
Abstract
Background
The endocannabinoid (eCB) system, an endogenous lipid signaling system, appears to be dysregulated in depression. The role of endocannabinoids (eCBs) as potent immunomodulators, together with the accumulating support for a chronic low-grade inflammatory profile in depression, suggests a compelling hypothesis for a fundamental impairment in their intercommunication, in depression.
Objective
We aim to review previous literature on individual associations between the immune and eCB systems and depression. It will focus on peripheral and central mechanisms of crosstalk between the eCB and immune systems. A potential dysregulation in this crosstalk will be discussed in the context of depression.
Results
Investigations largely report a hypoactivity of the eCB system and increased inflammatory markers in individuals with depression. Findings depict a multifaceted communication whereby immunocompetent and eCB-related cells can both influence the suppression and enhancement of the other’s activity in both the periphery and central nervous system. A dysregulation of the eCB system, as seen in depression, appears to be associated with central and peripheral concentrations of inflammatory agents implicated in the pathophysiology of this illness.
Conclusion
The eCB and immune systems have been individually associated with and implicated in pathogenic mechanisms of depression. Both systems tightly regulate the other’s activity. As such, a dysregulation in this crosstalk has potential to influence the onset and maintenance of this neuropsychiatric illness. However, few studies have investigated both systems and depression conjointly. This review highlights the demand to consider joint eCB-immune interactions in the pathoetiology of depression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An increasing number of pathways external to the brain are gaining support in explaining the pathogenesis of major depressive disorder (MDD). In particular, findings suggest an involvement of immunedysregulation (Maes et al. 1995a; Raison et al. 2006; Dantzer et al. 2008; Dowlati et al. 2010; Zunszain et al. 2013) and alterations of the endocannabinoid (eCB) signalling system (Hillard and Liu 2014) in the onset and maintenance of this mood disorder.
Research has supported the involvement of immunedysregulation and more specifically, a low-grade inflammation, in depression (Berk et al. 2013). Findings reveal heightened levels of inflammatory markers present in the peripheral blood and cerebral spinal fluid (CSF) of depressed individuals, compared to healthy controls. The influences of this chronic inflammatory state have been implicated in virtually every neuropathophysiologic abnormality associated with major depression (Miller et al. 2009).
The involvement of the eCB system in MDD has been suggested more recently. The eCB system is a homeostatic mechanism identified following the discovery of the main psychoactive constituent in marijuana, ∆9-tetrahydrocannabinol (THC) (Gaoni and Mechoulam 1964). This system refers to three main components: (1) the endogenous lipid transmitters: endocannabinoids (eCBs) anandamide (AEA) and 2-arachidonoylglycerol (2-AG) (Devane et al. 1992; Mechoulam et al. 1995); (2) the target of these eCBs: G-protein-coupled receptors, cannabinoid receptor type 1 (CB1R) and 2 (CB2R) (Matsuda et al. 1990; Munro et al. 1993); and (3) enzymes involved in the synthesis, namely N-acylphosphatidylethanolamine-hydolyzing phospholipase D (NAPE-PLD) and diacylglycerol lipase (DAGL) (Bisogno et al. 2003; Okamoto et al. 2004) and degradation enzymes including fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) (Cravatt et al. 1996; Karlsson et al. 1997). Generated from membrane-bound glycerophospholipids (Wang and Ueda 2009), eCBs refer to the wide spectrum of lipid-derived fatty acid structural analogues (Alexander and Kendall 2007) of which AEA and 2-AG are the best described (Fig. 1). These naturally occurring cannabinoids present similar psychoactive properties to THC. They act on both receptors CB1R and CB2R. There is an increasing interest into eCB-like molecules that converge on common catabolic and metabolic pathways, notably non-cannabinoid AEA structural analogues, palmitoylethanolamine and oleoylethanolamine (Alexander and Kendall 2007). Involvement of alternative targets, non-classical receptors, such as peroxisome proliferator-activated receptor (PPAR)-α and transient receptor potential cation channel sub-family V member 1 (TRPV1), are also being investigated. However, investigations into associations between the aforementioned and depression are scarce. As such, influences of 2-AG and AEA will make up the evidence base for this discussion.

AEA and 2-AG are synthesised from enzymes, NAPE-PLD and DAGL, respectively, from glycerophospholipids in the phospholipid bilayer of the cellular membrane. Both eCBs bind to intracellular and extracellular CB1R and CB2R. Intracellular degradation of AEA and 2-AG occurs when bound to FAAH and MAGL, respectively. Products of this are AA and either glycerol, from 2-AG, or ethanolamine, from AEA
Interestingly, there is a clear divide in expression levels between tissues, with CB1R being expressed most commonly in the brain and CB2R principally located across an array of immune cells in the periphery (Malfitano et al. 2014). CB1R and CB2R have thus shown differential involvement in neuromodulation and immunoregulation, respectively (Castillo et al. 2012).
This review will focus on the possible associations between the immune and eCB systems and depression, both independently of each other and interdependently. It will highlight pathophysiological changes associated with chronic activation of the immune system that may be mechanistically relevant in facilitating the pathogenesis and potentiation of this neuropsychiatric disorder. As such, individual involvement of eCB activity and inflammation in depression will be outlined initially, followed by an examination of the evidence relevant to their intercommunication, in the context of depression.
The inflammatory model of depression
Recent meta-analyses provide prominent support for an association between immune dysfunction and depression. Patients with MDD showed elevated levels of several inflammatory signalling proteins, such as IL-6, IL-1, tumour necrosis factor-alpha (TNF-α) and C-reactive protein (CRP), in both serum and plasma (Howren et al. 2009; Dowlati et al. 2010). Moreover, a dose-response relationship has been observed between severity of symptoms and levels of pro-inflammatory cytokines as measured in a population continuum from community-based individuals to those clinically depressed (Howren et al. 2009). Furthermore, exogenous administration of the pro-inflammatory cytokine interferon (IFN)-α, a recommended immunotherapeutic treatment for chronic hepatitis C viral infection, leads to the onset of depression in around 30 % of these patients (Asnis and De La Garza 2006). This is believed to be as a result of the increased inflammatory response (McNamara and Lotrich 2012). A close relationship has also been reported between dose of IFN-α and subsequent changes in mood (Baraldi et al. 2012).
Further support comes from reports depicting an increasingly evident comorbidity between depression and inflammatory conditions, particularly those of an autoimmune-nature (Iseme et al. 2014). A study measuring the prevalence of comorbidity between the inflammatory disease, psoriasis and depression, found that 40 % of individuals diagnosed score positively for depressed mood and 27 % were diagnosed with MDD (Zaher et al. 2010). Interestingly, in a subsequent investigation, etanercept, a biopharmaceutical with anti-inflammatory properties, significantly reduced the depressive symptomatology presented in the same patient group, independently of any improvements in the condition (Tyring et al. 2006). Other anti-inflammatory drugs, including celecoxib (a cyclooxygenase-2 inhibitor), minocycline and acetyl salicylic acid, have been similarly effective as adjunctive therapies in reducing depressive symptoms (Mendlewicz et al. 2006; Soczynska et al. 2012; Maciel et al. 2013; Köhler et al. 2014). Interestingly, current psychotropic treatments have also been seen to possess anti-inflammatory qualities, suggesting a possible contribution of immunoregulation in their efficacy (Maes et al. 1999; Cattaneo et al. 2013; Horowitz et al. 2014).
Microglia activation is one of the mechanisms by which peripheral immune challenges can alter brain functioning (Dantzer et al. 2008). Increased microglia cell activation in the hippocampus and a concomitant increase in central TNF-α levels have been observed upon a peripheral immune challenge (Riazi et al. 2008). Similar results were obtained following peripheral administration of IL-2 (Schneider et al. 2012). Alongside elevated peripheral concentrations of inflammatory markers, both preclinical and clinical studies in depression elude to microglial activation. Investigations involving animal models of depression delineate altered expression of microglial activation markers, as well as chronicity-dependent fluctuations in microglial concentration in areas of the brain associated with mood regulation (Hinwood et al. 2012; Wohleb et al. 2012; Couch et al. 2013; Kreisel et al. 2014). Additionally, a post-mortem study also observed significant microgliosis in depressed suicide victims (Steiner et al. 2008). Not all studies agreed, as a PET scan revealed no significant differences in microglia activation in depressed individuals (Hannestad et al. 2013). Interestingly, the recent discovery of the central nervous system lymphatic system sheds new light into the possible mechanisms that may be involved in the pathogenesis of depression via neuroinflammation (Louveau et al. 2015). Regardless of the mechanisms responsible for the resulting increase in CNS inflammation following peripheral inflammation, elevated inflammatory markers within the CNS are a common finding in the pathophysiology of depression (Levine et al. 1999; Raison et al. 2006, 2009; Howren et al. 2009; Miller et al. 2009; Dowlati et al. 2010). Such unabated inflammation results in several molecular cascades associated with alterations in neurotransmitter metabolism and neuroendocrine function typically present in depression (Bakunina et al. 2015).
Dysregulation of the endocannabinoid system in depression
Preclinical findings
Receptors of the eCB system are widely distributed in regions of the brain markedly involved in mood regulation and mediate physiological systems associated with emotional reward, cognition and response to stress, dysregulations in all of which are frequently reported in individuals with major depression (Gorzalka and Hill 2011; Hill et al. 2012a).
The eCB system exerts its neuromodulatory effects through mediating short- and long-term forms of synaptic plasticity. Primarily involved in retrograde signalling, eCBs are synthesised and released post-synaptically in a neurotransmitter-dependent, depolarisation-induced manner. Cannabinoid receptors (CBR)s, principally located on axon terminals of glutamatergic and GABAergic neurons, are targeted by the endogenous ligands, instigating a cascade of chemical events that inhibit influx of Ca2+ via voltage-gated calcium channels, thereby discontinuing presynaptic release of neurotransmitters. Essential in the control of spatiotemporal eCB signalling is the intracellular enzymatic degradation of the eCBs in the presynaptic terminals, thereby terminating eCB-induced suppression of depolarisation. Key enzymes responsible for this finalising step are FAAH and MAGL (Castillo et al. 2012).
A hypoactivity of the eCB system has been observed in animal models of depression, where genetic manipulation and pharmacological interventions have provided the greatest insight into how behaviour may be affected by eCB signalling. Disruption of the eCB system resulting from CB1R knockout mice presented significant increases in depressive-like behaviour (Valverde and Torrens 2012). Similar results have been replicated with pharmacological antagonism of CB1Rs, resulting in a depressive-like phenotype (Beyer et al. 2010). Reduction in depressive-like behaviour have also been reported following FAAH inhibition (Vinod et al. 2012). Pioneering work in this field, conducted by the Piomelli group, includes the potent antidepressant-like effects elicited by a FAAH inhibitor, seemingly due to elevated levels of AEA, in the mouse models of depression, tail suspension and forced swim tests (Gobbi et al. 2005). This effect was again replicated in the chronic mild stress test in rats, while investigating the effect of chronic treatment with a FAAH inhibitor, observing a similar concomitant increase in AEA (Bortolato et al. 2007). Furthermore, overexpression of CB2Rs in mice resulted in resistance to depression following exposure to stressful situations compared to a control group (García-Gutiérrez et al. 2010). Antidepressant and anxiolytic effects ensued subsequent to intraperitoneal administration of a CB2R agonist, and this was reversed with administration of a pharmacological CB2R antagonist (Bahi et al. 2014).
Attenuation of eCB signalling is reported in chronic unpredictable stress (CUS), a well-established animal model that evokes a symptomatic profile similar to a human depressive phenotype (Willner 2005). Consequential effects of CUS in mice have delineated a decrease in CB1R function with resulting impairment of eCB-mediated retrograde signalling in the nucleus accumbens (Wang et al. 2010). Reduced eCB signalling is further supported by a significant decrease in levels of AEA in regions associated with behaviour, such as the hippocampus, hypothalamus, amygdala and ventral striatum (Hill et al. 2008a). Interestingly, despite previous reports of diminished CB1R functioning, CUS produces a contradictory increase in CB1R protein and mRNA expression in the prefrontal cortex (PFC) (Hill et al. 2008a; McLauglin et al. 2013). This increase however is suggested to be a compensatory effect of reduced eCB signalling as demonstrated by studies that reported decreased PFC AEA content (Hill et al. 2008a) and exacerbated depression following CB1R antagonist administration (McLauglin et al. 2013). Interestingly, CB1R upregulation has been documented in post-mortem studies of depressed individuals and depressed suicide victims compared to controls (Hungund et al. 2004; Choi et al. 2012).
Of note is the widely investigated involvement of eCB activity in the hypothalamic-pituitary-adrenal (HPA) axis. Dysregulation of the HPA axis is a commonly observed attribute of depression and has been implicated in its pathophysiology (Pariante and Lightman 2008; Zunszain et al. 2011). Through tonic regulation of the HPA-axis, eCB activity, mediated via CB1Rs, is responsible for maintaining basal tone (Hill and Tasker 2012b). Pharmacological blockade of the CB1Rs disrupts this tonic regulation and results in increased corticosterone levels (Patel et al. 2004; Wade et al. 2006). Likewise, CB1R KO mice exhibited depressive-like symptoms and a concomitantly higher basal adrenocorticotropic-releasing hormone level, in comparison to wild-type mice (Barna et al. 2004).
Further indication of eCB dysregulation in the aetiology of depression stems from its influence on hippocampal neurogenesis. CB2R functionality has been implicated in all key stages of neural progenitor cell cycle proliferation, differentiation and migration (Palazuelos et al. 2012; Galve-Roperh et al. 2013). Pharmacological blockade of CB1R suppressed dentate gyrus progenitor proliferation and reduced brain derived neurotrophic factor (BDNF) content in the hippocampus (Lee et al. 2009; Beyer et al. 2010). The reverse is also true, with increased stimulation of neurogenesis due to elevated BDNF following increased eCB signalling (Jiang et al. 2005; Duman and Monteggia 2006; Wyrofsky et al. 2015). It is important to consider that whilst the neurogenic theory of depression is supported by promising results in preclinical investigations, heterogeneity of clinical findings has provoked scepticism. At present, measures of neurogenesis in the human population are indirect and improvements in methodology are necessary to verify this hypothesis (Miller and Hen 2015). A reduction in hippocampal neuronal density resulted from consistent use of marijuana, the occurring process suggested to be mediated by the increased levels of THC (Landfield et al. 1988). Persistent use of marijuana also led to decreased CB1R expression (Hirvonen et al. 2012). Of relevance to depression is the aforementioned neural atrophy associated reductions in BDNF in the hippocampus, as this feature has been observed in major depression and associated animal models (Sheline et al. 1996; Santarelli et al. 2003; Lee and Kim 2010).
Clinical findings
Clinical investigations of eCB-associated effects in the area of depression are relatively sparse. Nonetheless, findings have, for the most part, verified the eCB-depression association reported in preclinical data. For example, rimonabant, a CB1R antagonist developed for use as an anti-obesity drug, was withdrawn from the market due to serious adverse effects on mood that resulted in the onset of depression and suicidal ideation (Topol et al. 2010). Several other pharmaceuticals, namely taranabant, otenabant, ibipinabant and surinabant, also CBR antagonists, were removed earlier on, during clinical trials, for similar reasons (Wyrofsky et al. 2015). Moreover, abnormally reduced levels of ethanolamine, a precursor and metabolite of AEA, are present in the CSF of a significant proportion of individuals diagnosed with MDD, compared to healthy controls (Ogawa et al. 2015). Comparable reductions in circulating levels of 2-AG have been found in depressed patients compared to controls, which progressively decreased over the duration of the disorder (Hill et al. 2008b). Furthermore, associations have been found between gene polymorphisms in CNR1, CNR2 (genes coding for CB1R and CB2R, respectively) and FAAH genes and behaviours characteristic of depression (Zajkowska et al. 2014). However, due to the promiscuous involvement of eCB activity in numerous physiological pathways, it is important to acknowledge the great number of confounding factors, in clinical investigations of an eCB-depression association. Variances in eCB concentrations can arise from a multitude of lifestyle modifications such as exercise, diet and changes in weight as well as intake of tobacco, coffee, alcohol and cannabis. Use of pharmaceuticals is also known to influence fluctuations in eCB levels. Most critically, the use of anti-depressants, anti-psychotics and anxiolytics can upregulate the eCB system (McPartland et al. 2014).
Crosstalk and dysregulation in the periphery
Current research delineates a complex bidirectional interaction between the peripheral eCB and immune systems, where circulating eCBs are not only affected by the immune system but also mediate transient effects on the immune response. Predominantly, CB2Rs and, to a lesser extent, CB1Rs are expressed with high and varying degrees of prevalence across the spectrum of haematopoietic cells, with B cells having the highest expression and CD4 T cells the lowest (Malfitano et al. 2014). Mechanisms that underlie eCB-mediated effects can manifest directly through communication with immune cells or indirectly via modulation of eicosanoid signalling (Rouzer and Marnett 2011). Both pathways utilise molecular cascades to manipulate activation, proliferation, secretion and apoptosis, with eventual immunoregulatory and inflammatory outcomes. Moreover, immune cells also contribute to coordinating eCB signalling through regulation of transcription, synthesis, uptake and degradation of eCB components (Pandey et al. 2009).
Influences of eCB signalling on the immune system
Evidence suggests an involvement of eCB signalling primarily in immunosuppression. For instance, CB2R activation has been shown to attenuate inflammation in a range of inflammatory conditions from injury, inflammatory pain, hepatic injury and intestinal inflammatory disorders (Pandey et al. 2009). Furthermore, levels of circulating protein and mRNA encoding for IL-1, IL-6 and TNF-α are reduced by the administration of synthetic cannabinoids in a preclinical model of multiple sclerosis (MS) treatment (Croxford and Miller 2003). Furthermore, in an animal model of autoimmune hepatitis, Concanavalin (Con)A-induced acute hepatitis, administration of AEA has been shown to diminish hepatic injury, and this is correlated with a significant reduction in inflammatory cytokines such as TNF-α, IL-1B, IL-6, IL-9 and IL-17. The therapeutic effects of AEA are CB1 and CB2 dependent, as blockade of the receptors independently ameliorate the immunosuppressive effects (Hegde et al. 2008). However, a recent study suggests that this effect is not limited to CBR activity. Cannabidiol (CBD) is another component in marijuana; whilst it displays low affinity for CBRs, it possesses an affinity with TRPV1. CBD reduces inflammatory cytokines TNF-α, IL-2, IL-6, IL-12 and IL-17 in ConA treated mice, but this is inhibited in TPRV1 knockout (KO) mice (Hegde et al. 2011). Interestingly, the inflammatory effects exhibited in this disorder, and its preclinical model, are specifically mediated by the polyclonal activation of T cells. Hence, findings from these studies may support evidence for the expanding literature detailing eCB influences on inflammation through mediating suppression of T cell proliferation. In accordance, the eCB system is heavily implicated in regulating activation and subsequent proliferation of the lymphocytes, B cells and T cells. Evidence suggests a possible role of tonic eCB signalling that may provide an inhibitory control over spontaneous immune activation of lymphoid tissues (Pandey et al. 2009). CBRs and eCB ligands are collectively involved in the suppression of adenylate cyclase (AC) activity and by extension, cyclic adenosine monophosphate (cAMP) regulation. Lymphocyte activation requires stimulation of the cAMP signalling pathway; consequently, eCB intervention may counteract this process (Pandey et al. 2009). For example, AEA suppresses human T lymphocyte proliferation and this is dependent on CB2R action, as shown by replication of the aforementioned effect by administration of CB2R agonist and amelioration following use of a CB2R antagonist (Cencioni et al. 2010). A similar result has been demonstrated with 2-AG in mouse splenocytes, where the ligand produces a marked dose-dependent inhibition of anti-CD3 mAb-induced T cell proliferation and lipopolysaccharide (LPS)-induced B cell proliferation (Lee et al. 1995).
Therefore, application of this knowledge to reports of eCB hypoactivity in depressed individuals elicits expectation of findings depicting associated increases in lymphocyte concentrations. Surprisingly, the opposite is largely described in the literature, with reports of reduced T cell concentration in peripheral bloods of depressed patients compared to controls (Maes et al. 1995b; Zorrilla et al. 2001; Irwin and Miller 2007). However, research has yet to clearly map species-specific interactions of eCB signalling on divisions within the T cell taxonomy. Elucidation of pathways associated with non-classical receptor activation within lymphocytes is also required. Future avenues of research must work to address these issues in order understand eCB-mediated effects on lymphocyte activity in the context of depression.
Eicosanoid signalling in eCB-mediated effects on the immune system
In part, eicosanoid signalling facilitates the intercommunication between eCB signalling and immunoregulation. Eicosanoids are bioactive molecules that exert a complex control over many homeostatic functions within the body; of relevance is their influence on inflammation. Biosynthesis of eicosanoids initially requires oxidation of the polyunsaturated fatty acid, arachidonic acid (AA), by the enzymes cyclooxygenase (COX) and lipoxygenase (LOX). Prostaglandins such as prostaglandin E2 (PGE2) or leukotrienes are metabolised by COX and LOX, respectively (Alhouayek et al. 2014). Both 2-AG and AEA are derivatives of AA, making them subject to the same oxidative metabolic pathways key to eicosanoid biosynthesis (Fig. 2). By extension, fluctuations in eCB signalling can impact eicosanoid signalling. As a result, eicosanoids provide a collateral method of eCB-mediated control of immune functioning (Rouzer and Marnett 2011). For example, an analogue of CBD, HU-308, a CB2R agonist that binds with high specificity, exhibits a significant anti-inflammatory effect in mice with AA-induced ear edema. Subsequent CB2R antagonist administration reverses this immunosuppression (Hanus et al. 1999; Burstein 2015). In another study, LPS-induced IL-6 production was decreased by THC, indomethacin morpholinylamide (IMMA), AEA and 2-AG in a dose-dependent manner. Interestingly, despite previously described anti-inflammatory properties of eCBs, both AEA and 2-AG did not inhibit COX-2 production of PGE2; moreover, 2-AG, contrary to THC, IMMA and AEA, caused a subsequent increase in iNOS-dependent NO (itself an inflammatory agent) production, thus providing an immunoenhancing effect. Discrepancies within the results seen in 2-AG, PGE2 and NO production were understood to be the result of increased metabolic degradation of 2-AG to AA, which in turn served as a substrate for catalysing increased COX-2 production of PGE2, known to potentiate NO production (Chang et al. 2001). This study demonstrates that inflammation is a constant balance between pro-inflammatory and anti-inflammatory substances and eCB and eicosanoid signalling play homeostatic roles in its regulation.

Eicosanoids, PGEs and leukotrienes, can be metabolised from two differential eCB-related pathways. 2-AG and AEA can be metabolised directly into either PGEs or leukotrienes by COX and LOX, respectively. Alternatively, degradation of eCBs can occur via enzymes, either MAGL or FAAH, into AA, which is subsequently oxidised by COX and LOX to produce PGEs and leukotrienes, respectively. Therefore, fluctuations in eCB concentration can influence eicosanoid levels
An alternative pathway of enzymatic oxidation of AA by LOX provides immunosuppressing properties that mitigate those observed by COX-produced metabolites. N-arachindonoylglycine, another endogenous eicosanoid, is an AEA metabolite (Bradshaw et al. 2009). This cannabinoid analogue has anti-inflammatory effects, probably thanks to its ability to promote the release of AA, successively increasing LOX-catalysis of AA to lipoxin A4 (LXA4) (Burstein et al. 2011). Increased concentrations of LXA4 are associated with the resolution phase of an acute inflammatory response, in which it induces inflammatory cell apoptosis (Bannenberg and Serhan 2010). Given the elevated inflammatory profile observed in depression and the immunoregulating properties of lipoxins and their relationship with eCBs, it would be interesting to explore a potential association between depression and lipoxins. No study to date has directly investigated this avenue of research.
Interestingly, PGE2 has been implicated in depression. Elevated levels of PGE2 in plasma, serum and saliva have been measured in patients with MDD (Lieb et al. 1983; Ohishi et al. 1988; Song et al. 1998). PGE2 has been demonstrated in inflammatory conditions to upregulate phosphodiesterase-4 (Oger et al. 2015), which in turn inhibits downstream formation of BDNF (Logan 2003), which itself is implicated in depression.
Influences of immunocompetent cells on eCB signalling
Biochemical evidence also suggests the immune system plays a profound role in the regulation of eCB signalling. Immune stimuli, such as LPS, have demonstrated that immune cell activation can also modulate eCB signalling by increasing expression of CBRs (Klein et al. 2003). Of relevance, lymphoid CBR and AEA are also elevated in patients suffering from MS, where pro-inflammatory cytokines are a pathogenic component of the disease (Sospedra and Martin 2005); following 1 year of treatment with the anti-inflammatory IFN-β, levels of AEA and expression of CBR progressively decrease (López et al. 2014).
Elucidation of underlying mechanisms in immunocompetent cells demonstrates an endogenous control and modulation over key stages in eCB functioning. A preclinical study of murine macrophages demonstrates their capacity to produce both AEA and 2-AG, intriguingly through differential pathways. AEA synthesis was observed to be dependent on LPS-induced transcription of proteins either directly responsible for the synthesis of AEA or involved in the activation of proteins responsible for the synthesis of AEA. NF-кB appeared to be a key signalling molecule in the LPS-induced transcriptional pathways. In contrast, LPS-induced increases in platelet-activating factor (PAF) are the primary intermediate of 2-AG synthesis in macrophages, dependent on PAF receptor activation (Liu et al. 2003).
Immune cells are also involved in termination of eCB signalling through direct uptake and degradation of AEA and 2-AG. AEA in vitro has been shown to accumulate in peripheral immune cells in a concentration-dependent manner (Hillard and Jarrahian 2003). Following LPS administration, increased peripheral lymphatic AEA levels are seen, probably as the result of reduced FAAH expression, thus demonstrating the presence of degradative enzymes within human peripheral lymphocytes (Maccarrone et al. 2001).
Crosstalk between the eCB and immune systems in the CNS
Peripheral eCB signalling can be reflected in the brain through transportation of eCBs across the blood-brain barrier (BBB) via endothelial cell membranes of brain microvessels. Studies report a differential expression of CB1R and CB2R on the luminal and abluminal membranes, respectively, as well as opposing function: CB1R increases NO production and thus augments AEA membrane transporter (AMT) activity, and CB2R supresses AMT activity by negating NO production (Maccarrone et al. 2005). Taken together, these findings indicate a possible unidirectional transportation of AEA across the BBB (Centonze et al. 2008) (Fig. 3). A similar neuroprotective role has been observed following administration of two highly selective CB2R agonists, which reduce TNF-α-induced upregulation of several genes within the primary human brain microvascular endothelial cells that constitute the BBB. Using functional assays, CB2R stimulation results in BBB protection proceeding inflammatory insult and diminished monocyte migration across the BBB (Persidsky et al. 2015).

Differential expression of CB1R and CB2R on the luminal and abluminal membranes, respectively, may lead to unidirectional transport of AEA. CB1R increases nitric oxide (NO) production, which in turn induces activity of a selective AEA membrane transporter (AMT). Increased levels of AEA are then transported across the BBB. CB2R negates NO production and therefore suppresses AMT activity. Thus, transport of abluminal AEA across the BBB is reduced
Whilst the presence of CB1Rs in the CNS has been well characterised, the role and expression of CB2Rs has been the object of controversy. However, recent understanding of methodological flaws in detecting CB2R expression in the CNS has improved current methods of detection. As such supporting evidence has been on the rise. Since identifying the presence of CBRs on immunocompetent cells within the CNS, implications of intercommunication between the eCB and immune systems have become increasingly prominent. Microglia, the resident immune cells of the CNS, is of particular relevance due to their considerable neuroimmune regulatory properties. CB1Rs are located on microglia, neuronal terminals and astrocytes, with CB2Rs largely expressed in microglial as well as glial cells (Stella 2010). Moreover, microglial cells are not only the primary source and target for inflammatory mediators (Czirr and Wyss-coray 2012) but cultured astrocytes and microglia are also capable of 2-AG and AEA production (Stella 2009). Chronically activated microglial cells have been increasingly associated with neurodegenerative disorders and neuroinflammatory progression (Streit et al. 2004). Likewise, upregulation of CB2Rs has been found in activated microglia and in neuroinflammatory disorders, such as MS, motor neurone disease, Down syndrome and Alzheimer’s disease (Rom and Persidsky 2013). Such a coexisting dysregulation observed in both systems suggests an underlying communication and implicates potential impairment of its interaction in neurological disorders where inflammation is central to its pathogenesis, a feature now growing in recognition as relevant to depression.
Influences of eCB signalling on the immune system
Through use of CB2R agonists and transgenic mice, CB2Rs have been described as proficient neuroprotective mediators and are involved in reduced BBB endothelial cell stimulation and inflammatory responses (Fernández-ruiz et al. 2006). Their role in neuroinflammation has been characterised as largely suppressive, consistent with notions of neuroprotective properties, through actions that directly alter the balance of pro-inflammatory and anti-inflammatory mediators (Rom and Persidsky 2013). In accordance, recent studies have established a predominantly anti-inflammatory effect of microglial CB2R stimulation (Fernández-ruiz et al. 2006). In vitro and in vivo studies report similar findings of cannabinoid-induced increases in glial cell secretion of the anti-inflammatory cytokines IL-4 and IL-10 (Molina-Holgado et al. 1998). Successive investigation also found that CB2R signalling inhibits IFN-γ-induced mouse microglial cell expression of the pro-inflammatory mediators, iNOS and CCR2, a gene encoding for a receptor associated with monocyte infiltration (Racz et al. 2008). Two states of microglial cell activation are documented, a classic activated state (M1) and the alternative state (M2) (Cherry et al. 2014). M1 polarisation is associated with cytotoxic pro-inflammatory secretion of TNF-α, IL-1β and IL-6, whereas M2 is considered to have anti-inflammatory and neurotrophic properties, releasing IL-10 and BDNF (Ma et al. 2015). Recent indications of a CB2R role in mediating a shift from M1 state to M2 have been described. Following pretreatment with the CB2R agonist AM1241, inflammation is attenuated, with a concomitant enhanced release of anti-inflammatory and neurotrophic factors. Moreover, expression of iNOS, a marker for M1 activity, decreases, whilst expression of M2 marker, Arg-1, increases. AM1241 effects are ameliorated after administration of CB2R antagonist and protein kinase C (PKC) inhibitor, indicating firstly that the effects are CB2R mediated but also that PKC activity may facilitate the effects (Ma et al. 2015). Such results support the immunosuppressive observations seen of CB2R activation in the brain. Similarly, exogenous 2-AG administration in acute experimental autoimmune encephalomyelitis, a preclinical model, produces increased microglia population as well as M2 shifting. A recent mechanism proposed by Nakagawa and Chiba (2014) alludes to a differential immunomodulatory action of 2-AG dependent on the state of microglial activation and CBR. 2-AG binding to CB1R in M1-activated state microglia results in increased proinflammatory mediators, whereas 2-AG binding to CB2R on M2-polarised microglia elevates the anti-inflammatory cytokine IL-10 and the proresolving factor lipoxin A4 (Nakagawa and Chiba 2014) (Fig. 4). Further examination into the influences of the eCB system on microglial polarisation need to be carried out in order to provide a more comprehensive support.

Differential binding of 2-AG to CBRs on microglia elicits differential immunomodulatory effects, dependent on the state of microglial activation. Binding of CB2R on M2 state microglia is associated with increased concentrations of anti-inflammatory agents. CB2R-2AG binding inhibits IFN-γ-induced microglial cell expression of proinflammatory agents. 2-AG binding to CB1R in M1-activated state microglia, results in increased pro-inflammatory mediators
Impaired functionality of 2-AG-CB2R binding may contribute, in part, to the onset of depression. A study of note, conducted by Onaivi et al. (2008), found that a particular polymorphism (Q63R) of the CB2R gene is of significantly higher incidence in Japanese depressed patients than controls. Interestingly, this polymorphism has been shown to compromise the activation of the receptor in mice, with reports that 2-AG binding with CB2-Q63R produce significantly lesser maximum responses than in wild-type mice (Carrasquer et al. 2010). Additionally, chronic administration of minocycline, which has anti-inflammatory action, attenuates depressive-like behaviours in the olfactory bulbectomised animal model of depression. It also shows a concomitant downregulation of CD11, a marker of M1 activation, increased expression of the M2 microglia state marker, MRC2, and a shift from pro-inflammatory to anti-inflammatory cytokines (Burke et al. 2014). Taken together, the aforementioned evidence provides support for the hypothesis of impaired M2 polarisation in individuals suffering with depression, possibly due to reduced 2-AG-CB2R binding. Nonetheless, more research is needed to determine the effects of various eCB-CBR binding on microglial secretion of inflammatory markers, in the context of depression. Further to this, despite substantial evidence to support the dynamic plasticity in microglial function, only two studies, to date, have investigated an associated microglia polarisation skew in depression. More critically, reported findings were contradictory (Hannestad et al. 2013; Burke et al. 2014). However, a recent compelling study reported significant anti-inflammatory effects of commonly prescribed antidepressants, fluoxetine and escitalopram, mediated by a shift from M1 microglia polarisation to M2 alternative state activation (Su et al. 2015). This suggests the efficacy of these therapeutic interventions may, in part, be attributable to altered microglial plasticity. Further investigation is greatly warranted in order to ascertain the implications posed, by evidence of microglia polarisation, to our understanding of the pathophysiology underlying depression.
Additional implications of 2-AG in the suppression of neuroinflammation are supported by the observation that 2-AG, as well as AEA, suppresses LPS-induced TNF-α secretion by microglial cells (Facchinetti et al. 2003). Microglial cells secrete 2-AG in response to elevated neuroinflammation caused by injury (Panikashvili et al. 2001; Stella 2004). Furthermore, 2-AG increases microglial cell proliferation in a CB2R-dependent manner (Carrier et al. 2004).
Interestingly, the eicosanoid system seems to be implicated in CNS endocannabinoid immune-related communication. Increases in 2-AG and attenuation of free AA concentrations have been reported in mice treated with the MAGL inhibitor, JZL184 (Long et al. 2009). Genetic and pharmacological inhibition of MAGL in LPS administered mice resulted in reduced prostaglandins PGE2, PGD2, PGF2a as well as pro-inflammatory cytokines IL-1α, IL-1β, IL-6 and TNF-α in the brain, despite LPS administration. Consistent with the notion of eicosanoid involvement, these immunosuppressive effects are replicated by COX-1 blockade but are not reversed by a CB1R antagonist (Nomura et al. 2011).
Influences of the immune system on eCB signalling
The communication in the CNS, as in the periphery, is bidirectional, and extensive research has demonstrated not only effects of the eCB system on immunomodulation but also immunoregulatory effects of the immune system on eCB signalling. For example, upon LPS administration, CB1R expression is significantly reduced, probably due to reduced CB1R protein (Hsieh et al. 1999). In particular, CB1R immunoreactivity is diminished in the hippocampus and dorsal bed nucleus of the terminal stria terminalis in mice, areas associated with major depression and stress/reward pathways. However, CB1R mRNA is increased after LPS administration, indicating inflammation-induced adaptive regulation at the protein level as opposed to transcriptional dysregulation (Hu et al. 2012). The same study reported that LPS decreases expression of CB1R in CA3 pyramidal layer glutamatergic nerve terminals of the hippocampus. These findings may indicate a reduction in CB1R dependent suppression of glutamatergic synaptic transmission. Such overactivation of glutamatergic pathways can result in increased excitoxic damage and subsequent hippocampal atrophy, changes that have associations with mood disorders (Jorge et al. 2007; Zunszain et al. 2011).
Concluding remarks
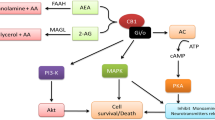
This nascent field of research implicates an intricate intercommunication between the peripheral and central eCB and immune systems in the pathogenesis of depression. Findings depict a multifaceted communication whereby immunocompetent and eCB-related cells can both influence the suppression and enhancement of the other’s activity. Typical functioning of the peripheral eCB system involves a tonic signalling control over spontaneous immune cell activation, mediated by cAMP pathways, thereby suppressing an inflammatory response. Similarly, eCB interactions with neural constituents provide key neuroprotective effects, suggested to be, in part, due to increased anti-inflammatory microglial M2-state cells through 2-AG-CB2R functioning. Moreover, an interaction with the eicosanoid signalling system influences the balance of pro- and anti-inflammatory agents: prostaglandin and leukotrienes, respectively. By extension, dysfunction of the eCB system has potential to chronically activate the inflammatory response and exacerbate neurodegeneration, both characteristics frequently observed in depression (Fig. 5).

Peripheral: eCB action on CB2Rs results in suppression of immune cell activation by inhibiting AC conversion of adenosine tri-phosphate (ATP) to cAMP. eCBs can be metabolised in to arachidonic acid (AA) which can subsequently be converted to inflammatory molecule, prostaglandins (PGE), or anti-inflammatory agent, leukotrienes (LOX) CNS: 2-AG induces M2-state microglia to secrete anti-inflammatory agents; BDNF, LXA4 and IL-10, mediated by CB2Rs
Comprehensive understanding of the biochemistry that manifests with depression warrants clarification on the impact of cell type, concentration and cellular environment and receptor density on eCB-immune processing. Elucidation of these factors will increase the potential for effectual therapeutic intervention towards a more personalised approach.
Summary |
|---|
• Inflammatory component in depression - Elevated levels of inflammatory signalling proteins in plasma and serum taken from individuals with depression - Increased inflammatory markers within the CNS - Altered microglial activity in preclinical and clinical studies of depression - Associated changes in inflammation with typically reported abnormalities in physiological processes implicated in depression; neurotransmitter metabolism, neuroendocrine function and neuroprogression |
• Role of the endocannabinoid (eCB) system in depression - Reported hypoactivity of the eCB system in animal models of depression - Manipulation of the eCB system results in associated changes in depressive-like behaviour in preclinical studies - HPA axis dysregulation, a commonly documented attribute of depression, can be produced by alterations of the eCB system - eCB system is implicated in stages of neurogenesis |
• Peripheral crosstalk and dysregulation of the eCB-immune system and its associations with depression - eCB activity is involved in immunosuppression through tonic regulation of cAMP production - 2-AG and AEA (eCBs) can be metabolised to produce eicosanoids. Dependent on the degradative enzyme, inflammatory or anti-inflammatory molecules, PGE or LOX, respectively, are produced - Abnormal concentrations of PGE2 have been associated with depression - Activated immune cells modulate eCB levels through their production, uptake and degradation |
- Central crosstalk and dysregulation of the eCB-immune system and its associations with depression - eCBs are largely neuroprotective, due to their suppression of neuroinflammation - Suppression of neuroinflammation is suggested to be the result of eCB influences on microglial state polarisation. - Possible association between reduced 2-AG-CB2R binding on M2 (alternative state microglia) and depression - Immune insult to the CNS alters levels of CB1R expression in areas associated with major depression - Reduced CB1R expression can lead to hippocampal atrophy, a feature reported in mood disorders. |
• Future directions Peripheral focus - Studies that address depression and eCB and immune system crosstalk, conjointly, both in the periphery and CNS - Clarification of eCB and lymphocyte interaction, in the context of depression - Outline specific interactions of eCB signalling with different species included in the T cell taxonomy. - Elucidate potential disparities in pathways associated with non-classical receptor activation of lymphocytes. - Further study of eicosanoid, in particular lipoxin, concentrations in individuals with depression CNS focus - Further examination into influences of the eCB system on microglial polarisation - Investigations into an association between microglial polarisation state and depression - Effects of various eCB-CBR binding on microglia secretion of inflammatory markers and whether this effect is associated with depression. |
References
Alexander SPH, Kendall D a (2007) The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol 152:602–623. doi:10.1038/sj.bjp.0707456
Alhouayek M, Masquelier J, Muccioli GG (2014) Controlling 2-arachidonoylglycerol metabolism as an anti-inflammatory strategy. Drug Discov Today 19:295–304. doi:10.1016/j.drudis.2013.07.009
Asnis GM, De La Garza R (2006) Interferon-induced depression in chronic hepatitis C: a review of its prevalence, risk factors, biology, and treatment approaches. J Clin Gastroenterol 40:322–35. doi:10.1097/01.mcg.0000210099.36500.fe
Bahi A, Al Mansouri S, Al Memari E et al (2014) β-Caryophyllene, a CB2 receptor agonist produces multiple behavioral changes relevant to anxiety and depression in mice. Physiol Behav 135:119–24. doi:10.1016/j.physbeh.2014.06.003
Bakunina N, Pariante CM, Zunszain PA (2015) Immune mechanisms linked to depression via oxidative stress and neuroprogression. J cells, Mol Syst Technol 144:365–373. doi:10.1111/imm.12443
Bannenberg G, Serhan CN (2010) Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta - Mol Cell Biol Lipids 1801:1260–1273. doi:10.1016/j.bbalip.2010.08.002
Baraldi S, Hepgul N, Mondelli V, Pariante CM (2012) Symptomatic treatment of interferon-α-induced depression in hepatitis C: a systematic review. J Clin Psychopharmacol 32:531–43. doi:10.1097/JCP.0b013e31825d9982
Barna I, Zelena D, Arszovszki AC, Ledent C (2004) The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci 75:2959–70. doi:10.1016/j.lfs.2004.06.006
Berk M, Williams LJ, Jacka FN et al (2013) So depression is an inflammatory disease, but where does the inflammation come from ? BMC Med 11:1. doi:10.1186/1741-7015-11-200
Beyer CE, Dwyer JM, Piesla MJ et al (2010) Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol Dis 39:148–155. doi:10.1016/j.nbd.2010.03.020
Bisogno T, Howell F, Williams G et al (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–8. doi:10.1083/jcb.200305129
Bortolato M, Mangieri R a, Fu J et al (2007) Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry 62:1103–1110. doi:10.1016/j.biopsych.2006.12.001
Bradshaw HB, Rimmerman N, Hu SS-J et al (2009) The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem 10:1–11. doi:10.1186/1471-2091-10-14
Burke NN, Kerr DM, Moriarty O et al (2014) Minocycline modulates neuropathic pain behaviour and cortical M1–M2 microglial gene expression in a rat model of depression. Brain Behav Immun 42:147–156. doi:10.1016/j.bbi.2014.06.015
Burstein S (2015) Cannabidiol (CBD) and its analogs: a review of their effects on inflammation [published online ahead of print January 30 2015]. Bioorg Med Chem. doi: 10.1016/j.bmc.2015.01.059
Burstein S, Mcquain C, Ross A et al (2011) Resolution of inflammation by N-arachidonoylglycine. J Cell Biochem 112:3227–3233. doi:10.1002/jcb.23245.Resolution
Carrasquer A, Nebane NM, Williams WM, Song Z (2010) Functional consequences of nonsynonymous single nucleotide polymorphisms in the CB2 cannabinoid receptor. Pharmacogenet Genomics 20:157–166. doi:10.1097/FPC.0b013e3283367c6b
Carrier EJ, Kearn CS, Barkmeier AJ et al (2004) Cultured Rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB 2 receptor-dependent mechanism. Mol Pharmacol 65:999–1007. doi:10.1124/mol.65.4.999
Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. doi:10.1016/j.neuron.2012.09.020
Cattaneo A, Gennarelli M, Uher R et al (2013) Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline “predictors” and longitudinal “targets”. Neuropsychopharmacology 38:377–85. doi:10.1038/npp.2012.191
Cencioni MT, Chiurchiù V, Catanzaro G et al (2010) Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB 2 receptors. PLoS One 5:2–11. doi:10.1371/journal.pone.0008688
Centonze D, Battistini L, Maccarrone M (2008) The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr Pharm Des 14:2370–2342. doi:10.2174/138161208785740018
Chang Y, Lee ST, Lin W (2001) Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem 81:715–723. doi:10.1002/jcb.1103
Cherry JD, Olschowka JA, O’Banion MK (2014) Neuroinflammation and M2 microglia : the good, the bad, and the inflamed. J Neuroinflammation 11:1–15. doi:10.1186/1742-2094-11-98
Choi K, Le T, McGuire J et al (2012) Expression pattern of the cannabinoid receptor genes in the frontal cortex of mood disorder patients and mice selectively bred for high and low fear. J Psychiatr Res 46:882–9. doi:10.1016/j.jpsychires.2012.03.021
Couch Y, Anthony DC, Dolgov O et al (2013) Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress-altered behaviour in mice susceptible to anhedonia. Brain Behav Immun 29:136–146. doi:10.1016/j.bbi.2012.12.017
Cravatt BF, Giang DK, Mayfield SP et al (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–7. doi:10.1038/384083a0
Croxford JL, Miller SD (2003) Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R (+) WIN55, 212. 111:1231–1240. doi: 10.1172/JCI200317652.Introduction
Czirr E, Wyss-coray T (2012) The immunology of neurodegeneration. J Clin Invest 122:1156–1163. doi:10.1172/JCI58656.1156
Dantzer R, O’Connor JC, Freund GG et al (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. doi:10.1038/nrn2297
Devane W, Hanus L, Breuer A et al (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science (80-) 258(80-):1946–1949. doi:10.1126/science.1470919
Dowlati Y, Herrmann N, Swardfager W et al (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67:446–57. doi:10.1016/j.biopsych.2009.09.033
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127. doi:10.1016/j.biopsych.2006.02.013
Facchinetti F, Del Giudice E, Furegato S et al (2003) Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia 41:161–8. doi:10.1002/glia.10177
Fernández-ruiz J, Romero J, Velasco G et al (2006) Cannabinoid CB 2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci 28:39–45. doi:10.1016/j.tips.2006.11.001
Galve-Roperh I, Chiurchiù V, Díaz-Alonso J et al (2013) Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res 52:633–650. doi:10.1016/j.plipres.2013.05.004
Gaoni Y, Mechoulam R (1964) Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646–1647. doi:10.1021/ja01062a046
García-Gutiérrez MS, Pérez-Ortiz JM, Gutiérrez-Adán a, Manzanares J (2010) Depression-resistant endophenotype in mice overexpressing cannabinoid CB 2 receptors. Br J Pharmacol 160:1773–1784. doi:10.1111/j.1476-5381.2010.00819.x
Gobbi G, Bambico FR, Mangieri R et al (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A 102:18620–5. doi:10.1073/pnas.0509591102
Gorzalka BB, Hill MN (2011) Putative role of endocannabinoid signaling in the etiology of depression and actions of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry 35:1575–85. doi:10.1016/j.pnpbp.2010.11.021
Hannestad J, Dellagioia N, Gallezot J-D et al (2013) The neuroinflammation marker translocator protein is not elevated in individuals with mild-to-moderate depression: a [11C]PBR28 PET study. Brain Behav Immun 33:131–138. doi:10.1016/j.bbi.2013.06.010
Hanus L, Breuer A, Tchilibon S et al (1999) HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A 96:14228–14233. doi:10.1073/pnas.96.25.14228
Hegde VL, Hegde S, Cravatt BF et al (2008) Attenuation of experimental autoimmune hepatitis by exogenous and endogenous cannabinoids: involvement of regulatory T cells. Mol Pharmacol 74:20–33. doi:10.1124/mol.108.047035.cytokines
Hegde VL, Nagarkatti PS, Nagarkatti M (2011) Role of Myeloid-derived suppressor cells in amelioration of experimental autoimmune hepatitis following activation of TRPV1 receptors by cannabidiol. PLoS One 6:1–12. doi:10.1371/journal.pone.0018281
Hill MN, Carrier EJ, Mclaughlin RJ et al (2008a) Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem 106:2322–2336. doi:10.1111/j.1471-4159.2008.05567.x
Hill MN, Hellemans KGC, Verma P et al (2012) Neurobiology of chronic mild stress: parallels to major depression. Neurosci Biobehav Rev 36:2085–2117. doi:10.1016/j.neubiorev.2012.07.001
Hill MN, Tasker JG (2012) Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 204:5–16. doi:10.1016/j.neuroscience.2011.12.030
Hill N, Miller GE, Ho WSV et al (2008b) Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry 41:48–53. doi:10.1055/s-2007-993211
Hillard CJ, Jarrahian A (2003) Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol 140:802–808. doi:10.1038/sj.bjp.0705468
Hillard CJ, Liu Q (2014) Endocannabinoid signalling in the etiology and treatment of major depressive illness
Hinwood M, Morandini J, Day T a, Walker FR (2012) Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex 22:1442–1454. doi:10.1093/cercor/bhr229
Hirvonen J, Goodwin R, Li C-T et al (2012) Reversible and regionally selective downregulation of brain cannabinoid CB(1) receptors in chronic daily cannabis smokers. Mol Psychiatry 17:642–649. doi:10.1016/j.biotechadv.2011.08.021
Horowitz M, Wertz J, Zhu D, et al (2014) Antidepressant compounds can be both pro- and anti- inflammatory in human hippocampal cells [published online ahead of print October 31 2014]. Int J Neuropsychopharmacol. doi: 10.1016/j.sna.2011.03.022
Howren MB, Lamkin DM, Suls J (2009) Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71:171–86. doi:10.1097/PSY.0b013e3181907c1b
Hsieh C, Brown S, Derleth C, Mackie K (1999) Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem 73:493–501. doi:10.1046/j.1471-4159.1999.0730493.x
Hu H, Ho W, Mackie K et al (2012) Brain CB(1) receptor expression following lipoplysaccharide-induced inflammation. Neuroscience 227:211–222. doi:10.1016/j.neuroscience.2012.09.067
Hungund BL, Vinod KY, Kassir S a et al (2004) Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol Psychiatry 9:184–190. doi:10.1038/sj.mp.4001376
Irwin MR, Miller AH (2007) Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun 21:374–383. doi:10.1016/j.bbi.2007.01.010
Iseme RA, McEvoy M, Kelly B et al (2014) Autoantibodies and depression: evidence for a causal link? Neurosci Biobehav Rev 40:62–79. doi:10.1016/j.neubiorev.2014.01.008
Jiang W, Zhang Y, Xiao L et al (2005) Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic-and antidepressant-like effects. J Clin Invest 115:3104–3116. doi:10.1172/JCI25509.3104
Jorge RE, Acion L, Starkstein SE, Magnotta V (2007) Hippocampal volume and mood disorders after traumatic brain injury. Biol Psychiatry 62:332–338. doi:10.1016/j.biopsych.2006.07.024
Karlsson M, Contreras JA, Hellman U et al (1997) cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase: evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem 272:27218–27223. doi:10.1074/jbc.272.43.27218
Klein TW, Newton C, Larsen K et al (2003) The cannabinoid system and immune modulation. J Leukoc Biol 74:486–496. doi:10.1189/jlb.0303101
Köhler O, Benros ME, Nordentoft M et al (2014) Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71:1381–1391. doi:10.1001/jamapsychiatry.2014.1611
Kreisel T, Frank MG, Licht T et al (2014) Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 19:699–709. doi:10.1038/mp.2013.155
Landfield PW, Cadwallader LB, Vinsant S (1988) Quantitative changes in hippocampal structure following long-term exposure to delta 9-tetrahydrocannabinol: possible mediation by glucocorticoid systems. Brain Res 443:47–62. doi:10.1016/0006-8993(88)91597-1
Lee BH, Kim YK (2010) The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig 7:231–235. doi:10.4306/pi.2010.7.4.231
Lee M, Yang KH, Kaminski NE (1995) Effects of putative cannabinoid receptor anandamide in B6C3F1 mouse Splenocytes 1 on immune function. J Pharmacol Exp Ther 275:529–536
Lee S, Kim DH, Yoon SH, Ryu JH (2009) Sub-chronic administration of rimonabant causes loss of antidepressive activity and decreases doublecortin immunoreactivity in the mouse hippocampus. Neurosci Lett 467:111–116. doi:10.1016/j.neulet.2009.10.017
Levine J, Barak Y, Chengappa KNR et al (1999) Cerebrospinal cytokine levels in patients with acute depression. Biol Psychiatry 40:171–176. doi:10.1159/000026615
Lieb J, Karmali R, Horrobin D (1983) Elevated levels of prostaglandin E2 and thromboxane B2 in depression. Prostaglandins Leukot Med 10:361–367. doi:10.1016/0262-1746(83)90048-3
Liu J, Pacher P, Harvey-white J et al (2003) Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kB independently of platelet-activating factor. J Biol Chem 278:45034–45039. doi:10.1074/jbc.M306062200
Logan AC (2003) Neurobehavioral aspects of omega-3 fatty acids : possible mechanisms and therapeutic value in major depression. Altern Med Rev 8:410–425
Long JZ, Li W, Booker L et al (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44. doi:10.1038/nchembio.129
López AJS, Roman-Vega L, Tojeiro ER, Giuffrida A (2014) Regulation of cannabinoid receptor gene expression and endocannabinoid levels in lymphocyte subsets by interferon-β: a longitudinal study in multiple sclerosis patients. J Transl Immunol 179:119–127. doi:10.1111/cei.12443
Louveau A, Smirnov I, Keyes TJ et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. doi:10.1038/nature14432
Ma L, Jia J, Liu X et al (2015) Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem Biophys Res Commun 458:92–97. doi:10.1016/j.bbrc.2015.01.073
Maccarrone M, Fiori A, Bari M et al (2005) Regulation by cannabinoid receptors of anandamide transport across the blood-brain barrier and through other endothelial cells. Thromb Haemost 95:117–127. doi:10.1160/TH05-06-0413
Maccarrone M, De Petrocellis L, Bari M et al (2001) Lipopolysaccharide downregulates fatty acid amide hydrolase expression and increases anandamide levels in human peripheral lymphocytes. Arch Biochem Biophys 393:321–328. doi:10.1006/abbi.2001.2500
Maciel IS, Silva RBM, Morrone FB et al (2013) Synergistic effects of celecoxib and bupropion in a model of chronic inflammation-related depression in mice. PLoS One 8:e77227. doi:10.1371/journal.pone.0077227
Maes M, Meltzer HY, Buckley P, Bosmans E (1995a) Plasma-soluble interleukin-2 and transferrin receptor in schizophrenia and major depression. Eur Arch Psychiatry Clin Neurosci 244:325–329
Maes M, Smith R, Scharpe S (1995b) The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology 20:111–116. doi:10.1016/0306-4530(94)00066-J
Maes M, Song C, Lin A-H et al (1999) Negative immunoregulatory effects of antidepressants: inhibition of interferon-gamma and stimulation of interleukin-10 secretion. Neuropsychopharmacology 20:370–9. doi:10.1016/S0893-133X(98)00088-8
Malfitano AM, Basu S, Maresz K et al (2014) What we know and do not know about the cannabinoid receptor 2 (CB2). Semin Immunol 2:1–11. doi:10.1016/j.smim.2014.04.002
Matsuda LA, Lolait SJ, Brownstein MJ et al (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–4. doi:10.1038/346561a0
McLauglin RJ, Hill MN, Dang SS et al (2013) Upregulation of CB1 receptor binding in the ventromedial prefrontal cortex promotes proactive stress-coping strategies following chronic stress exposure. Behav Brain Res 237:333–337. doi:10.1016/j.bbr.2012.09.053
McNamara R, Lotrich F (2012) Elevate immune-inflammatory signaling in mood disorders: a new therapeutic target? Expert Rev Neurother 12:1143–1161. doi:10.1586/ern.12.98
McPartland JM, Guy GW, Di Marzo V (2014) Care and feeding of the endocannabinoid system: a systematic review of potential clinical interventions that upregulate the endocannabinoid system. PLoS One 9:e89566. doi:10.1371/journal.pone.0089566
Mechoulam R, Ben-Shabat S, Hanus L et al (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90
Mendlewicz J, Kriwin P, Oswald P et al (2006) Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol 21:227–231. doi:10.1097/00004850-200607000-00005
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–41. doi:10.1016/j.biopsych.2008.11.029
Miller BR, Hen R (2015) The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol 30:51–58. doi:10.1016/j.conb.2014.08.012
Molina-Holgado F, Molina-Holgado E, Guaza C (1998) The endogenous cannabinoid anandamide potentiates interleukin-6 production by astrocytes infected with Theiler’s murine encephalomyelitis virus by a receptor-mediated pathway. FEBS Lett 433:139–142. doi:10.1016/S0014-5793(98)00851-5
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–5. doi:10.1038/365061a0
Nakagawa Y, Chiba K (2014) Role of microglial M1/M2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 7:1028–1048. doi:10.3390/ph7121028
Nomura DK, Morrison BE, Blankman JL et al (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334(80-):809–814. doi:10.1126/science.1209200
Ogawa S, Hattori K, Sasayama D et al (2015) Reduced cerebrospinal fluid ethanolamine concentration in major depressive disorder. Sci Rep 5:1–8. doi:10.1038/srep07796
Oger S, Méhats C, Dallot E et al (2015) Expression in human myometrial cells through a prostaglandin E 2- and cyclic adenosine 3′,5′-monophosphate-dependent pathway. J Clin Endocrinol Metab 87:5524–5531. doi:10.1210/jc.2002-020575
Ohishi K, Ueno R, Nishino S et al (1988) Increased level of salivary prostaglandins in patients with major depression. Biol Psychiatry 23:326–334. doi:10.1016/0006-3223(88)90283-1
Okamoto Y, Morishita J, Tsuboi K et al (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–305. doi:10.1074/jbc.M306642200
Onaivi ES, Ishiguro H, Gong J-P et al (2008) Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One 3:e1640. doi:10.1371/journal.pone.0001640
Palazuelos J, Ortega Z, Diaz-Alonso J et al (2012) CB 2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem 287:1198–1209. doi:10.1074/jbc.M111.291294
Pandey R, Mousawy K, Nagarkatti M, Nagarkatti P (2009) Endocannabinoids and immune regulation. Pharmacol Res 60:85–92. doi:10.1016/j.phrs.2009.03.019
Panikashvili D, Simeonidou C, Ben-shabat S et al (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413:527–531. doi:10.1038/35097089
Pariante CM, Lightman SL (2008) The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31:464–468. doi:10.1016/j.tins.2008.06.006
Patel S, Roelke CT, Rademacher DJ et al (2004) Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 145:5431–8. doi:10.1210/en.2004-0638
Persidsky Y, Fan S, Dykstra H, et al (2015) Activation of Cannabinoid Type Two Receptors (CB 2) Diminish Inflammatory Responses in Macrophages and Brain Endothelium [Published ahead of print February 11 2015]. J Neuroimmune Pharmacol. doi: 10.1007/s11481-015-9591-3
Racz I, Nadal X, Alferink J et al (2008) Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J Neurosci 28:12136–12145. doi:10.1523/JNEUROSCI.3402-08.2008
Raison CL, Borisov AS, Majer M et al (2009) Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry 65:296–303. doi:10.1016/j.biopsych.2008.08.010
Raison CL, Capuron L, Miller AH (2006) Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27:24–31. doi:10.1016/j.it.2005.11.006
Riazi K, Galic M a, Kuzmiski JB et al (2008) Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U S A 105:17151–17156. doi:10.1073/pnas.0806682105
Rom S, Persidsky Y (2013) Cannabinoid receptor 2: potential role in immunomodulation and neuroinflammation. J Neuroimmune Pharmacol 8:608–620. doi:10.1007/s11481-013-9445-9
Rouzer C a, Marnett LJ (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev 111:5899–921. doi:10.1021/cr2002799
Santarelli L, Saxe M, Gross C et al (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(80-):805–809. doi:10.1126/science.1083328
Schneider P, Weber-Fahr W, Schweinfurth N et al (2012) Central metabolite changes and activation of microglia after peripheral interleukin-2 challenge. Brain Behav Immun 26:277–283. doi:10.1016/j.bbi.2011.09.011
Sheline YI, Wang PW, Gado MH et al (1996) Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A 93:3908–3913. doi:10.1073/pnas.93.9.3908
Soczynska JK, Mansur RB, Brietzke E et al (2012) Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav Brain Res 235:302–17. doi:10.1016/j.bbr.2012.07.026
Song C, Lin A, Bonaccorso S et al (1998) The inflammatory response system and the availability of plasma tryptophan in patients with primary sleep disorders and major depression. J Affect Disord 49:211–219. doi:10.1016/S0165-0327(98)00025-1
Sospedra M, Martin R (2005) Immunology of multiple sclerosis. Annu Rev Immunol 23:683–747. doi:10.1146/annurev.immunol.23.021704.115707
Steiner J, Bielau H, Brisch R et al (2008) Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42:151–157. doi:10.1016/j.jpsychires.2006.10.013
Stella N (2010) Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 58:1017–30. doi:10.1002/glia.20983
Stella N (2009) Endocannabinoid signaling in microglial cells. Neuropharmacology 56:244–253. doi:10.1016/j.neuropharm.2008.07.037
Stella N (2004) Cannabinoid signaling in glial cells. Glia 48:267–277. doi:10.1002/glia.20084
Streit WJ, Mrak RE, Griffin WST (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:1–4. doi:10.1186/1742-2094-1-14
Su F, Yi H, Xu L, Zhang Z (2015) Fluoxetine and S-citalopram inhibit M1 activation and promote M2 activation of microglia in vitro. Neuroscience 294:60–68. doi:10.1016/j.neuroscience.2015.02.028
Topol EJ, Bousser MG, Fox KA et al (2010) Rimonabant for prevention of cardiovascular events (CRESCENDO): a randomised, multicentre, placebo-controlled trial. Lancet 376:517–523. doi:10.1016/S0140-6736(10)60935-X
Tyring S, Gottlieb A, Papp K et al (2006) Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367:29–35. doi:10.1016/S0140-6736(05)67763-X
Valverde O, Torrens M (2012) CB1 receptor-deficient mice as a model for depression. Neuroscience 204:193–206. doi:10.1016/j.neuroscience.2011.09.031
Vinod KY, Xie S, Psychoyos D et al (2012) Dysfunction in fatty acid amide hydrolase is associated with depressive-like behavior in Wistar Kyoto rats. PLoS One 7:e36743. doi:10.1371/journal.pone.0036743
Wade MR, Degroot A, Nomikos GG (2006) Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol 551:162–167. doi:10.1016/j.ejphar.2006.08.083
Wang J, Ueda N (2009) Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat 89:112–9. doi:10.1016/j.prostaglandins.2008.12.002
Wang W, Sun D, Pan B et al (2010) Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology 35:2249–2261. doi:10.1038/npp.2010.99
Willner P (2005) Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology 52:90–110. doi:10.1159/000087097
Wohleb ES, Fenn AM, Pacenta AM et al (2012) Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37:1491–1505. doi:10.1016/j.psyneuen.2012.02.003
Wyrofsky R, Mcgonigle P, Van Bockstaele EJ (2015) Drug discovery strategies that focus on the endocannabinoid signaling system in psychiatric disease. Expert Opin 10:17–36. doi:10.1517/17460441.2014.966680
Zaher HAEM, Amin ME, Rakhawy MY (2010) Coping with depression and anxiety in patients with psoriasis. Egypt J Psychiatry 31:57–63
Zajkowska ZE, Englund A, Zunszain P a (2014) Towards a personalized treatment in depression: endocannabinoids, inflammation and stress response. Pharmacogenomics 15:687–98. doi:10.2217/pgs.14.40
Zorrilla EP, Luborsky L, McKay JR et al (2001) The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 15:199–226. doi:10.1006/brbi.2000.0597
Zunszain PA, Anacker C, Cattaneo A et al (2011) Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychopharmacol Biol Psychiatry 35:722–9. doi:10.1016/j.pnpbp.2010.04.011
Zunszain PA, Hepgul N, Pariante CM (2013) Inflammation and depression. Curr Top Behav Neurosci 14:135–51. doi:10.1007/7854_2012_211
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare the following conflict of interest: Prof. Pariante has received fees as a speaker or as a member of advisory board, as well as research funding, from pharmaceutical companies that commercialize or are developing antidepressants in the last 3 years, such as Lilly, Servier, and Janssen. Dr. Zunszain has received speaker fees from Servier.
Prof. Pariante and Dr. Zunszain are supported by the National Institute of Health Research Biomedical Research Centre in Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London, and by the Medical Research Council UK (MR/J002739/1 and MR/L014815/1). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Boorman, E., Zajkowska, Z., Ahmed, R. et al. Crosstalk between endocannabinoid and immune systems: a potential dysregulation in depression?. Psychopharmacology 233, 1591–1604 (2016). https://doi.org/10.1007/s00213-015-4105-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-015-4105-9