
Abstract
Rationale
Second-generation antipsychotics occupy dopamine D2 receptors and act as antagonists or partial agonists at these receptors. While these drugs alleviate positive symptoms in patients with schizophrenia, they are less effective for treating cognitive deficits and negative symptoms. Dopamine D3 receptors are highly expressed in areas of the brain thought to play a role in the regulation of motivation and reward-related behavior. Consequently, the dopamine D3 receptor has become a target for treating negative symptoms in combination with D2 antagonism to treat positive symptoms in patients with schizophrenia.
Objective
The purpose of this study was to determine the cariprazine receptor occupancies in brain for D2 and D3 receptors in patients with schizophrenia.
Methods
Using [11C]-(+)-PHNO as a radioligand, positron emission tomography (PET) scans were performed in eight patients at baseline and postdose on days 1, 4, and 15. Plasma and cerebrospinal fluid (CSF) samples were analyzed for cariprazine concentrations.
Results
A monotonic dose-occupancy relationship was observed for both receptor types. After 2 weeks of treatment, near complete (∼100 %) occupancies were observed for both receptors at a dose of 12 mg/day. At the lowest cariprazine dose (1 mg/day), mean D3 and D2 receptor occupancies were 76 and 45 %, respectively, suggesting selectivity for D3 over D2 receptors at low doses. An exposure-response analysis found a ∼3-fold difference in EC50 (D3 = 3.84 nM and D2 = 13.03 nM) in plasma after 2 weeks of dosing.
Conclusion
This PET imaging study in patients with schizophrenia demonstrated that cariprazine is a D3-preferring dual D3/D2 receptor partial agonist.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cariprazine (RGH-188) is an orally active, high-affinity dopamine D3 and D2 receptor partial agonist with preferential binding to D3 receptors (Kiss et al. 2010). A D3 strategy for treatment of schizophrenia is based on the brain distribution and putative role of D3 receptors (Gross and Drescher 2012); these receptors have high density in the ventral striatum (Gurevich and Joyce 1999), one of the core areas in the pathology of schizophrenia, as well as depression and anxiety. In preclinical models, dopamine D3 receptor antagonists reversed cognitive impairment (Laszy et al. 2005; Millan et al. 2007; Sigala et al. 1997) and enhanced locomotor activity of habituated rats (Gyertyán and Sághy 2004; Sautel et al. 1995; Waters et al. 1993), suggesting potential for improving negative symptoms. However, highly selective D3 receptor antagonists failed to show antipsychotic efficacy in preclinical models of schizophrenia (Millan et al. 2000; Reavill et al. 2000) suggesting that D3 receptor antagonism alone is not sufficient for antipsychotic efficacy.
It has been shown that ∼60–80 % occupancy of dopamine D2 receptors is necessary to achieve antipsychotic action in the clinic (Nyberg et al. 1996; Seeman 2001). It has therefore been hypothesized that combined occupancy of dopamine D3 and D2 receptors may offer distinct advantages over existing antipsychotics with primarily D2 receptor antagonism in the treatment of schizophrenia (Gyertyán et al. 2008; Kiss et al. 2008).
In rodent models, cariprazine pretreatment significantly diminished scopolamine- and PCP-induced cognitive deficits (Zimnisky et al. 2013) and stress-induced anhedonia (Gyertyán and Sághy 2004; Sautel et al. 1995; Waters et al. 1994); in knockout mice, these effects were found to be D3-receptor dependent (Zimnisky et al. 2013). Cariprazine has also shown clinical potential for treating negative symptoms based on results from a post hoc analysis of a phase II study in patients with schizophrenia (Debelle et al. 2014) as well as recent results from a prospectively defined schizophrenia study of patients with persistent and predominant negative symptoms (Debelle et al. 2015). One study found negative results of D3 receptor antagonists in schizophrenia (Redden et al. 2011); however, this study may have been limited by low occupancies at the tested doses (Graff-Guerrero et al. 2010).
Previous in vivo imaging studies exploring the binding profile of cariprazine in humans and nonhuman primates characterized the D2 receptor binding profile of cariprazine but were not informative about its D3 receptor-binding profile (Seneca et al. 2011). [11C]-(+)-4-propyl-9-hydroxynaphthoxazine ([11C]-(+)-PHNO) is a recently developed D3/D2 agonist positron emission tomography (PET) radioligand with preferential in vivo selectivity for dopamine D3 over D2 receptors (Gallezot et al. 2012; Ginovart et al. 2007; Graff-Guerrero et al. 2009; Willeit et al. 2006; Wilson et al. 2005). In contrast to previously used tracers like [11C]raclopride, [11C]-(+)-PHNO binding is due mostly to D3 receptors in the substantia nigra and ventral tegmental area, D2 receptors in the dorsal striatum, and a mixture of both types in globus pallidus, ventral striatum, and thalamus. This mixed binding profile can be exploited to infer the occupancy by antipsychotic drugs at D2 and D3 receptors separately (Girgis et al. 2011; Rabiner et al. 2009; Searle et al. 2010).
The pharmacokinetic (PK) properties of cariprazine in humans are characterized by relatively slow absorption, multi-exponential disposition, and slow elimination with a long terminal elimination half-life (T½). Two major active metabolites, desmethyl cariprazine (DCAR) and di-desmethyl cariprazine (DDCAR), were identified and monitored in clinical studies (Kapás et al. 2008; Mészáros et al. 2007). At steady state, DDCAR is the predominant active moiety, with systemic exposure that is about 2- to 3-fold higher than cariprazine; DCAR systemic exposure is around 30–40 % of parent drug exposure (Nakamura et al. 2016). DCAR and DDCAR have exhibited partial agonist activity at both D2 and D3 receptors and have displayed 21- and 25-fold selectivity, respectively, for cloned human D3 versus D2L receptors (unpublished data) compared with a 6- to 8-fold D3 selectivity for the parent compound (Kiss et al. 2010). Moreover, DDCAR also showed somewhat lower intrinsic activity at cloned human D2 receptors compared with cariprazine (Tadori et al. 2011). The activity of cariprazine after multiple dosing is, therefore, expected to be attributable to the parent drug and its two active metabolites.
This study was designed to assess the binding and occupancy of cariprazine, DCAR, and DDCAR to D3 and D2 receptors in various regions of the brain in adult patients with schizophrenia using PET imaging with [11C]-(+)-PHNO and to assess the relationship between occupancy and plasma concentrations of cariprazine and its two active metabolites.
Materials and methods
Clinical study design
This was an open-label, multiple-dose study in patients (ages 18–55 years, inclusive) with a primary diagnosis of schizophrenia according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) (APA 2000) criteria based on the Structured Clinical Interview for the DSM-IV (First et al. 2007). There were three different dosing cohorts with three patients each (Fig. S1); all had initial up-titration to the final dose to improve tolerability. Cohort 1 received 1.5 mg on day 1, 3 mg on day 2, 6 mg once daily (QD) on days 3–4, 9 mg QD on days 5–6, and 12 mg QD on days 7–15. Cohort 2 received 0.5 mg on day 1, 1 mg QD on days 2–4, and 3 mg QD on days 5–15. Cohort 3 received 0.5 mg QD on days 1–4 and 1 mg QD on days 5–15. The study was conducted at the New York State Psychiatric Institute (NYSPI) at Columbia University Medical Center and the Clinical Neuroscience Research Unit (CNRU)/Schizophrenia Research Clinic at Yale University. All PET scans and pharmacokinetic and cerebrospinal fluid (CSF) sample collections were performed at Yale University. PET data analysis was done at Columbia University. The study protocol was approved by the Yale and NYSPI Institutional Review Boards and was conducted in compliance with the ICH Good Clinical Practice guidelines and the Declaration of Helsinki. Patients provided signed informed consent at screening and had the capacity to understand it. Participants were compensated for their participation.
Dynamic PET scans were acquired with [11C]-(+)-PHNO. A maximum of five PET scans were performed for each patient to evaluate D3 and D2 receptor occupancy on day 1 at predose (baseline) and 4 h postdose (after plasma PK sampling), on day 4 at 4 h postdose (after plasma PK sampling), on day 15 at 4 h postdose (after plasma PK sampling), and on day 24 after the plasma PK sampling. The average injected radioactivity was 564 ± 157 MBq, specific activity was 80 ± 33 MBq/nmol, and injected mass was 2.0 ± 0.48 μg.
Safety was assessed by adverse event (AE) recording, clinical laboratory tests, vital sign assessments, electrocardiography, physical and psychiatric examinations, and Columbia–Suicide Severity Rating Scale (C-SSRS) (Posner et al. 2011), Clinical Global Impressions–Severity scale (CGI-S) (Guy, 1976), and extrapyramidal symptoms scales (Barnes Akathisia Rating Scale (Barnes 1989), Abnormal Involuntary Movement Scale (Guy 1976), and Simpson-Angus Scale (Simpson and Angus 1970)).
Blood sample collection and processing
Blood samples for determining cariprazine, DCAR, and DDCAR concentrations in plasma were collected using a prechilled 4-mL Vacutainer tube (containing K2EDTA as an anticoagulant) on days 1/2, 4/5, and 15/16 at 0 h (predose) and 1, 2, 4, 6, 8, 19, and 24 h post days 1, 4, and 15 dosing and on day 24 at 216 h after the day-15 dose; plasma was also collected predose on day 3. Blood samples for PK evaluation were centrifuged within 30 min of drawing at ≥2500g for 10 min at 4 °C and the plasma was harvested. After centrifugation, plasma samples were transferred into prechilled, coded polypropylene tubes. Samples were flash-frozen in an isopropyl alcohol/dry ice bath and stored at approximately −70 °C. Additional blood samples were collected for clinical laboratory analysis.
Cerebrospinal fluid collection
CSF samples were collected on day 3 (predose) and day 16 (approximately 24 h after day-15 dose). Lumbar punctures were performed using standard aseptic technique and a 20-mL Quincke or 25-mL Sprotte needle. A maximum of 10 mL of CSF was collected for PK analyses.
Bioanalytical method
Plasma and CSF concentrations of cariprazine, DCAR, and DDCAR were measured using a liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method, validated to demonstrate the accuracy, linearity, reproducibility, and precision of the analytical procedure. The lower limit of quantitation (LLOQ) for the plasma assay was 20:20:50 pg/mL for cariprazine/DCAR/DDCAR using a plasma sample volume of 200 μL; the LLOQ for the CSF assay was 5:5:10 pg/mL, respectively, using a sample volume of 250 μL.
PET and radiochemistry methods
[11C]-(+)-PHNO was prepared by N-acylation of the despropyl precursor with [11C]-propionyl chloride followed by reduction of the resulting amide with lithium aluminum hydride and purification by reverse-phase high-performance liquid chromatography (HPLC) (Wilson et al. 2005). All scans were 120 min, following a single bolus injection of [11C]-(+)-PHNO. Data were acquired on a Siemens High-Resolution Research Tomograph (HRRT, Siemens/CTI, Knoxville, TN). Data were acquired in list mode, reconstructed using the MOLAR algorithm (Carson et al. 2003) including correction for subject motion using an optical detection system (Vicra, NDI Systems, Waterloo, Ontario, Canada) and binned into a sequence of 33 frames of increasing duration (6 × 30 s, 3 × 1 min, 2 × 2 min, 22 × 5 min). A T1-weighted anatomical magnetic resonance imaging (MRI) scan was acquired for each subject. Regions of interest (ROIs) were drawn individually for each subject on their MRI; they included caudate and putamen, ventral striatum, globus pallidus, thalamus, substantia nigra/ventral tegmental area, and cerebellum as a reference tissue. Left and right sides were averaged. PET data were coregistered to subjects’ MRIs; ROIs were applied to the coregistered PET images. Time activity curves were generated as the average ROI activity in each frame.
Statistical analysis
Pharmacokinetics
The principal parameters describing the PK of cariprazine, DCAR, and DDCAR were derived from plasma concentrations using noncompartmental analysis with the validated software program Phoenix WinNonlin version 6.1 (Pharsight, St. Louis, MO). The plasma PK parameters for cariprazine and its metabolites were calculated after dosing on days 1, 4, and 15. Reported parameters included area under the curve (AUC) over 24 h, peak concentration (C max), time of maximum concentration (T max), and trough concentration (C min). Concentration in CSF (C csf) at 24 h after last dose was also determined.
PET analysis
Reference tissue-based kinetic modeling was performed on all data using the basis function version of the simplified reference tissue method (SRTM) (Gunn et al. 1997; Lammertsma and Hume 1996), with cerebellum as the reference tissue. The primary outcome measure was the binding potential relative to the nondisplaceable compartment (BPND) (Innis et al. 2007). The fractional change in BPND across conditions (∆BPND) was computed as the relative change between baseline BPND and BPND during each condition:
∆BPND data from each condition were entered into a nonlinear regression model to extract the specific contribution of D2 and D3 receptors (Girgis et al. 2011; Rabiner et al. 2009; Searle et al. 2010). In this model, BPND in each region is parsed into its D2 and D3 components from each scan and condition. We defined f D3 as the fraction of total BPND attributable to D3 binding, which in general, varies across regions but is similar across subjects. Under these assumptions, ∆BPND following cariprazine is:
where OCCD3 and OCCD2 are the occupancies at D3 and D2 receptors by cariprazine and its metabolites. For a given scan, OCCD3 and OCCD2 are treated as equal across brain regions (though not across receptor types), but their values vary across scans. The [11C]-(+)-PHNO BPND data were fitted to this equation with ∆BPND for each scan and region as input to the model. Two methods were applied. In method 1, f D3 for each region was treated as a fixed parameter according to the values in (Searle et al. 2010), and OCCD3 and OCCD2 were estimated for each subject and day, obtained as parameter estimates using ordinary linear least squares fitting. In method 2, in addition to the two occupancy parameters, f D3 was treated as a fitted parameter and all parameters were estimated by nonlinear least squares fitting. In each region, f D3 was treated as equal across subjects. These fits were done for each day leading to 54 or 60 ∆BPND dependent variables (10 subjects on day 1, 9 for all other days, 6 regions per subject) and 18 or 20 estimated parameters for method 1 (two occupancies for each subject on that day) or 24 or 26 estimated parameters for method 2 (six f D3s estimated for the day and two occupancies for each subject on that day). In theory, f D3 may vary across subjects but estimating it for each subject might require a richer data set (e.g., multiple doses within subject under each condition) to allow reliable estimation. Thus, single values across subjects, whether fixed or fitted, were considered more parsimonious, given the available data and the general consistency of f D3 observed across primate species and subjects (Rabiner et al. 2009; Searle et al. 2010; Slifstein et al. 2012).
Pharmacokinetic-pharmacodynamic PET analysis
The relationships between D3 and D2 receptor occupancy and plasma levels of cariprazine and its metabolites at the time of PET were explored by fitting the plasma concentration of total active cariprazine (cariprazine + DCAR + DDCAR) versus occupancy data using an E max model from the library of pharmacodynamic (PD) models provided within Phoenix WinNonlin (version 6.1):
where EC50 is the expected concentration for 50 % occupancy and E max is the maximum occupancy. E max upper bounds were set to 100 % for this analysis. Additionally, administered doses were fitted to a similar equation to obtain ED50, the expected dose leading to 50 % occupancy according to:
Results
Patients
Nine patients (seven men, two women) aged 24 to 55 years were enrolled. One patient (cohort 1) experienced emesis on day 4 and withdrew; this subject’s day 1 data were included in the analysis.
Pharmacokinetics
Plasma concentrations of cariprazine, DCAR, and DDCAR in cohort 1 are presented in Fig. 1. Total active cariprazine during the 24-h interval after day-1 dosing consists primarily of parent drug; exposure of the DCAR metabolite slowly increased, while plasma concentration of the DDCAR metabolite was below the LLOQ of the bioanalytical assay (Fig. 1a). After subchronic dosing (15 days), plasma active drug level consisted primarily of cariprazine and DDCAR, while DCAR level was substantially lower (AUC ∼25–40 % of parent drug exposure) (Fig. 1b). On day 24, 9 days following the final dose, plasma levels of cariprazine and DCAR decreased substantially, while DDCAR level decreased to a smaller extent (from 21.22 ng/ml at 24 h post day-15 dose to 13.11 ng/mL on day 24), suggesting a longer half-life (Fig. 1c).

Plasma concentration (mean ± SD) of cariprazine and its metabolites as a function of time in cohort 1. Cohort 1 received one 1.5 mg tablet on day 1, 3 mg on day 2, 6 mg once daily on days 3–4, 9 mg once daily on days 5–6, and 12 mg once daily on days 7–15. The concentration versus time profiles for all three active moieties of cariprazine are shown during the 24-h interval after dose on day 1 (a), during the 24-h interval after dose on day 15 (b), and interval from days 1 to 24 (with last dose administered on day 15) (c)
Because the three cohorts had different periods of dose up-titration to improve drug tolerability, results are presented separately by day of treatment (Supplemental Table 1). Plasma exposure (C max, C min, and AUC) of all three components, in general, increased with dose and were consistent with dose proportional PK by day 15.
In all three cohorts, mean CSF concentrations of DCAR and DDCAR 24 h after the final dose ranged from 18 to 35 and 94–120 %, respectively, of those of cariprazine (Supplemental Table 1); corresponding mean plasma trough (C min) concentrations of DCAR and DDCAR (based on nM concentrations) were 29–53 and 139–203 %, respectively, of those of cariprazine. On average, concentrations of cariprazine, DCAR, and DDCAR in CSF were approximately 5–10, 4–6, and 3–8 % of the respective concentrations in plasma.
PET results
Figure 2 shows ∆BPND by condition and dose for each region. Under each dosing condition (acute, day 4/5, subchronic), there was a clear dose response, with the magnitude of ∆BPND increasing as a function of dose. Both occupancy models gave similar results for subchronic dosing, but model 2 did not reliably estimate f D3 on days 1 and 4 due to the similarity of occupancy at both receptor types at these times; therefore, we report method 1 results only (f D3 fixed to literature values). Figure 3 depicts the receptor occupancies at D3 and D2 receptors in each cohort and at each time of the PET measurement. In all three cohorts, receptor occupancies increased from days 1 to 4 to 15 as the doses were increased with initial slow up-titration, and decreased from days 15 to 24 following cessation of cariprazine administration. A monotonic dose-occupancy relationship was observed at both receptor types, with days 4, 15, and 24 following the order occupancy (cohort 1) > occupancy (cohort 2) > occupancy (cohort 3). Near-complete (100 %) receptor occupancy at both receptor types was observed at 12 mg/day on day 15. At the lowest dose of 1 mg/day, mean D3 and D2 receptor occupancy on day 15 was 76 % (range 58–89 %) and 45 % (range, 14–64 %), respectively. At the intermediate 3-mg/day dose, mean D3 and D2 receptor occupancies were 92 % (range, 86–96 %) and 79 % (range, 68–88 %), respectively. Occupancy was reduced in all brain regions in all three cohorts 9 days after the last dose of cariprazine, suggesting reversibility of receptor binding. Mean receptor occupancy decreased from 76 % (range, 58–89 %) to 20 % (range, −2 to 31 %) for D3 and 45 % (range, 14–64 %) to 32 % (range, 23–48 %) for D2 between days 15 and 24 in cohort 3 after 1 mg of final daily dosing. Similarly, at the final dose of 3 mg in cohort 2, mean receptor occupancy decreased from 92 to 54 % for D3 and 79 to 40 % for D2. At the highest dose of 12 mg, drug exposure resulted in complete occupancy for both receptor types and did not decrease substantially even after 9 days of drug washout (D3 from 99 to 97 % and D2 from 95 to 77 %) suggesting that high levels of active drug were still present 9 days after the 12-mg dose on day 15.

The percent decrease in binding potential (∆BPND) by day and dose. Error bars are standard deviations. Acute doses were 1.5 mg (n = 3) and 0.5 mg (n = 6). Day 4/5 doses (day 4, n = 7; day 5, n = 1) were 6 mg (n = 2), 1 mg (n = 3), and 0.5 mg (n = 3). Subchronic doses (day 15, n = 6; day 11, n = 1, day 17, n = 1) were 12 mg (n = 2), 3 mg (n = 3), and 1 mg (n = 3). GP globus pallidus, PUT putamen, CAD caudate nucleus, VST ventral striatum, THAL thalamus, SN/VTA substantia nigra/ventral tegmental area

Occupancy estimates on different days of dosing using fixed literature values for the D3 fraction of regional [11 C]-(+)-PHNO BPND (method 1)
ED50 estimates and 95 % confidence intervals (CIs) for acute (day 1/4) and subchronic (day 15) doses were 1.52 (0.76, 3.05) and 0.30 (0.15, 0.59) mg for D3 occupancy and 1.84 (1.41, 2.40) and 1.03 (0.54, 2.05) mg for D2 occupancy using method 1. One subject taking 12 mg/day had a D3 occupancy estimate that exceeded 100 %. As a result, the dose-occupancy relationship for subchronic administration was also fitted with the D3 occupancy estimate from this subject constrained to 100 % and with the subject’s data dropped; this had a negligible effect on the estimated D3 ED50 for cariprazine, with values of 0.3049 and 0.3064 mg, respectively, compared to 0.3018 mg using the original data. The D3 versus D2 receptor selectivity ratio of cariprazine treatment was more apparent following subchronic dosing (range, 3.43–5.75) than after an acute dose (range, 1.21–1.31), and was similar for both methods. Selectivity estimates for D3 compared with D2 overall ranged from about 3- to 6-fold after subchronic dosing.
Pharmacokinetic-pharmacodynamic analyses
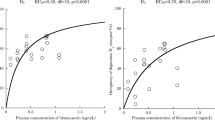
Occupancy at D3 and D2 receptors and plasma concentrations of total active cariprazine (cariprazine + DCAR + DDCAR) at the time of the PET scans (4 h postadministration) following acute (days 1 and 4) and subchronic treatment (day 15) were fitted to an E max model (Fig. 4).

Receptor occupancy versus total active cariprazine in plasma after dosing on days 1, 4, and 15 fitted to an E max model. D3 and D2 receptor occupancies versus total active cariprazine plasma concentration are shown after acute treatment (a, b) and subchronic treatment (c, d)
The slope for receptor occupancy was quite steep after acute dosing for both receptors suggesting relatively small drug concentration necessary to occupy both receptors. The EC50 (± SE) was similar between the two receptors (D3 = 1.42 [0.37] nM and D2 = 1.77 [0.39] nM) with a ratio of D3/D2 of about 1.25, consistent with that observed from ED50 analysis.
PK-PD analysis of data collected after subchronic administration (15-day dosing) allowed more time for equilibration between plasma and brain regions, as well as accumulation of slow-forming active metabolite(s), DDCAR in particular. This demonstrated EC50 (± SE) estimates for D3 and D2 receptor-rich regions of 3.84 (1.85) and 13.03 (6.8) nM, respectively. The ratio of EC50 for D3/D2 receptor occupancy of 3.4 suggests preferred affinity at D3 compared to D2 receptors after multiple-daily dosing of cariprazine for about 2 weeks.
Discussion
The objectives of this study were to assess D3 and D2 receptor occupancy of cariprazine and its two major active metabolites, DCAR and DDCAR, in patients with schizophrenia using [11C]-(+)-PHNO PET imaging, and to explore the relationship between plasma concentrations of cariprazine/DCAR/DDCAR and D3 and D2 receptor occupancy.
Cariprazine was well absorbed following oral administration. Following the first dose, cariprazine was the most prominent moiety in plasma. The mean plasma AUC of cariprazine on day 1 was 5- to 10-fold higher than the desmethyl metabolite DCAR. The di-desmethyl metabolite DDCAR was not measurable after the first dose on day 1. After 2 weeks of multiple-dose administration, DDCAR and cariprazine were the most prominent moieties in plasma; hence, DDCAR may be responsible for a significant portion of the pharmacological activity of cariprazine following chronic oral administration. In vitro plasma protein binding in various species (including human) is about 96 % (data on file). The concentrations of cariprazine, DCAR, and DDCAR in CSF in this study were 5–10, 4–6, and 3–8 % of the respective concentrations in plasma. The CSF/plasma ratio observed in this study was comparable to the free fraction in plasma (∼4 %), indicating equilibration of free drug and its active metabolites across the blood-brain barrier between plasma and CSF compartments.
The observed PET data show that in patients with schizophrenia, cariprazine robustly binds to both D3 and D2 receptors, and is moderately D3-preferring following subchronic (15-day) dosing with ED50 and EC50 ratios in the three to six range. These results are consistent with preclinical studies showing binding to both D3 and D2 receptors in vivo (Gyertyán et al. 2011) and preferential binding to D3 receptors in vitro (Kiss et al. 2010) as well as PET studies in nonhuman primates (Seneca et al. 2011; Toth et al. 2013) and humans (Keator et al. 2009; Laszlovszky et al. 2007; Potkin et al. 2009) showing that cariprazine binds to D2 receptors. However, these prior PET studies were performed with [11C]raclopride and [18F]fallypride, and did not allow for separate assessment of D3 and D2 receptor binding. In addition, the maximum cariprazine doses in the two prior human studies (1.0 and 3.0 mg) were lower than doses investigated in clinical efficacy studies.
Both ED50 and EC50 demonstrated comparable D3/D2 receptor selectivity after acute drug administration (days 1 and 4), while 2 weeks of daily dosing showed 3- to 6-fold higher selectivity (lower EC50 and ED50) for D3 over D2 receptors. The D3 receptor-preferring nature of cariprazine and its metabolites following subchronic dosing was similar using either method that was tested (methods 1 or 2). Lower selectivity following acute dosing may be due in part to rapid changes in drug and metabolite levels in the body during this transient period, but additionally, the metabolites of cariprazine are more D3 receptor preferring than cariprazine itself. In vitro, DCAR is approximately 2-fold more potent than cariprazine at human D3 receptors and DDCAR displayed somewhat lower affinity (∼2-fold) than cariprazine at human D2 receptors (Tadori et al. 2011). Considering the contribution of DDCAR to total active cariprazine was greater after 2 weeks of dosing than after the initial dose on day 1, the differential affinities of these moieties may help explain the greater selectivity for D3 receptors after subchronic dosing. The active metabolite, DDCAR, is formed slowly and takes a few weeks to reach steady state. In order to improve tolerability in patients with schizophrenia, clinical studies with longer treatment duration utilized a dose up-titration during the first week of treatment. During up-titration, cariprazine levels increased rapidly and contributed to the immediate pharmacological effect. It took about 3 weeks to achieve 90 % of steady state for total active cariprazine concentration and 4 weeks for DDCAR; at steady state, about 70 % of the total active concentration comprised the metabolite DDCAR with cariprazine and DCAR contributing the remaining 20 and 10 %, respectively (Nakamura et al. 2016). Hence, the complex PK and differential receptor affinities of cariprazine and its two major metabolites result in rapid onset of pharmacological effect mostly by the parent cariprazine during the first week of treatment followed by a sustained maintenance of anti-D3/D2 activity by the longer acting metabolite, DDCAR, following chronic dosing. The D3/D2 binding profiles of the metabolites may also explain why D3 receptor binding of cariprazine is sustained and increases with subchronic dosing, while currently available antipsychotic agents seem to bind to D3 receptors in expected proportions after acute dosing (Girgis et al. 2015) but not after subchronic dosing (Graff-Guerrero et al. 2009; McCormick et al. 2010; Mizrahi et al. 2011).
Limitations of this study include the relatively small sample size, different dosing schedules, and the fact that we could only measure occupancy for cariprazine and its metabolites combined without estimating their individual contributions. However, these limitations do not prevent clear interpretation of the main results.
In conclusion, this PET imaging study using [11C]-(+)-PHNO in patients with schizophrenia demonstrated that cariprazine and/or its metabolites bind robustly to dopamine D2 and D3 receptors in vivo; this binding is moderately D3 receptor-preferring, with ED50 and EC50 ratios in the 3- to 6-fold range. All currently available antipsychotic medications in the USA are more selective for D2 than D3 receptors. Evidence from animal models suggests that combining potent D3 with D2 receptor antagonism resulting in high brain occupancy and effective modulation of both receptors may yield antipsychotic compounds with a better profile for improving negative symptoms as well as cognitive deficits (Gyertyán et al. 2008; Kiss et al. 2008). Cariprazine, with its D3-preferring profile, may provide a new treatment option for patients with schizophrenia, with a potential for advantages in treating negative symptoms and cognitive impairment.
References
APA (2000) Diagnostic and statistical manual of mental disorders, fourth edition, text revision. American Psychiatric Association, Washington DC
Barnes TR (1989) A rating scale for drug-induced akathisia. Br J Psychiatry 154:672–676
Carson R, Barker W, Liow JS (2003) Design of a motion-compensation OSEM list-mode algorithm for resolution-recovery reconstruction for the HRRT. In: Metzier S (ed) 2003 I.E. nuclear science symposium (IEEE NUCLEAR SCIENCE SYMPOSIUM - CONFERENCE RECORD), Portland, OR, USA, pp 3281–3285
Debelle M, Faradzs-zade S, Szatmari B, Nagy K (2014) Cariprazine in negative symptoms of schizophrenia: post hoc analyses of a fixed-dose, placebo- and active-controlled trial. Eur Neuropsychopharmacol 24:S534
Debelle M, Németh G, Szalai E, Szatmári B, Harsányi J, Barabássy A, Laszlovszky I (2015) Cariprazine as monotherapy for the treatment of schizophrenia patients with predominant negative symptoms: a double-blind, active controlled trial. Eur Neuropsychopharamcol 25:S510
First MB, Williams JBW, Spitzer RL, Gibbon M (2007) Structured clinical interview for DSM-IV-TR Axis I disorders, clinical trials version (SCID-CT). Biometrics Research, New York State Psychiatric Institute, New York
Gallezot JD, Beaver JD, Gunn RN, Nabulsi N, Weinzimmer D, Singhal T, Slifstein M, Fowles K, Ding YS, Huang Y, Laruelle M, Carson RE, Rabiner EA (2012) Affinity and selectivity of [(1)(1)C]-(+)-PHNO for the D3 and D2 receptors in the rhesus monkey brain in vivo. Synapse 66:489–500
Ginovart N, Willeit M, Rusjan P, Graff A, Bloomfield PM, Houle S, Kapur S, Wilson AA (2007) Positron emission tomography quantification of [C-11]-(+)-PHNO binding in the human brain. J Cereb Blood Flow Metab 27:857–871
Girgis R, Xu X, Miyake N, Easwaramoorthy B, Gunn R, Rabiner E, Abi-Dargham A, Slifstein M (2011) In vivo binding of antipsychotics to D3 and D2 receptors: a PET study in baboons with [11]-(+)-PHNO. Neuropsychopharmacology:887–895
Girgis RR, Xu X, Gil RB, Hackett E, Ojeil N, Lieberman JA, Slifstein M, Abi-Dargham A (2015) Antipsychotic binding to the dopamine-3 receptor in humans: a PET study with [(11)C]-(+)-PHNO. Schizophr Res 168:373–376
Graff-Guerrero A, Mizrahi R, Agid O, Marcon H, Penny B, Rusjan P, Wilson AA, Zipursky RB, Kapur S (2009) The dopamine D2 receptors in high-affinity state and D3 receptors in schizophrenia: a clinical [11C]-(+)-PHNO PET study. Neuropsychopharmacology 34:1078–1086
Graff-Guerrero A, Redden L, Abi-Saab W, Katz DA, Houle S, Barsoum P, Bhathena A, Palaparthy R, Saltarelli MD, Kapur S (2010) Blockade of [11C](+)-PHNO binding in human subjects by the dopamine D3 receptor antagonist ABT-925. Int J Neuropsychopharmacol 13:273–287
Gross G, Drescher K (2012) The role of dopamine D(3) receptors in antipsychotic activity and cognitive functions. Handb Exp Pharmacol:167–210
Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ (1997) Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. NeuroImage 6:279–2787
Gurevich EV, Joyce JN (1999) Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology 20:60–80
Guy W (1976) The abnormal movement scale. ECDEU assessment manual for psychopharmacology. National Institute of Mental Health, Rockville, MD, pp. 218–222 DHEW Publication No. 76-338
Gyertyán I, Kiss B, Sághy K, Laszy J, Szabó G, Szabados T, Gémesi LI, Pásztor G, Zájer-Balázs M, Kapás M, Csongor E, Domány G, Tihanyi K, Szombathelyi Z (2011) Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int 59:925–935
Gyertyán I, Sághy K (2004) Effects of dopamine D3 receptor antagonists on spontaneous and agonist-reduced motor activity in NMRI mice and Wistar rats: comparative study with nafadotride, U 99194A and SB 277011. Behav Pharmacol 15:253–262
Gyertyán I, Sághy K, Laszy J, Elekes O, Kedves R, Gémesi LI, Pásztor G, Zájer-Balázs M, Kapás M, Ágai Csongor E, Domány G, Kiss B, Szombathelyi Z (2008) Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: II. Behavioural characterisation of RG-15. Naunyn Schmiedeberg’s Arch Pharmacol 378:529–539
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27:1533–1539
Kapás M, Mészáros G, Yu B, Shi J, Knoche B, Laszlovszky I, Bolton G (2008) Comparison of the pharmacokinetic behavior of RGH 188 in schizophrenic patients and healthy volunteers. Eur Neuropsychopharmacol 18:S433
Keator D, Mukherjee J, Preda A, Highum D, Gage A, Potkin S (2009) Dopamine D2 and D3 receptor occupancy of cariprazine in schizophrenic patients. Schizophr Bull 35:154
Kiss B, Horváth A, Némethy Z, Schmidt E, Laszlovszky I, Bugovics G, Fazekas K, Hornok K, Orosz S, Gyertyán I, Ágai-Csongor E, Domány G, Tihanyi K, Adham N, Szombathelyi Z (2010) Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther 333:328–340
Kiss B, Laszlovszky I, Horváth A, Némethy Z, Schmidt E, Bugovics G, Fazekas K, Gyertyán I, Ágai-Csongor E, Domány G, Szombathelyi Z (2008) Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: I. Neurochemical characterisation of RG-15. Naunyn Schmiedeberg’s Arch Pharmacol 378:515–528
Lammertsma AA, Hume SP (1996) Simplified reference tissue model for PET receptor studies. NeuroImage 4:153–158
Laszlovszky I, Németh G, Mészáros GP, Kapás M, Brooks DJ, Pavese N, SZombathelyi Z (2007) Dopamine D2/D3 receptor occupancy of RGH-188, a D3/D2, antagonist/partial agonist antipsychotic, in healthy volunteers. Eur Neuropsychopharmacol 17(Suppl 4):S455
Laszy J, Laszlovszky I, Gyertyán I (2005) Dopamine D3 receptor antagonists improve the learning performance in memory-impaired rats. Psychopharmacology 179:567–575
McCormick PN, Kapur S, Graff-Guerrero A, Raymond R, Nobrega JN, Wilson AA (2010) The antipsychotics olanzapine, risperidone, clozapine, and haloperidol are D2-selective ex vivo but not in vitro. Neuropsychopharmacology 35:1826–1835
Mészáros G, Kapás M, Borsos M, Laszlovszky I, Németh G, Tihanyi K, Szombathelyi Z (2007) Pharmacokinetics of RGH 188, a novel dopamine D3/D2 antagonist/partial agonist atypical antipsychotic, in healthy male volunteers. Eur Neuropsychopharmacol 17:S451
Millan MJ, Dekeyne A, Rivet JM, Dubuffet T, Lavielle G, Brocco M (2000) S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther 293:1063–1073
Millan MJ, Di Cara B, Dekeyne A, Panayi F, De Groote L, Sicard D, Cistarelli L, Billiras R, Gobert A (2007) Selective blockade of dopamine D(3) versus D(2) receptors enhances frontocortical cholinergic transmission and social memory in rats: a parallel neurochemical and behavioural analysis. J Neurochem 100:1047–1061
Mizrahi R, Agid O, Borlido C, Suridjan I, Rusjan P, Houle S, Remington G, Wilson AA, Kapur S (2011) Effects of antipsychotics on D3 receptors: a clinical PET study in first episode antipsychotic naive patients with schizophrenia using [11C]-(+)-PHNO. Schizophr Res 131:63–68
Nakamura T, Kubota T, Iwakaji A, Imada M, Kapas M, Morio Y (2016) Clinical pharmacology study of cariprazine (MP-214) in patients with schizophrenia (12-week treatment). Drug Des Devel Ther 10:327–338
Nyberg S, Nakashima Y, Nordstrom AL, Halldin C, Farde L (1996) Positron emission tomography of in-vivo binding characteristics of atypical antipsychotic drugs. Review of D2 and 5-HT2 receptor occupancy studies and clinical response. Br J Psychiatry Suppl 29:40–44
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, Currier GW, Melvin GA, Greenhill L, Shen S, Mann JJ (2011) The Columbia-suicide severity rating scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277
Potkin S, Keator D, Mukherjee J, Preda A, Highum D, Gage A, Xie J, Ghahramani P, Laszlovszky I (2009) Dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacol 19:S316
Rabiner E, Slifstein M, Nobrega J, Plisson C, Huiban M, Raymond R, Diwan M, Wilson AA, McCormick P, Gentile G, Gunn RN, Laruelle M (2009) In vivo quantification of regional dopamine-D3 receptor binding potential of (+)PHNO: studies in non-hman primates and transgenic mice. Synapse 63:782–793
Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, Boyfield I, Branch CL, Cilia J, Coldwell MC, Hadley MS, Hunter AJ, Jeffrey P, Jewitt F, Johnson CN, Jones DN, Medhurst AD, Middlemiss DN, Nash DJ, Riley GJ, Routledge C, Stemp G, Thewlis KM, Trail B, Vong AK, Hagan JJ (2000) Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J Pharmacol Exp Ther 294:1154–1165
Redden L, Rendenbach-Mueller B, Abi-Saab WM, Katz DA, Goenjian A, Robieson WZ, Wang Y, Goss SL, Greco N, Saltarelli MD (2011) A double-blind, randomized, placebo-controlled study of the dopamine D(3) receptor antagonist ABT-925 in patients with acute schizophrenia. J Clin Psychopharmacol 31:221–225
Sautel F, Griffon N, Sokoloff P, Schwartz JC, Launay C, Simon P, Costentin J, Schoenfelder A, Garrido F, Mann A, et al. (1995) Nafadotride, a potent preferential dopamine D3 receptor antagonist, activates locomotion in rodents. J Pharmacol Exp Ther 275:1239–1246
Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, Mugnaini M, Griffante C, Wilson AA, Merlo-Pich E, Houle S, Gunn R, Rabiner EA, Laruelle M (2010) Imaging dopamine D-3 receptors in the human brain with positron emission tomography, [C-11]PHNO, and a selective D-3 receptor antagonist. Biol Psychiatry 68:392–399
Seeman P (2001) Antipsychotic drugs, dopamine receptors, and schizophrenia. Clin Neurosci Res 1:53–60
Seneca N, Finnema SJ, Laszlovszky I, Kiss B, Horvath A, Pasztor G, Kapas M, Gyertyan I, Farkas S, Innis RB, Halldin C, Gulyas B (2011) Occupancy of dopamine D(2) and D(3) and serotonin 5-HT(1)A receptors by the novel antipsychotic drug candidate, cariprazine (RGH-188), in monkey brain measured using positron emission tomography. Psychopharmacology 218:579–587
Sigala S, Missale C, Spano P (1997) Opposite effects of dopamine D2 and D3 receptors on learning and memory in the rat. Eur J Pharmacol 336:107–112
Simpson GM, Angus JW (1970) A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 212:11–19
Slifstein M, Rabiner EA, Gunn RN (2012) Imaging dopamine D3 receptors in vivo. In: Seeman P, Madras B, Johnson JE (eds) Imaging of the human brain in health and disease. Neuroscience-Net
Tadori Y, Forbes RA, McQuade RD, Kikuchi T (2011) In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3 receptors. Eur J Pharmacol 668:355–365
Toth M, Varrone A, Steiger C, Laszlovszky I, Horvath A, Kiss B, Gyertyan I, Adham N, Halldin C, Gulyas B (2013) Brain uptake and distribution of the dopamine D3 /D2 receptor partial agonist [11 C]cariprazine: an in vivo positron emission tomography study in nonhuman primates. Synapse 67:258–264
Waters N, Lofberg L, Haadsma-Svensson S, Svensson K, Sonesson C, Carlsson A (1994) Differential effects of dopamine D2 and D3 receptor antagonists in regard to dopamine release, in vivo receptor displacement and behaviour. J Neural Transm Gen Sect 98:39–55
Waters N, Svensson K, Haadsma-Svensson SR, Smith MW, Carlsson A (1993) The dopamine D3-receptor: a postsynaptic receptor inhibitory on rat locomotor activity. J Neural Transm Gen Sect 94:11–19
Willeit M, Ginovart N, Kapur S, Houle S, Hussey D, Seeman P, Wilson AA (2006) High-affinity states of human brain dopamine D2/3 receptors imaged by the agonist [C-11]-(+)-PHNO. Biol Psychiatry 59:389–394
Wilson AA, McCormick P, Kapur S, Willeit M, Garcia A, Hussey D, Houle S, Seeman P, Ginovart N (2005) Radiosynthesis and evaluation of [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J Med Chem 48:4153–4160
Zimnisky R, Chang G, Gyertyan I, Kiss B, Adham N, Schmauss C (2013) Cariprazine, a dopamine D(3)-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology 226:91–100
Acknowledgments
Editorial support for manuscript preparation was provided by Paul Ferguson, M.S., of Prescott Medical Communications Group, Chicago, Illinois, a contractor of Forest Research Institute, an Allergan affiliate. The clinical study was monitored by Dahlia Henry of Forest Research Institute, an Allergan affiliate. The authors would like to acknowledge the nursing staff of the Clinical Neuroscience Research Unit, the Schizophrenia Neuropharmacology Research Group at Yale (SNRGY), and the Yale PET Center for their critical contributions towards the success of this study. All experiments in this study comply with the current laws of the country in which they were performed.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Support for this research and publication was funded by Forest Laboratories, LLC, Jersey City, New Jersey, USA, an Allergan affiliate, and Gedeon Richter, Plc, Budapest, Hungary. Forest Laboratories and Gedeon Richter were involved in the study design, analysis, interpretation of data, and decision to present these results. Antonia Periclou, Suresh Durgam, and Nika Adham acknowledge a potential conflict of interest as employees of Forest Research Institute, an Allergan affiliate. Yih Lee, Parviz Ghahramani, and Ashok Rakhit acknowledge a potential conflict of interest as employees of Forest Research Institute at the time of the study. István Laszlovszky, Béla Kiss, and Margit Kapás acknowledge a potential conflict of interest as employees of Gedeon Richter Plc. Ragy Girgis has received research funding from Otsuka and Genentech. Mark Slifstein has served as a consultant to Amgen and has received research funding from Otsuka. Anissa Abi-Dargham has served as a consultant for Roche, Otsuka, Forum, and Amgen and has received research support from Takeda, Forest Laboratories, Pierre Fabre, and CHDI Foundation. Nabeel Nabulsi had received royalty payments from the University of Texas M.D. Anderson Cancer Center from patented work. Deepak D’Souza, Richard Carson, and Yiyun Huang have no disclosures to report pertaining to this work.
Electronic supplementary material
Fig. S1
Study Design and Cariprazine Dosing Schedule (GIF 28 kb)
Supplemental Table 1
(DOCX 30 kb)
Supplemental Table 2
(DOCX 25 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Girgis, R.R., Slifstein, M., D’Souza, D. et al. Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [11C]-(+)-PHNO. Psychopharmacology 233, 3503–3512 (2016). https://doi.org/10.1007/s00213-016-4382-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-016-4382-y