes and IS were separated on a DB-20 (WAX) capillary column (30 m × 0.25 mm, 0.50 μm film thickness) from Agilent. Injector temperature and initial oven temperature were 260 °C and 65 °C, respectively. The oven temperature was raised from 65 °C to 70 °C at a rate of 5 °C/min, then from 70 °C to 200 °C at a rate of 10 °C/min, maintained at 200 °C for 4 min, raised further to 220 °C at a rate of 10 °C/min, and finally maintained at 220 °C for 4 min. Detector temperature was set at 320 °C all over the run. Helium was from SIAD GmbH (Vienna, Austria) and used in a purity of 6.0 as a carrier gas, applying a flow rate of 1.0 ml/min. A sample volume of 1.0 μl was injected in splitless mode by means of the autosampler. Solvent delay was set with 8.0 min. MS was operated in scan mode (50–550 m/z) for qualitative analysis, testing the method specificity, screening of blanks, and confirmation of compound identity. Compound identification was based on matching experimental spectra with references available from Wiley library (version 2005, Wiley, New York, USA). Selected ion monitoring (SIM) mode was used for quantitative analysis. For each analyte and IS, two ions, i.e., the highest fragment (HF) for quantification and the nominal mass (NM) as a control, were selected in the SIM mode. Selected ions were also extracted from chromatograms recorded in scan mode and obtained from a combined CP stock solution with both IS in order to confirm their uniqueness. The selected ions (HF/NM) were 71/154 m/z for Lin, 137/152 m/z for CC, 159/174 m/z for TMT, 177/192 m/z for β-Io, 178 m/z (HF and NM) for Mie, and 111/180 m/z for DHA (Fig. 2). Peak areas were integrated by MSD ChemStation (v. D.03.00.611) and utilized for quantification subsequent to their correction by both IS.

Chromatograms and MS spectra of CPs and IS. a Chromatogram for a CP standard solution (50 μg/ml of each CP dissolved in 10% (v/v) THF in n-hexane). b Chromatogram for CP standard solution after SPE (expected concentrations for individual CPs correspond to a. 1–6 MS spectra of CPs derived from a: 1 linalool (IS), 2 cyclocitral (CC), 3 1,1,6-trimethyltetraline (TMT), 4 β-ionone (β-IO), 5 methylisoeugenol (IS), and 6 dihydroactinidiolide (DHA)
Standard solutions for GC-MS instrumental validation
A combined stock solution was prepared gravimetrically in n-hexane containing all analytes and both IS at 100 μg/ml and stored in darkness at −20 °C. From this stock solution, standard solutions for calibration were freshly prepared immediately prior to their use in GC-MS in ranges of 1.0–50.0 and 0.5–4.5 μg/ml in n-hexane for linearity testing.
Standard solutions for SPE GC-MS validation
Solutions of 50 mmol/l Tween 20 containing all CPs and Mie, which was applied as IS to correct for differences in the SPE performance, at 20 mg/ml were dissolved in MEM at a ratio of 1:200 (v/v) resulting in final concentrations of 100 μg/ml of either CP and 0.25 mmol/l Tween 20. This working solution was prepared immediately prior to SPE extraction and employed for determination of SPE recoveries as well as the inter- and intra-day precision of the finally selected SPE phenyl adsorbent. For testing the SPE linearity, MEM was spiked with the Tween 20-CP stock solution to give nominal concentrations between 0.5 and 4.5 μg/ml for each CP after extraction.
SPE extraction procedure
Strata-X 200 mg/3 ml, Strata-X-AW 200 mg/3 ml, Strata CN 500 mg/3 ml, and Strata Phenyl 500 mg/3 ml SPE-columns were obtained from Phenomenex (Torrance, CA, USA) and applied in optimizing the SPE procedure. SPE columns were conditioned with 3 ml methanol followed by 3 ml ultrapure water before 1.0 ml of the spiked MEM working solution was loaded. The column was washed with 2 ml ultrapure water subsequently. Elution of CPs (and Mie) was done by 2.0 ml 10% (v/v) THF in n-hexane at a flow rate ≥2 ml/min. The eluate was frozen at −20 °C to facilitate the separation of the organic and aqueous phase. The hydrophobic fraction of the eluate was collected and transferred to a 2-ml volumetric flask. After addition of Lin, i.e., the IS for correcting the instrumental performance, and adjustment of the final volume with n-hexane, the extract was injected into GC-MS.
Treatment solution and CP solubility in cell culture media
To simulate cultivation conditions of primary hepatocytes and pneumocytes, 5 ml MEM supplemented with non-essential amino acids, pyruvate (1 mmol/l), aspartate (0.2 mmol/l), serine (0.2 mmol/l), and penicillin (100 U)/streptomycin (100 μg/ml) were placed in 60-mm diameter plastic culture dishes with subsequent incubation for 3 h at 37 °C, 5% CO2, and 95% relative humidity. The optimization of treatment solutions for primary cells addressed solubility and stability of CPs over the incubation interval testing THF and Tween 20 as suitable solvents. Appropriate volumes of the treatment solutions with CPs either dissolved in pure THF or in 50 mmol/l Tween 20 in ultrapure water were spiked to MEM to achieve nominal concentrations of 50 μg/ml for individual CPs after SPE, respectively. Final contents of THF and Tween 20 in MEM were 1.0% (v/v) and 0.25 mmol/l, respectively. Losses of CPs were determined over 180 min incubation. Cytotoxic concentrations of either solvent were determined in cell cultures of primary hepatocytes or pneumocytes, which were prepared from female Fischer 344 rats as outlined elsewhere [38–40].
Statistical treatment
Statistical evaluation of data, e.g., analysis of variance (ANOVA) and Welch test, was performed by use of the SPSS Statistical Software Package version 16. Tests for comparing slopes of regression equations and the Mandel’s fitting test are not implemented in the standard operations of SPSS. Thus, calculation algorithms were established by employing the SPSS syntax function. When necessary, details of calculation are specified in the respective section.
Results and discussion
The GC-MS method with preceding SPE was developed for volatiles previously identified as relevant CPs of BC under in vitro HClO degradation conditions as well as by activated macrophages (Fig. 1) [31]. Currently applied methods for determination of volatile CPs in cell cultures are not validated and fail in their quantification but also in standardizing application solutions. Both aspects are essential in geno- and cytotoxicity studies. Since some of these volatiles have been postulated to be reactive especially in a highly oxidative environment, e.g., in cell cultures exposed to oxidative stress, the extraction procedure should be rapid in order to depict the true analyte profile prevailing in the culture media.
Instrumental validation of GC-MS
The in-house developed GC-MS method was subject of a single-laboratory validation according to the IUPAC and the relevant ICH guideline Q2(R1) [41, 42]. Instrumental basis validation addressed intra-day and inter-day precision of retention times (t r) and peak areas, linearity testing for two concentration ranges, homoscedasticity testing, limit of detection (LOD), and limit of quantification (LOQ). Peak areas were corrected by IS for determination of precision and linearity testing. All data were based on CP standard solutions prepared in n-hexane including both IS. A representative chromatogram including MS spectra of CPs and both IS is given in Fig. 2.
Instrumental repeatability and inter-day precision
Repeatability (intra-day precision) for t r and corrected peak areas were evaluated from five replicate injections of a 50-μg/ml CP standard solution. The inter-day precision was calculated from three replicate injections of this standard solution on five different days. The respective intra- and inter-day precision given as coefficient of variation (CV) for corrected peak areas was from 0.20% to 0.57% and from 0.70% to 1.02%, respectively. Precision of t r was <0.03% (CV) in either case.
Instrumental linearity
Calibration was done in the SIM mode and performed over the GC-MS measurement step based on CP standard solutions covering low and high concentration ranges, i.e., 0.5–4.5 and 1.0–50.0 μg/ml, respectively. Six concentration levels were evenly distributed over the tested range and injected in triplicate, respectively. The low concentration range refers to concentrations of volatile target CPs produced, for example, by activated macrophages (data not shown). Solutions derived from in vitro degradation of BC in the presence of HClO and intended as standardized CP mixtures for cell treatment have therefore also to cover this concentration domain. The high concentration range addressed concentrations of CP spiking solutions applied for cell culture treatment in dose–response testing of genotoxic effects. Calibration curves for both concentration ranges were calculated for individual CPs by the least square approach on the basis of peak areas corrected by IS. Intercepts were tested by an ANOVA approach and were neglected in case test results were not significant (Table 1). Although coefficients of determination (R 2) were ≥0.995 in either case, this cannot be considered an ultimate proof of linearity, but merely refers to the goodness of correlation between corrected peak areas and related concentrations [43]. Therefore, linearity was confirmed by Mandel’s fitting test [43, 44]. Briefly, residual errors for a linear first-order regression line and an alternative quadratic regression model were calculated and compared by means of an F-test at a statistical significance of 99%. For all concentration ranges and analytes, the calculated F value (F calc) did not exceed the tabulated reference value (F tab). Thus, none of the tests was significant, which proved the validity of linear response models, since quadratic modeling delivered no improved data fitting. Furthermore, an ANOVA approach as available by SPSS was applied to evaluate the linear regression model [45]. Both statistical strategies provided corresponding results (Table 1).
Testing for homogeneity of variance
Additionally, calibration data were tested for absence of heteroscedasticity, since this would entail the uncertainty of analytical results to vary in a concentration-dependent manner. Homogeneity of variance was tested by analyzing standard solutions of CPs (including IS) at concentration levels of 1.0 and 50.0 μg/ml by ten injection replicates, respectively. F calc was derived by dividing the variance of the higher by the variance of the smaller concentration. In all tested cases F calc < F tab (5.34), confirming homogeneity of variance over the linear range at a 99.0% confidence level (Table 1) [44].
Instrumental limit of detection and limit of quantification
LOD and LOQ of individual analytes have to be calculated from the standard deviation of signals derived either from the noise of a blank or from low analyte concentrations [41, 42]. LOD and LOQ both for the GC-MS measurement step but also for the entire method (data in subsequent section) were considered to address possible differences. Instrumental LOD and LOQ were determined from a combined CP standard solution of 0.5 μg/ml. Baseline fluctuations required for the calculation of the noise were recorded in SIM mode and determined over time intervals equivalent to the twofold base peak width, which were selected on either side of the respective CP peak. LODs were calculated as the 3.3-fold of the standard deviation of the noise divided by the slope of the calibration curve for low concentrations (0.5–4.5 μg/ml) for the respective CP. Naturally, this refers to a calibration addressing signal heights. Again, linearity of these calibration curves was confirmed prior to their application in LOD calculation (data not shown). For reasons of completeness, LOQ was calculated according to the ICH guideline dividing the tenfold standard deviation of the signal fluctuation of the noise by the slope of the calibration curve for peak heights. DHA possessed the highest LOD and LOQ of all CPs with 131 and 399 ng/ml, respectively. LOD and LOQ of all other CPs were <18 and <53 ng/ml, respectively (Table 2).
Appropriate solvents for cell culture treatment
The hydrophobicity of the volatile target CPs (Fig. 1) required appropriate solvents to allow for a preparation of CP solutions in sufficiently high concentrations to avoid substantial changes in the composition of cell culture media in the course of their fortification. Furthermore, the solvent has to be miscible with the aqueous medium and should preferably ensure (bio)availability over the incubation period of 180 min as well as uptake of CPs by cells. For this purpose, THF, a standard solvent for the application of carotenoids, and Tween 20 were tested as solvents. Cells were incubated in MEM spiked with increasing concentrations of THF and Tween 20. These controls were compulsory to reveal possible onset of cytotoxic/genotoxic effects. Culture media containing up to 0.5% (v/v) THF induced no adverse effects at the cellular level. In case of Tween 20, even at the highest tested concentration of 1.0 mmol/l, adverse effects were absent for both primary hepatocytes and pneumocytes (data not shown).
Application to cell cultures
Losses of CPs from culture media without cells were determined over the intended incubation duration of 180 min. For this purpose, MEM was spiked with CP stock solutions prepared either in THF or Tween 20. Aliquots of the media were analyzed immediately at beginning of incubation and after incubation for 15, 30, 60, 120, and 180 min (Fig. 3). The reduction in CP concentrations was more pronounced in culture media spiked with CPs prepared in THF than in Tween 20. This is most likely related to the volatility of THF, since major CP losses were observed over the first 30 min, with a less prominent reduction (for CC and TMT) and a quasi-stable situation (for β-IO and DHA) afterwards. Losses within the first half hour were CP specific, which is obviously related to a combined effect of analyte volatility and hydrophobicity. In this context, TMT showed the highest loss with only 3% of the initial amount persistent after 3 h (Fig. 3). Although CC possesses a higher vapor pressure, its residual amount was considerably higher which is due to its lower hydrophobicity in comparison to TMT. Although the hydrophobicity of β-IO was intermediate, it was less affected than TMT and CC, which is due to its lower volatility. Subsequent to an initial reduction, the DHA concentration was maintained, clearly proving that the observed initial loss is related to the solubility diminution accompanying the THF evaporation but scarcely affected by the analyte volatility itself. The high percentage of remaining DHA was related to its high boiling point and low hydrophobicity (Figs. 1 and 3).

Time-dependent losses of individual CPs in spiked cell culture medium (MEM) over an incubation period of 180 min. Spiked MEM contained either 1.0% (v/v) THF or 0.25 mmol/l Tween 20. Spiked MEM was extracted prior to incubation (referred to as 100%) and after 30, 60, 120, and 180 min of incubation and analyzed by GC-MS. Graphs depict CP relative (percent) to the initially applied CP amount. Each data point represents the mean of three independent measurement series, each analyzed in triplicate by SPE-GC-MS. Error bars give standard deviations
The application of Tween 20 resulted in reduced losses of all CPs, most evident for TMT. The reduction of β-IO, CC, and TMT followed a linear trend over 120 min. Again, TMT and CC showed highest losses, but more than 40% of the initial amount was still present when incubation was accomplished, whereas DHA remained nearly unaffected. Contrary to THF, Tween 20 remained in solution over the entire incubation interval. A comparison of both data sets thus underpins more pronounced solubility losses of CPs (probably in combination with evaporation) in THF due to its release (Fig. 3). Both extended availability of CPs and the lower toxicity of the solvent per se clearly proved Tween 20 more appropriate for its application as solvent in cell cultures than THF. Additionally, THF is known to form peroxides in the presence of air and light and has increased toxicity for certain cell lines. Moreover, recent data also indicate promotion of tumor formation and an influence of THF on P450 proteins, which would induce interfering effects and aggravate unambiguous statements about CP mechanisms [46].
Optimization of extraction
LLE has been used as a standard strategy in CP extraction from cell cultures so far, although recovery data are missing in literature [31]. Thus, in an initial approach, recoveries for LLE were determined. Therefore, spiked MEM was extracted with a threefold excess either of toluene or n-hexane, modifying a method of Sommerburg et al. [31]. Recovery for either selected target CP was <10%. Consequently, SPE was selected as an alternative to ensure a rapid extraction process and prevent artificial product generation in highly reactive mixtures over extended analysis time. A pool of four different SPE phases, i.e., Strata-X, Strata-X-AW, Strata CN, and Strata Phenyl, was selected for SPE optimization. However, Mie was not implemented for recovery corrections at this stage to prevent artificial distortions by possible variable adsorption of this IS to different adsorbents (Fig. 4). Highest recoveries were achieved with the reversed-phase phenyl stationary phase, whereby extraction is primarily based on π–π interactions available via the phenyl ring. Consequently, this stationary phase is appropriate for extraction of aromatic hydrophobic compounds such as TMT but also for hydrophobic compounds with π-conjugated systems present in all target CPs as well as BC itself.

Comparison of recovery for different SPE columns with CPs at 50 μg/ml. Mean recovery was calculated from three independent extractions each injected in triplicate. Error bars annotate standard deviation of the respective mean recovery
Recoveries from model solutions with a nominal CP concentration of 50 μg/ml after extraction were between 82% and 105% for β-IO, DHA, and TMT, which proves the superiority of Strata Phenyl over all tested columns. CC showed a slightly lower recovery of 67%. These high recoveries were related to the compliance of several essential extraction parameters. Samples have to be percolated through the column at 2.0 ml/min or faster. For elution, a THF content of 10% (v/v) in n-hexane is required since otherwise recoveries were reduced by more than 30%. THF contents >10% (v/v) provided no increase in recovery. Furthermore, a minimum elution volume of 2.0 ml is recommended to ensure repeatable recoveries. In addition, the residual water remaining in the SPE columns from the previous loading of the aqueous sample interfered with the final collection of the entire organic phase. Freezing the eluate at −20 °C facilitated the complete transfer of the CP containing n-hexane fraction.
Validation of SPE
Intra-day and inter-day precision of SPE recovery
The precision of SPE within single-laboratory validation was determined at two levels, i.e., by (a) intra-day assay which was based on five extraction replicates within 1 day and (b) an inter-day approach based on three extractions daily performed over five consecutive days, respectively. In all cases, MEM was spiked with a CP stock solution prepared in Tween 20 to give nominal analyte concentrations of 50 μg/ml in the eluates. Each eluate was analyzed in triplicate by GC-MS. A representative chromatogram after SPE is given in Fig. 1b. Data were subject to statistical evaluation for differences in variance by means of a one-factor ANOVA. Since variances of recoveries were not homogenous for β-IO and DHA over the tested period, Welch test was additionally applied for confirmation. Both tests were not significant for DHA and TMT, proving that their fluctuations in recoveries were random. In case of CC and β-IO, both tests indicated significant differences between daily mean recoveries. However, it has to be considered that the inter-day 95% confidence intervals of recovery were exceptionally low (<2.7%). Furthermore, the inter-day assay implements not only differences in the actual extraction performance but also deviations related to the extracted working solutions, which are prepared freshly every day. Precisions of the entire method are expressed as CV (Table 2).
Linearity of SPE recovery
Linearity testing of the entire analytical method considers the independent preparation of CP working solutions and SPE differences which might be related to CP concentrations but also to the individual extraction performance of columns. The tested range was confined to the low concentration domain previously addressed, thus covering CP concentrations expected in real samples but also in in vitro degradation solutions of BC and CP solutions intended for genotoxicity testing. Adequate to instrumental linearity, MEM working solutions spiked with CPs and Mie were prepared at five concentration levels to give nominally 0.5 to 4.5 μg/ml after SPE extraction. Calculations of calibration curves were based on corrected peak areas considering also a correction for the actual recovery. Mandel’s fitting test and also ANOVA proved linearity over the tested range and, except from CC results for intercept testing, were not significant. Therefore, a zero intercept was assumed for β-Io, DHA, and TMT (Table 1).
Specificity
According to the IUPAC, selectivity is defined as the degree to which a method can quantify the analyte accurately in the presence of interferents [41]. Due to the complex composition of culture media, the currently unknown portfolio of products released by cells and the possible elution of contaminants from SPE columns, a single interferent testing—as proposed by the IUPAC—is inapplicable. Instead, specificity was evaluated by extracting a blank matrix (0.25 mmol/l Tween 20 in MEM), which was collected after 3 h of incubation in the presence of either primary hepatocytes or pneumocytes (without and with bovine serum added to the respective cell culture). For analytes and IS, retention time windows were constructed based on the previously calculated precision data to implement possible t r shifts in the evaluation of specificity. No interfering peaks were encountered at the t r of CPs and the internal standards neither in SIM nor in scan mode (data not shown). In addition, the slopes derived by the least square approach both for the instrumental and the SPE calibration (compare previous section) over the low concentration range were compared for significance of differences. Briefly, this was performed via a modified t-statistics, calculated by the difference between both slopes divided by the standard error of the difference between the slopes, with (N-4) degrees of freedom [47]. For all CPs, significant differences were revealed between the slopes of respective regression curves. Consequently, for CP quantification in real samples, the instrumental regression equation has to be replaced by the SPE-derived counterpart to avoid error-prone results.
LOD and LOQ derived from SPE
Both LOD and LOQ were recalculated as described in the instrumental validation, but by means of the slope derived from the first-order linearity calibration function for peak heights considering the entire SPE GC-MS method. Linearity for peak heights in the low concentration range (0.5–4.5 μg/ml) was confirmed by Mandel’s fitting test (data not shown). Derived LOD and LOQ data were slightly higher than their instrumental counterparts (Table 2).
Trueness
The trueness, which is quantitatively given by the bias, refers to an accepted reference value derived from a certified reference material or a reference method. Since unfortunately none of these references is currently available for the volatile target CPs in cell culture media, afore given recovery data from spiked MEM have to serve as a preliminary assessment of the bias [41].
Method application to cell cultures
The validated SPE GC-MS method was applied under cell treatment conditions incubating hepatocyte cultures for 3 h with a combination of all four volatile CPs covering 1.53 to 2.00 μg/mL for individual analytes (corresponding to nominal 10 μM in the medium). These concentrations are centered within the confirmed SPE linearity (Table 1). Culture media with hepatocytes were analyzed prior to and after accomplished incubation. Quantification of CPs was based on the determined SPE linearity considering peak areas corrected by internal standards. This way, aforementioned matrix effects were eliminated. After 3 h of incubation, 5.0% to 47.4% of the initially applied amounts were recovered for β-IO, CC, and DHA, whereas the concentration of TMT was below the LOD (Table 3). The observed reduction of CPs in the presence of cells had to be corrected for volatility losses to derive cell-related uptake. However, CP concentrations in hepatocyte cultures were lower than those applied for monitoring evaporation-related bioavailability (see Fig. 3). Therefore, evaporation losses without cells had to be re-estimated after 3 h incubation for the concentrations applied to hepatocytes in order to prevent concentration-related mismatches. Recoveries of CPs at 10 μM were between 50.9% and 83.5% of the initially applied concentration in cell-deficient media (N = 4). The deviation from previous recovery data (Fig. 3) was only 3.5–13.5% for β-IO, CC, and DHA. For TMT, recovery was 25.2% higher with the lower concentration, which might be concentration related. Consequently, the pronounced reduction of CP concentrations encountered in the presence of hepatocytes cannot be explained exclusively by evaporative losses. Therefore, residual amounts of volatile CPs in hepatocyte cultures were corrected by evaporative losses determined at equivalent CP concentrations to derive cell-related CP uptake. However, one has to take into consideration that these data address a presumed uptake and may actually comprise CPs entering hepatocytes as well as CPs adhered to or incorporated into plasma membranes. Furthermore, hepatocytes secrete proteins, e.g., albumin, into the medium, which might also interact with CPs [48]. However, at the current state, a detailed distribution budget of CPs is beyond the scope of this article. Evaporation and cellular uptake of CPs from culture media are concurrent events. Consistently, corrected CP data may reflect only the minimum cellular uptake/adhesion over the incubation period (Table 3).
Conclusion
A GC-MS method with preceding SPE has been successfully developed for the determination of volatile CPs of BC in cell culture media. The rapid extraction intends to prevent changes in the analyte profile due to possible conversions in a reactive environment. Therefore, the method was validated for spiked culture media. Recoveries were between 63.7% and 108.2% and between 71.7% and 95.7% for all CPs at 50.0 μg/mL and 1.0 μg/ml, respectively. Intra-day and inter-day precisions of the entire analytical method were determined <2.0% and <4.8% (CV), respectively. Limits of detection were between 4.6 and 156 ng/ml. Linearity was tested over a low concentration range intended for monitoring cell culture media as well as a high concentration domain for standardization of treatment solutions. Both Mandel’s fitting test and ANOVA confirmed linearity over the selected ranges. A comparison of calibration slopes derived either from direct injection of standard solutions or from spiked media which passed through the entire method revealed significant differences for either analyte and indicated matrix effects. Consequently, quantification has to be based on the regression equation calculated for the entire method. The method has also been employed to monitor CP availability for cells over the incubation period and assisted in the selection of an appropriate application solvent. The presented method allows for the first time a quantitative determination of biologically relevant volatile CPs of BC in in vitro cell systems. Future strategies aim to quantify target CPs in differently treated cell cultures and address their genotoxicity. Therefore, this validated method represents a crucial pre-requisite for revealing dose-dependent genotoxic effects either by treating cell cultures of primary hepatocytes and pneumocytes with individual or combined CP application solutions or by BC degradation induced directly in the cell culture.
References
Mayne S (1996) Beta-carotene, carotenoids, and disease prevention in humans. FASEB J 10(7):690–701
Bonrath W, Netscher T (2005) Catalytic processes in vitamins synthesis and production. Appl Catal A Gen 280(1):55–73
Siems W, Wiswedel I, Salerno C, Crifò C, Augustin W, Schild L, Langhans C-D, Sommerburg O (2005) β-Carotene breakdown products may impair mitochondrial functions—potential side effects of high-dose β-carotene supplementation. J Nutr Biochem 16(7):385–397
Leo MA, Lieber CS (1999) Alcohol, vitamin a, and β-carotene: adverse interactions, including hepatotoxicity and carcinogenicity. Am J Clin Nutr 69(6):1071–1085
Lin Y, Burri B, Neidlinger T, Muller H, Dueker S, Clifford A (1998) Estimating the concentration of beta-carotene required for maximal protection of low-density lipoproteins in women. Am J Clin Nutr 67(5):837–845
Paolini M, Abdel-Rahman SZ, Sapone A, Pedulli GF, Perocco P, Cantelli-Forti G, Legator MS (2003) β-Carotene: a cancer chemopreventive agent or a co-carcinogen? Mutat Res, Rev Mutat Res 543(3):195–200
Schmitz HH, Poor CL, Wellman RB, Erdman JW Jr (1991) Concentrations of selected carotenoids and vitamin a in human liver, kidney and lung tissue. J Nutr 121(10):1613–1621
Siems W, Sommerburg O, Schild L, Augustin W, Langhans C-D, Wiswedel I (2002) β-Carotene cleavage products induce oxidative stress in vitro by impairing mitochondrial respiration. FASEB J 16(10):1289–1291
Albanes D, Heinonen OP, Taylor PR, Virtamo J, Edwards BK, Rautalahti M, Hartman AM, Palmgren J, Freedman LS, Haapakoski J, Barrett MJ, Pietinen P, Malila N, Tala E, Liippo K, Salomaa E-R, Tangrea JA, Teppo L, Askin FB, Taskinen E, Erozan Y, Greenwald P, Huttunen JK (1996) α-Tocopherol and β-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst 88(21):1560–1570
Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Valanis B, Williams JH, Barnhart S, Cherniack MG, Brodkin CA, Hammar S (1996) Risk factors for lung cancer and for intervention effects in caret, the beta-carotene and retinol efficacy trial. J Natl Cancer Inst 88(21):1550–1559
Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Valanis B, Williams JH, Barnhart S, Hammar S (1996) Effects of a combination of beta carotene and vitamin a on lung cancer and cardiovascular disease. N Engl J Med 334(18):1150–1155
Paolini M, Cantelli-Forti G, Perocco P, Pedulli GF, Abdel-Rahman SZ, Legator MS (1999) Co-carcinogenic effect of β-carotene. Nature 398(6730):760–761
Paolini M, Antelli A, Pozzetti L, Spetlova D, Perocco P, Valgimigli L, Pedulli GF, Cantelli-Forti G (2001) Induction of cytochrome p450 enzymes and over-generation of oxygen radicals in beta-carotene supplemented rats. Carcinogenesis 22(9):1483–1495
Arora A, Willhite CA, Liebler DC (2001) Interactions of β-carotene and cigarette smoke in human bronchial epithelial cells. Carcinogenesis 22(8):1173–1178
Palozza P, Serini S, Di Nicuolo F, Boninsegna A, Torsello A, Maggiano N, Ranelletti FO, Wolf FI, Calviello G, Cittadini A (2004) β-Carotene exacerbates DNA oxidative damage and modifies p53-related pathways of cell proliferation and apoptosis in cultured cells exposed to tobacco smoke condensate. Carcinogenesis 25(8):1315–1325
Burton G, Ingold K (1984) Beta-carotene: an unusual type of lipid antioxidant. Science 224(4649):569–573
Palozza P (2005) Can β-carotene regulate cell growth by a redox mechanism? An answer from cultured cells. Biochim Biophys Acta Mol Basis Dis 1740(2):215–221
Liu C, Wang X-D, Bronson RT, Smith DE, Krinsky NI, Russell RM (2000) Effects of physiological versus pharmacological β-carotene supplementation on cell proliferation and histopathological changes in the lungs of cigarette smoke-exposed ferrets. Carcinogenesis 21(12):2245–2253
Dally H, Gassner K, Jäger B, Schmezer P, Spiegelhalder B, Edler L, Drings P, Dienemann H, Schulz V, Kayser K, Bartsch H, Risch A (2002) Myeloperoxidase (mpo) genotype and lung cancer histologic types: the mpo −463 a allele is associated with reduced risk for small cell lung cancer in smokers. Int J Cancer 102(5):530–535
London SJ, Lehman TA, Taylor JA (1997) Myeloperoxidase genetic polymorphism and lung cancer risk. Cancer Res 57(22):5001–5003
Hawkins CL, Davies MJ (1999) Hypochlorite-induced oxidation of proteins in plasma: formation of chloramines and nitrogen-centred radicals and their role in protein fragmentation. Biochem J 340(2):539–548
Hawkins CL, Davies MJ (2002) Hypochlorite-induced damage to DNA, RNA, and polynucleotides: formation of chloramines and nitrogen-centered radicals. Chem Res Toxicol 15(1):83–92
Hawkins CL, Davies MJ (1998) Degradation of hyaluronic acid, poly- and mono-saccharides, and model compounds by hypochlorite: evidence for radical intermediates and fragmentation. Free Radical Biol Med 24(9):1396–1410
Hawkins CL, Rees MD, Davies MJ (2002) Superoxide radicals can act synergistically with hypochlorite to induce damage to proteins. FEBS Lett 510(1–2):41–44
Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brümmer J, Rudolph V, Münzel T, Heitzer T, Meinertz T, Baldus S (2005) Myeloperoxidase mediates neutrophil activation by association with cd11b/cd18 integrins. Proc Natl Acad Sci USA 102(2):431–436
Siems W, Capuozzo E, Crifò C, Sommerburg O, Langhans C-D, Schlipalius L, Wiswedel I, Kraemer K, Salerno C (2003) Carotenoid cleavage products modify respiratory burst and induce apoptosis of human neutrophils. Biochim Biophys Acta Mol Basis Dis 1639(1):27–33
Handelman GJ, van Kuijk FJGM, Chatterjee A, Krinsky NI (1991) Characterization of products formed during the autoxidation of [beta]-carotene. Free Radical Biol Med 10(6):427–437
Alija AJ, Bresgen N, Sommerburg O, Siems W, Eckl PM (2004) Cytotoxic and genotoxic effects of β-carotene breakdown products on primary rat hepatocytes. Carcinogenesis 25(5):827–831
Alija A, Bresgen N, Sommerburg O, Langhans C, Siems W, Eckl P (2005) Cyto- and genotoxic potential of β-carotene and cleavage products under oxidative stress. Biofactors 24(1–4):159–163
Alija AJ, Bresgen N, Sommerburg O, Langhans CD, Siems W, Eckl PM (2006) β-Carotene breakdown products enhance genotoxic effects of oxidative stress in primary rat hepatocytes. Carcinogenesis 27(6):1128–1133
Sommerburg O, Langhans C-D, Arnhold J, Leichsenring M, Salerno C, Crifò C, Hoffmann GF, Debatin K-M, Siems WG (2003) [beta]-carotene cleavage products after oxidation mediated by hypochlorous acid-a model for neutrophil-derived degradation. Free Radical Biol Med 35(11):1480–1490
Peng F, Sheng L, Liu B, Tong H, Liu S (2004) Comparison of different extraction methods: steam distillation, simultaneous distillation and extraction and headspace co-distillation, used for the analysis of the volatile components in aged flue-cured tobacco leaves. J Chromatogr A 1040(1):1–17
Beltran J, Serrano E, López F, Peruga A, Valcarcel M, Rosello S (2006) Comparison of two quantitative GC–MS methods for analysis of tomato aroma based on purge-and-trap and on solid-phase microextraction. Anal Bioanal Chem 385(7):1255–1264
Guedes De Pinho P, Gonçalves RF, Valentão P, Pereira DM, Seabra RM, Andrade PB, Sottomayor M (2009) Volatile composition of Catharanthus roseus (L.) G. Don using solid-phase microextraction and gas chromatography/mass spectrometry. J Pharm Biomed Anal 49(3):674–685
Bao M-L, Barbieri K, Burrini D, Griffini O, Pantani F (1997) Determination of trace levels of taste and odor compounds in water by microextraction and gas chromatography-ion-trap detection-mass spectrometry. Water Res 31(7):1719–1727
Fernandes F, Pereira DM, Guedes de Pinho P, Valentão P, Pereira JA, Bento A, Andrade PB (2010) Headspace solid-phase microextraction and gas chromatography/ion trap-mass spectrometry applied to a living system: Pieris brassicae fed with kale. Food Chem 119(4):1681–1693
Caven-Quantrill DJ, Buglass AJ (2006) Comparison of micro-scale simultaneous distillation-extraction and stir bar sorptive extraction for the determination of volatile organic constituents of grape juice. J Chromatogr A 1117(2):121–131
Eckl P, Bresgen N (2003) The cultured primary hepatocyte and its application in toxicology. J Appl Biomed 1:117–126
Eckl PM, Whitcomb WR, Michalopoulos G, Jirtle RL (1987) Effects of egf and calcium on adult parenchymal hepatocyte proliferation. J Cell Physiol 132(2):363–366
Richards R, Davies N, Atkins J, Oreffo V (1987) Isolation, biochemical characterization, and culture of lung type II cells of the rat. Lung 165(1):143–158
Thompson M, Ellison SLR, Wood R (2002) Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC technical report). Pure Appl Chem 74(5):835–855
ICH (2005) Validation of analytical procedures: text and methodology Q2(R1). ICH harmonized tripartite guideline. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, November 2005, ICH, UK
Einax J, Reichenbächer M (2006) Solution to quality assurance challenge 2. Anal Bioanal Chem 384(1):14–18
Funk W, Dammann V, Donnevert G (1992) Qualitätssicherung in der Analytischen Chemie. VCH, Weinheim
Searle SR (1971) Linear models. Wiley, New York
Rodríguez AM, Sastre S, Ribot J, Palou A (2005) Beta-carotene uptake and metabolism in human lung bronchial epithelial cultured cells depending on delivery vehicle. Biochim Biophys Acta Mol Basis Dis 1740(2):132–138
Sachs L (1999) Angewandte Statistik - Anwendung statistischer Methoden, 9th edn. Springer, Berlin
Bresgen N, Ohlenschläger I, Wacht N, Afazel S, Ladurner G, Eckl PM (2008) Ferritin and Fasl (CD95l) mediate density dependent apoptosis in primary rat hepatocytes. J Cell Physiol 217(3):800–808
Acknowledgement
This work was supported by the Austrian Science Fund (FWF) grant number P20096.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Analytical Sciences in Austria with Guest Editors G. Allmaier, W. Buchberger, and K. Francesconi.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Martano, G., Vogl, C., Bojaxhi, E. et al. Solid-phase extraction and GC-MS analysis of potentially genotoxic cleavage products of β-carotene in primary cell cultures. Anal Bioanal Chem 400, 2415–2426 (2011). https://doi.org/10.1007/s00216-011-4836-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4836-3