
Abstract
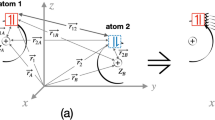
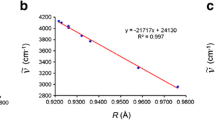
The molecular improved generator coordinate Hartree–Fock (MIGCHF) method is used to generate increasing size atom-centered Gaussian basis sets for the CO2 molecule. From these basis sets total HF energies and second-order correlation energies were calculated and compared with results obtained with other approaches. Considering our largest basis set, the HF energy is in error by 98 μhartree and the second-order correlation energy corresponds to ∼95.6% of an estimate of the limiting value. The relevance of the present calculations is to show the accuracy that can be achieved in studies of small polyatomic molecules with the MIGCHF method.
Similar content being viewed by others

Author information
Authors and Affiliations
Corresponding author
Additional information
Acknowledgement We acknowledge the financial support of CNPq (Brazilian Agency). We employed computational facilities at Universidade Federal do Espírito Santo and Universidade Estadual Paulista (IQ Araraquara).
Rights and permissions
About this article
Cite this article
Barreto, M., Muniz, E., Jorge, F. et al. Gaussian basis sets for CO2 molecule generated with the molecular improved generator coordinate Hartree–Fock method. Theor Chem Acc 113, 69–72 (2005). https://doi.org/10.1007/s00214-004-0617-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-004-0617-9