
Abstract
Rationale
Asenapine, a novel psychopharmacologic agent in the development for schizophrenia and bipolar disorder, has high affinity for serotonergic, α-adrenergic, and dopaminergic receptors, suggesting potential for antipsychotic and cognitive-enhancing properties.
Objectives
The effects of asenapine in rat models of antipsychotic efficacy and cognition were examined and compared with those of olanzapine and risperidone.
Materials and methods
Amphetamine-stimulated locomotor activity (Amp-LMA; 1.0 or 3.0 mg/kg s.c.) and apomorphine-disrupted prepulse inhibition (Apo-PPI; 0.5 mg/kg s.c.) were used as tests for antipsychotic activity. Delayed non-match to place (DNMTP) and five-choice serial reaction (5-CSR) tasks were used to assess short-term spatial memory and attention, respectively. Asenapine doses varied across tasks: Amp-LMA (0.01–0.3 mg/kg s.c.), Apo-PPI (0.001–0.3 mg/kg s.c.), DNMTP (0.01–0.1 mg/kg s.c.), and 5-CSR (0.003–0.3 mg/kg s.c.).
Results
Asenapine was highly potent (active at 0.03 mg/kg) in the Amp-LMA and Apo-PPI assays. DNMTP or 5-CSR performance was not improved by asenapine, olanzapine, or risperidone. All agents (P < 0.01) reduced DNMTP accuracy at short delays; post hoc analyses revealed that only 0.1 mg/kg asenapine and 0.3 mg/kg risperidone differed from vehicle. All active agents (asenapine, 0.3 mg/kg; olanzapine, 0.03–0.3 mg/kg; and risperidone, 0.01–0.1 mg/kg) significantly impaired 5-CSR accuracy (P < 0.05).
Conclusions
Asenapine has potent antidopaminergic properties that are predictive of antipsychotic efficacy. Asenapine, like risperidone and olanzapine, did not improve cognition in normal rats. Rather, at doses greater than those required for antipsychotic activity, asenapine impaired cognitive performance due to disturbance of motor function, an effect also observed with olanzapine and risperidone.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although antipsychotics are effective in controlling the psychotic or positive symptoms of schizophrenia, they have limited efficacy in the treatment of negative or cognitive symptoms. In addition, most of the currently used drugs are associated with poor tolerability, which contributes to high rates of treatment discontinuation (Lieberman et al. 2005). Thus, it is clear that there is a continued need for new therapeutic agents with improved efficacy and/or tolerability. Asenapine is a novel psychopharmacologic agent being developed for the treatment of schizophrenia and bipolar disorder. It has a unique signature of human receptor binding that is characterized by strong affinity and antagonism at serotonergic, dopaminergic, α-adrenergic, and histamine receptor subtypes (Shahid et al. 2009). In particular, it has high activity toward a broad range of serotonin (5-HT1A, 5-HT1B, 5-HT2C, 5-HT6, and 5-HT7) receptors, which may aid efficacy. Indeed, emerging clinical data show that asenapine is effective in treating both positive and negative symptoms of schizophrenia (Potkin et al. 2007), mania in bipolar disorder patients (McIntyre et al. 2008) with preliminary indications for cognitive deficits in patients with schizophrenia (Fleming et al. 2007, abstract).
The efficacy of antipsychotic agents in treating the positive symptoms of schizophrenia is related in large part to their antagonism of dopamine D2 receptors (Kapur and Mamo 2003; Remington 2003). As a result, reversal of amphetamine-stimulated locomotor activity (Amp-LMA) and reversal of apomorphine-induced deficits in prepulse inhibition (Apo-PPI) have been used as animal models of antipsychotic efficacy (Geyer et al. 2001; Nakai et al. 2008; Nordquist et al. 2008; Varty and Higgins 1995). In the Amp-LMA model, the magnitude of inhibition induced by atypical and typical antipsychotics differs as a function of the dose of amphetamine administered (Arnt 1995). Therefore, the differential inhibition of Amp-LMA by antipsychotics as a function of amphetamine dose can be used to assess the “atypicality” of novel antipsychotic agents, just as higher affinity for 5-HT receptors relative to D2 receptors can be used. The PPI model operationally indexes sensorimotor gating by assessing the ability of a weak stimulus to inhibit the response to a startle stimulus, which can be disrupted by dopamine receptor agonists. The ability of a pharmacologic treatment to reverse deficits in PPI induced by dopaminergic agents, such as amphetamine and apomorphine, has also been used to assess antipsychotic activity.
Schizophrenia is associated with an array of cognitive deficits that are evident and substantially unaltered even when positive symptoms of schizophrenia are effectively controlled (Sharma and Antonova 2003). Cognitive deficits are now recognized as a core element of the disorder and are believed to have significant bearing on the patient's recovery and reintegration into society (Addington et al. 2001; Cuesta et al. 2001). The importance of animal models for assisting in the identification of antipsychotic compounds with cognitive-enhancing properties is becoming increasingly recognized (Hagan and Jones 2005). However, preclinical investigations face challenges that include determining the most appropriate model, translating observations to the clinical setting, and addressing whether challenging a normal animal to the limits of its capabilities or inducing deficits thought to mimic a component of the pathologic condition is more appropriate. There are several well-established animal models that can be used to investigate cognitive function in the rodent. The delayed non-match to place (DNMTP) and five-choice serial reaction (5-CSR) tasks are commonly used to assess short-term spatial memory and sustained attention, respectively, and are sensitive to pharmacologic interventions that improve cognition (Amitai N and Markou 2009; Quarta et al. 2007; Sukhotina et al. 2008; Ward et al. 2004).
Given the potent antidopaminergic activity of asenapine and its relatively higher affinity for a broad range of serotonergic and α-adrenergic receptors, we hypothesized that asenapine may have a psychopharmacologic profile combining potent antipsychotic action with the potential to attenuate aspects of the cognitive deficits associated with schizophrenia. In the current report, the effects of asenapine were assessed in rats in tests of antipsychotic efficacy (Amp-LMA and Apo-PPI) and cognitive function (DNMTP and 5-CSR). The aim was to compare doses producing antipsychotic efficacy with those having effects on cognitive function.
Materials and methods
Animals
Studies were conducted in three laboratories using two strains of male rats. Sprague–Dawley rats were used in the Amp-LMA (130–150 g, Harlan, Inc., Indianapolis, IN, USA) and Apo-PPI studies (250–325 g, Harlan, Inc., San Diego, CA, USA). Lister Hooded rats (300–400 g, Harlan UK Limited, Oxon, UK) were used in the DNMTP and 5-CSR studies. All rats were group-housed (two to four per cage) and maintained on a 12-h light–dark cycle (lights on at 6–7 a.m.). Ad libitum food and water access was available during the Amp-LMA and Apo-PPI studies, except for intermittent 24-h periods before drug testing in the Amp-LMA study. In the DNMTP and 5-CSR studies, food availability was restricted in order to maintain the rats at approximately 85% of their predicted free-feeding body weight. To achieve this, animals were weighed daily, and the quantity of lab chow given to each cage adjusted. If an individual animal diverged significantly from the pattern of his cagemates the animal would be separated temporarily during the feeding period to allow either a proportionately higher or lower food allowance to be accessed.
The Amp-LMA and Apo-PPI studies were approved by the respective Institutional Animal Care and Use Committees and were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources 1996). The DNMTP and 5-CSR studies were conducted in accordance with the 1986 UK Animals (Scientific Procedures) Act.
Drugs
Asenapine (Schering-Plough, Newhouse, Lanarkshire, UK) was administered subcutaneously (s.c.) in all studies. The doses administered were consistent with previously published studies in rats (Franberg et al. 2008; Huang et al. 2008; Tarazi et al. 2008) and encompassed a range that approximates clinically relevant D2 receptor occupancy levels (Schotte et al. 1996). Olanzapine and risperidone (Pfizer Inc., Ann Arbor, MI, USA for the Amp-LMA studies; Schering-Plough for the DNMTP and 5-CSR studies) doses were selected based on previous publications (Abdul-Monim et al. 2006; Amitai et al. 2007; Arnt 1995; Didriksen et al. 2007; Grayson et al. 2007; Wolff and Leander 2003). Haloperidol and apomorphine were obtained from Sigma-Aldrich (St. Louis, MO, USA) for the Amp-LMA and Apo-PPI studies.
In the LMA study, asenapine (0.01–0.3 mg/kg s.c.) was suspended in 1% cremophor EL, 1% HCl 1 N, and 0.5% methocel in saline; olanzapine (1.0–10.0 mg/kg oral gavage) and risperidone (0.1–3.0 mg/kg oral gavage) were suspended in 1% cremophor EL, 1% HCl 1 N, and 0.5% methocel in water. Haloperidol (0.1–1.0 mg/kg oral gavage) was dissolved in 1% lactic acid in water. d-Amphetamine (1.0 or 3.0 mg/kg s.c.) was dissolved in 0.9% saline.
In the Apo-PPI studies, asenapine (0.001–3.0 mg/kg s.c.) was dissolved in 0.9% saline, and apomorphine (0.5 mg/kg s.c.) was dissolved in 0.9% saline containing 1 mg/mL ascorbic acid. Both were administered in a volume of 1 mL/kg.
In the DNMTP and 5-CST studies, asenapine (0.003–0.3 mg/kg s.c.), olanzapine (0.03–0.3 mg/kg s.c.), and risperidone (0.01–0.3 mg/kg s.c) were prepared in a 5% mulgofen/saline solution.
Procedures
Study 1: spontaneous and amphetamine-stimulated locomotor activity
Activity was assessed using 16-Beam Digiscan Animal Activity Monitors (Accuscan Instruments Inc.; Columbus, OH, USA). Each activity monitor consisted of a Plexiglas® box (16 × 16 in.) that was housed inside a stainless steel, ventilated, sound-attenuating chamber. Horizontal beam breaks were converted to distance traveled (centimeter).
Spontaneous LMA was assessed in 5-min intervals during a 30-min session. The chamber was darkened to maximize spontaneous activity. Rats were treated with asenapine 30 min before assessment; olanzapine, risperidone, or haloperidol was administered 60 min before assessment.
Amp-LMA was assessed in 5-min intervals during a 120-min session. The chamber was illuminated to minimize spontaneous activity. For asenapine testing, rats were placed into the chambers 30 min before asenapine or vehicle administration. For testing with haloperidol, olanzapine, and risperidone, rats were administered active treatment or vehicle immediately before being placed into the chambers. Thirty minutes after being placed into the chambers, low- or high-dose d-amphetamine was administered. Therefore, d-amphetamine injections were administered 30 min after haloperidol, olanzapine, and risperidone and immediately after asenapine.
Study 2: apomorphine-disrupted prepulse inhibition
Two experiments were conducted to examine the effects of asenapine on startle and PPI. The first experiment used high doses of asenapine (0.03–3.0 mg/kg). Based on the effects of 0.03 and 0.1 mg/kg asenapine in that study, a second was conducted to replicate these data and to determine the effects of lower asenapine doses.
Startle magnitude and PPI were assessed using apparatus (SR-LAB™ Startle System, San Diego Instruments, San Diego, CA, USA) and procedures similar to those described in previous studies (Swerdlow et al. 1998; Varty and Geyer 1998). Rats were handled before testing was initiated to reduce the stress of handling.
Each test was initiated 5 min after the rat was placed in the chamber, where background white noise (70 dB) was present during the acclimation period and for the duration of testing. Each test consisted of a series of trials, separated by a variable interval averaging 15 s, with two conditions: (1) presentation of a startle stimulus (118 dB, 40-ms noise burst) and (2) presentation of the startle stimulus 100 ms after the presentation of a 20-ms prepulse at 73, 76, or 82 dB. PPI was defined as the percentage reduction in startle amplitude in the presence vs the absence of the prepulse [100–(100 × (startle amplitude on prepulse trial/startle amplitude on startle-only trial))]. Sessions were divided into four blocks. Blocks 1 and 4 assessed only startle magnitude in response to four consecutively presented startle-only trials. Blocks 2 and 3 each included eight startle-only trials and five trials with each prepulse type. Stabilimeter recordings were made between each trial, and the mean value of these measures was subtracted from startle values to account for motor artifact.
All rats were assessed in an abbreviated session before drug testing to divide rats into PPI-matched treatment groups (n = 10). All tests took place between 0900 and 1600 hours. Rats were tested according to a predetermined sequence that counterbalanced treatment groups with respect to time of day and stabilimeter chamber. For drug testing, asenapine or vehicle pretreatment occurred 30 min before apomorphine or vehicle treatment. Rats were placed into individual startle chambers immediately after receiving apomorphine or vehicle treatment.
Study 3: delayed non-match to place
Behavior was assessed in operant boxes (Med Associates, Inc., St. Albans, VT, USA) equipped with two levers, a house light and cue lights, and a reinforcement magazine containing a cue light. Magazine entries (i.e., nose pokes) were monitored by an infrared beam. Operant boxes were monitored and controlled by Med Associates interfaces using MED-PC® for Microsoft Windows®, version 1.15 (Med Associates, Inc.).
DNMTP training was conducted in three phases. First, rats were trained to associate illumination of the magazine cue light with delivery of a 45-mg food pellet (Noyes Precision Pellets, Formula P, New Brunswick, NJ, USA). During the next phase, a single press of the lever whose associated cue light was illuminated was reinforced. During the final phase, the DNMTP task was introduced.
During a sample component, rats were required to nose poke into the food magazine after pressing a randomly presented lever. The first nose poke occurring after a variable delay (0.3–30 s) initiated the choice component. During the choice component, a response on the lever not presented during the sample component (i.e., a non-match to place) was reinforced and designated as a correct response. A response on the sample-component lever (i.e., a match to place) was designated as an incorrect response and was not reinforced. Incorrect responses were followed by a 3-s timeout. Each session lasted 126 trials or 40 min. Drug testing was initiated when accuracy, defined as the percentage of correct responses, was reliably ≥85%. Criterion levels of performance were attained after no more than 30 sessions.
Accuracy was the primary performance variable assessed. Total session accuracy and accuracy at each delay (short delays, 0.3–3.0 s; long delays, 5.6–30 s) were transformed using the arcsin square root function [ASIN(SQRT(((percentage correct/100) − 0.5) × 2))]. To facilitate this transformation, accuracy scores <50% were substituted with a value of 50%. Behavioral latencies, total trials completed, total session nose pokes, and nose-poke rates were also examined. To normalize the distribution of the mean behavioral latencies, these data were log10 transformed. All rats were included in the analysis of the total number of trials completed, but only rats completing ≥28 trials were included in the assessment of other performance analyses. Because nose-poke rates during the 0.3- and 1.0-s delay could not be captured, they were not assessed.
For drug testing, all agents were administered 30 min before placing the animal in the operant chamber. The sequence of drug administration followed a Latin square design, with baseline performance (Monday and Thursday) assessed the day before drug testing (Tuesday and Friday). At least 1 day elapsed between drug-test days. All rats received vehicle and all drug doses. All testing was performed between 0900 and 1600 hours with a particular animal's test time never deviating more than plus or minus 30 min across days.
Study 4: five-choice serial reaction task
Behavior was assessed in five-choice operant boxes (Med Associates, Inc.) that were housed inside ventilated, sound-attenuating chambers. Each box was equipped with a stainless steel grid floor and five wall-mounted response apertures and a reinforcement magazine into which 45-mg Noyes food pellets could be delivered. Operant boxes were monitored and controlled as described above.
Rats were first trained to retrieve food pellets from the illuminated magazine every 15 s during a 10-min session. During phase 2, a nose poke into the illuminated magazine initiated a trial. After a 5-s intertrial interval (ITI), all the response apertures were illuminated, and a nose poke in any aperture was reinforced. To encourage nose poking, three pellets were placed into each aperture at the beginning of each 25-min session.
At the start of each session during the last stage of training, a nose poke into the magazine initiated the 5-s ITI. Nose pokes during the ITI were designated premature responses and initiated a 5-s timeout. If the ITI period ended without another premature response, a response aperture was illuminated in a pseudorandom fashion for 10 s [i.e., stimulus duration (SD) of 10 s]. A nose poke into this aperture within the SD period or the ensuing 5-s limited hold (LH) was designated a correct response. A nose poke in any other aperture was designated an incorrect response. If there was no response within the SD + LH period, an omission was recorded. During training, the SD progressively decreased until an SD of 0.5 s was attained. Drug testing began when accuracy (i.e., the percentage of correct responses: [(correct/(correct + incorrect)) × 100]) averaged >80% and the percentage of omissions was <20% for two consecutive days. The latencies for correct responses, incorrect responses, and pellet collection were also recorded. Each training session lasted 30 min or 100 trials.
All test agents were administered 30 min before placing the animal in the operant chamber. The sequence of drug administration followed the same Latin square design described for the DNMTP studies. All rats received vehicle and all drug doses. Two task conditions were assessed: standard (SD = 0.5 s) and difficult (SD = 0.25 s). Asenapine (n = 12) and olanzapine (n = 8) were tested under the standard and difficult test conditions, whereas risperidone (n = 12) was tested only under difficult test conditions. Test sessions lasted 45 min or 400 trials. All testing was performed between 0900 and 1600 hours with a particular animal’s test time never deviating more than plus or minus 30 min across days.
Statistics
Data were analyzed using one-, two-, or three-way analysis of variance (ANOVA), with statistical significance set at P < 0.05. For the Amp-LMA studies, statistical analyses (Sigmaplot®, Systat Software Inc., San Jose, CA, USA) consisted of one-way ANOVA, with treatment group as a between-subject factor; post hoc Dunnett tests were used to assess dose effects compared with vehicle when appropriate. For the Apo-PPI studies, statistical analyses (Systat® and Statview, Systat Software Inc., San Jose, CA, USA) consisted mixed-design ANOVA. Asenapine pretreatment dose (between-subjects factor), apomorphine treatment (within-subjects factor), and trial blocks and/or types (repeated measures factors adjusted using Greenhouse-Geisser or Huynh-Feldt techniques) were included in the model. As appropriate, post hoc comparisons were made using Newman–Keuls, Dunnett, or Tukey tests. For DNMTP and 5-CSR studies, statistical analyses were conducted using Minitab release 14 (Minitab Inc., State College, PA, USA). DNMTP analyses consisted of two-way (treatment × day) or three-way (treatment × day × delay) repeated measures ANOVA. 5-CSR analyses consisted of two-way (treatment × time) repeated measures ANOVA. In these studies, all factors were treated as within-subject measures. Post hoc Dunnett tests compared active agent with vehicle, and main interactions were assessed using Tukey tests.
Results
Study 1: spontaneous and amphetamine-stimulated locomotor activity
d-Amphetamine, 1 and 3 mg/kg respectively, stimulated LMA, with peaks at around 45 and 20 min postdosing (Fig. 1a, b). Asenapine significantly reversed LMA stimulated by 1 and 3 mg/kg d-amphetamine (Table 1 and Fig. 1a, b). Post hoc analyses indicated that 0.03 and 0.1 mg/kg asenapine reversed low-dose Amp-LMA and 0.1 and 0.3 mg/kg asenapine reversed high-dose Amp-LMA (Dunnett tests, all P < 0.05 vs vehicle, Table 1). The minimal effective doses (MEDs) of asenapine (0.03 and 0.1 mg/kg after low- and high-dose d-amphetamine, respectively) produced 55% and 70% inhibitions of Amp-LMA, respectively (Table 1).

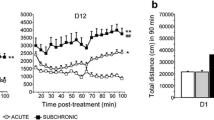
Effects of asenapine on locomotor activity stimulated by 1.0 mg/kg d-amphetamine (a), 3.0 mg/kg d-amphetamine (b), or on spontaneous locomotor activity (c). d -Amp d-amphetamine, s.c. subcutaneous
Spontaneous LMA in vehicle-treated rats was initially high but rapidly decreased to low levels after 30 min (Fig. 1c). Asenapine significantly inhibited spontaneous LMA (Table 1). Post hoc analyses indicated that 0.01–0.1 mg/kg asenapine reduced spontaneous LMA compared with vehicle (Dunnett’s tests, all P < 0.05). The MED of asenapine for spontaneous LMA (0.01 mg/kg) produced a 25% inhibition (Table 1).
Haloperidol, olanzapine, and risperidone significantly reduced spontaneous and Amp-LMA (Fig. 2 and Table 1). Post hoc analysis indicated that only the highest doses of haloperidol (0.3 and 1.0 mg/kg) and olanzapine (3.0 and 10.0 mg/kg) reduced LMA compared with vehicle (Dunnett’s tests, all P < 0.05), with neither agent having a differential MED across test conditions (Table 1). The percent reductions in LMA for the MED of haloperidol (0.3 mg/kg) were comparable across d-amphetamine doses (54.3% and 43.3% reductions after low- and high-dose d-amphetamine, respectively). In contrast, reductions in Amp-LMA produced by the MED of olanzapine (3.0 mg/kg) were more pronounced after low-dose d-amphetamine compared with high-dose d-amphetamine (29.9% vs 73.1%). Risperidone significantly reduced low-dose Amp-LMA after 0.3 and 1.0 mg/kg (53.4% reduction at MED of 0.3 mg/kg), high-dose Amp-LMA after 3.0 mg/kg (93.9% reduction), and spontaneous LMA after 1.0 and 3.0 mg/kg (64.7% reduction at MED of 1.0 mg/kg).

Effects of haloperidol (a, b), olanzapine (c, d), and risperidone (e, f) on locomotor activity stimulated by 1.0or 3.0 mg/kg d-amphetamine. d -Amp d-amphetamine, s.c. subcutaneous
Study 2: apomorphine-disrupted prepulse inhibition
Significant main effects of asenapine and apomorphine, as well as statistical interactions with prepulse intensity and trial block, were reported in the low- and high-dose experiments (see Table 2). Only post hoc analyses are summarized below.
Low-dose asenapine
Post hoc analysis revealed that startle magnitude significantly increased (P < 0.002) in apomorphine-treated rats pretreated with 0.1 mg/kg asenapine compared with rats pretreated with vehicle (Fig. 3a). A comparable nonsignificant increase was observed for 0.03 mg/kg asenapine. An overall habituation effect on startle magnitude between blocks 1 and 4 was reflected by a main effect of trial block (data not shown).

Effects of low-dose asenapine (a) or high-dose asenapine (b) on average startle magnitude during prepulse trails (blocks 2 and 3). PPI prepulse inhibition, s.c. subcutaneous, Veh vehicle. † P < 0.05 (vehicle pretreatment + apomorphine treatment vs asenapine treatment + apomorphine treatment). ‡ P < 0.05 (vehicle pretreatment + vehicle treatment vs asenapine treatment + vehicle treatment)
In post hoc analyses restricted to apomorphine-treated rats, asenapine 0.03 mg/kg (P < 0.006) and 0.1 mg/kg (P < 0.0002) reversed apomorphine-induced disruption of PPI when collapsed across prepulse intensity (Fig. 4a). Asenapine 0.01 mg/kg also demonstrated a near-significant effect (P = 0.054). In post hoc analysis restricted to vehicle pretreated rats and collapsed across prepulse intensity, PPI was significantly increased after asenapine 0.003 mg/kg and significantly decreased after 0.1 mg/kg compared with vehicle (both P < 0.05; Fig. 4a).

Effects of low-dose asenapine (a) or high-dose asenapine (b) on the inhibition of PPI induced by apomorphine collapsed across prepulse intensity and for each prepulse intensity (inset). PPI prepulse inhibition, s.c. subcutaneous, Veh vehicle. *P < 0.001; **P < 0.0001 (vehicle pretreatment + apomorphine treatment vs vehicle pretreatment + vehicle treatment). †a P < 0.05; †b P < 0.01; †c P < 0.001; †d P < 0.0001 (vehicle pretreatment + apomorphine treatment vs asenapine treatment + apomorphine treatment). ‡a P < 0.05; ‡b P < 0.01; ‡c P < 0.001(vehicle pretreatment + vehicle treatment vs asenapine treatment + vehicle treatment)
High-dose asenapine
Asenapine pretreatment (0.03 to 1.0 mg/kg) produced a trend toward increased startle compared with vehicle pretreatment. Post hoc analysis of individual doses of asenapine in vehicle-treated rats demonstrated that 1.0 mg/kg asenapine significantly increased startle magnitude compared with vehicle pretreatment (P < 0.05, Fig. 3b). In apomorphine-treated rats, 0.03 mg/kg asenapine significantly increased startle magnitude compared with vehicle pretreatment (P < 0.05, Fig. 3b). The significant apomorphine treatment effect was reflected by an inverted “U” shaped function of apomorphine on startle magnitude when data are collapsed across blocks 1 and 4. An overall habituation effect on startle magnitude between blocks 1 and 4 was reflected by a main effect of trial block (data not shown).
Post hoc assessment of apomorphine-treated rats collapsed across prepulse intensity revealed significant reversals of apomorphine-induced disruption of PPI with 0.03 mg/kg (P < 0.015), 0.1 mg/kg (P < 0.0001), and 3.0 mg/kg (P < 0.006) asenapine (Fig. 4b ). In vehicle-treated rats, asenapine at 0.1 (P < 0.045), 0.3 (P < 0.0004), 1.0 (P < 0.0015), and 3.0 mg/kg (P < 0.015) produced a significant decrease in PPI when compared with vehicle (Fig. 4b).
Study 3: delayed non-match to place
Asenapine
Assessment of accuracy reported no significant overall effects of asenapine (Fig. 5a). Although the treatment × delay interaction was not significant, post hoc analysis reported effects of asenapine on accuracy during short delays [F(3,96) = 12.22, P < 0.001], with 0.1 mg/kg asenapine decreasing accuracy (P < 0.0001 vs vehicle; Fig. 5b). Asenapine did not alter the total number of trials completed (Table 3).

Effects of asenapine (a, b), olanzapine (c, d), and risperidone (e, f) on DNMTP performance. Data are mean ± SEM. Ase asenapine, DNMTP delayed non-match to place, Olan olanzapine, Risp risperidone, s.c. subcutaneous. § P < 0.0001 (Dunnett test vs vehicle)
Significant main effects of asenapine were reported for correct and incorrect response latencies (Table 3). Post hoc analysis indicated that 0.1 mg/kg asenapine increased correct and incorrect response latencies (both P < 0.05 vs vehicle). Asenapine also tended to increase pellet collection latency. A main treatment effect of asenapine on nose-poke rate was also found. Post hoc analysis indicated that 0.056 and 0.10 mg/kg asenapine decreased nose-poke rate (P < 0.01 and P < 0.001, respectively).
Olanzapine
Assessment of accuracy reported no overall effects of olanzapine (Fig. 5c). A significant treatment effect [F(3,84) = 3.14, P < 0.05] and a treatment × delay interaction [F(9,84) = 2.33, P < 0.05] were reported on accuracy at short delays, but post hoc analyses did not identify significant differences between olanzapine and vehicle (Fig. 5d). Olanzapine did not alter the total number of trials completed (Table 3).
Analysis of response latencies reported main effects of treatment for correct response latency (Table 3). Post hoc analysis indicated that 0.5 and 1.0 mg/kg olanzapine increased correct response latency (both P < 0.05 vs vehicle). Olanzapine did not alter the total number of nose pokes, but nose-poke rate was decreased by olanzapine with post hoc analysis indicating that 1.0 mg/kg olanzapine decreased nose-poke rate (P < 0.001 vs vehicle).
Risperidone
Risperidone decreased overall accuracy [F(3,28) = 5.59, P < 0.005], but post hoc assessment did not identify significant differences between risperidone and vehicle (Fig. 5e). A significant main effect of risperidone during short delays was reported [F(3,84) = 26.10, P < 0.001], with accuracy being decreased by 0.3 mg/kg risperidone (Dunnett’s test, P < 0.0001 vs vehicle; Fig. 5f). Analysis of longer delays revealed a treatment × delay interaction [F(9,112) = 3.31, P < 0.001]. Post hoc analysis reported that 0.3 mg/kg risperidone improved accuracy at the 30-s delay (P < 0.05 vs vehicle; Fig. 5f). Risperidone decreased the total number of trials completed [F(3,28) = 24.80, P < 0.001], with total trials completed being decreased by 0.3 mg/kg risperidone (P < 0.0001 vs vehicle; Table 3).
Analysis of response latencies reported main effects of risperidone on correct response, incorrect response, and pellet collection latencies (Table 3). Post hoc analysis confirmed that 0.3 mg/kg risperidone increased these latencies (all P < 0.0001 vs vehicle). Main effects of risperidone treatment were also reported for total number of nose pokes and nose-poke rate per second of delay. Post hoc analysis indicated that 0.3 mg/kg risperidone decreased the total number of nose pokes compared with vehicle (P < 0.0001) and 0.1 and 0.3 mg/kg risperidone decreased nose-poke rate per second delay compared with vehicle (P < 0.01).
Study 4: five-choice serial reaction task
Standard condition–stimulus duration of 0.5 s
Asenapine
Asenapine did not significantly alter performance (Table 4).
Olanzapine
Main effects of treatment were reported for the percentage of omissions. Post hoc analyses revealed that 0.3 mg/kg olanzapine increased the percentage of omissions (P < 0.0001 vs vehicle; Table 4).
Difficult condition–stimulus duration of 0.25 s
Asenapine
Main effects of treatment on trials completed, the percentage of omissions, and incorrect response latency were reported with low-dose asenapine (Table 4). Post hoc comparisons revealed that 0.03 mg/kg asenapine decreased total trials completed (P = 0.003), increased the percentage of omissions (P = 0.001), and increased incorrect response latency (P = 0.011) compared with vehicle (Table 4).
Main effects of treatment were reported for trials completed, the percentages of correct trials and omissions, and correct and incorrect response latencies with high-dose asenapine (Table 3). Post hoc analysis revealed that 0.1 mg/kg asenapine decreased the percentage of correct responses (P < 0.0001 vs vehicle). Both 0.1 and 0.3 mg/kg asenapine increased the percentage of omissions (P = 0.005 and P < 0.0001 vs vehicle, respectively) and decreased total trials completed (both P < 0.0001 vs vehicle). Correct response and incorrect response latencies were increased by 0.3 mg/kg asenapine (P < 0.0001 and P = 0.006, respectively, vs vehicle). The effects of 0.03 mg/kg asenapine were not replicated in this experiment.
Olanzapine
Main effects of olanzapine were reported for trials completed, the percentage of correct responses and omissions, and pellet collection latency (Table 4). Post hoc comparisons revealed that 0.03 and 0.3 mg/kg olanzapine reduced the percentage of correct responses (P = 0.008 and P = 0.002, respectively, vs vehicle). Both 0.1 and 0.3 mg/kg olanzapine increased the percentage of omissions (P = 0.009 and P < 0.0001 vs vehicle, respectively). All olanzapine doses reduced total trials completed (all P < 0.05 vs vehicle). Despite the significant main effect of treatment on pellet collection latency, post hoc analysis did not report significant differences between olanzapine and vehicle.
Risperidone
Main effects of treatment were reported for trials completed, the percentage of omissions, correct and incorrect response latencies, and pellet collection latency. Risperidone did not significantly alter the percentage of correct responses (Table 4). Post hoc comparisons revealed that 0.03 and 0.1 mg/kg risperidone increased the percentage of omissions (both P = 0.001 vs vehicle) and reduced trials completed (both P < 0.02 vs vehicle). Incorrect response latency was increased by 0.03 and 0.1 mg/kg risperidone (P = 0.005 and P < 0.0001, respectively, vs vehicle). Only 0.1 mg/kg risperidone increased correct response latency (P < 0.0001 vs vehicle) and only 0.03 mg/kg risperidone increased pellet collection latency (P = 0.017 vs vehicle).
Discussion
The main findings of the studies described in the current report are that: (1) asenapine showed highly potent activity in two independent tests predictive of antipsychotic efficacy (Amp-LMA and Apo-PPI) and (2) neither asenapine nor the comparator drugs (olanzapine, risperidone) across a range of doses improved performance in cognitive tasks designed to assess effects on short-term spatial memory and attention in normal healthy rats. These data further increase the understanding of the behavioral psychopharmacologic properties of asenapine.
d-amphetamine-induced hyperactivity and Apo-PPI (Geyer and Ellenbroek 2003) are two of several behavioral animal paradigms that are used to detect antipsychotic activity. The potent reversal of Amp-LMA by asenapine supports its potential for antipsychotic activity. The potency of asenapine in this model is also consistent with its high affinity and antagonistic activity at the dopamine D2 receptor (Shahid et al. 2009). The effects of asenapine in this study were examined against two doses of d-amphetamine based on evidence indicating that low and high doses of the dopaminergic stimulant show differential sensitivity of inhibition by different classes of antipsychotic agents (Arnt and Skarsfeldt 1998). It has been speculated that this may be partially related to the preferential engagement of mesolimbic and striatal neuronal pathways by low and high doses of d-amphetamine, respectively (Arnt 1995). Asenapine showed a slightly higher potency for inhibiting the effects of 1 mg/kg d-amphetamine compared with 3 mg/kg d-amphetamine. Like asenapine, the effects of risperidone differed between the two doses of d-amphetamine. In contrast, haloperidol showed similar potency with both doses of d-amphetamine. The same could be argued for olanzapine, which showed a similar MED of 3 mg/kg against the two doses of d-amphetamine. However, it should be noted that olanzapine did show a much stronger inhibition of 1 mg/kg d-amphetamine compared with 3 mg/kg d-amphetamine, with respective reductions compared with vehicle of 73% and 30%. Thus, overall the current data are consistent with the findings reported by Arnt (1995).
The ability of second-generation antipsychotics (SGAs) to differentially reduce hyperactivity induced by low- vs high-dose d-amphetamine may also be partially attributed to their antagonism of 5-HT2A receptors (Arnt 1995). It has been reported that the effects of low-dose, but not high-dose, d-amphetamine are inhibited by the selective 5-HT2A antagonists ritanserin and MDL 100,151 (Feldman et al. 1997). Our findings support this hypothesis because the rank order of MEDs for inhibition of hyperactivity stimulated by low-dose amphetamine (0.03 mg/kg asenapine < 0.3 mg/kg risperidone < 3.0 mg/kg olanzapine) mirrors the affinity of these agents for the 5-HT2A receptor (asenapine, k i = 0.07 nM; risperidone, k i = 0.2 nM; olanzapine, k i = 1.3 nM; Shahid et al. 2009). As with asenapine, spontaneous LMA in non-habituated rats was inhibited by olanzapine, risperidone, and haloperidol.
The ability of asenapine to reverse disruptions in PPI induced by apomorphine is shared by most antipsychotic drugs (Geyer et al. 2001; Mansbach et al. 1988) and is primarily attributed to antagonism of D2 receptors. Given that asenapine has higher affinity for the D2 receptor (1.3 nM) than olanzapine and risperidone (21 and 6.2 nM, respectively; Shahid et al. 2009), it is not surprising that the potency of asenapine in this study was higher than in previous studies examining the effects of olanzapine and risperidone. For example, Auclair et al. (2006) reported that olanzapine and risperidone doses of at least 0.63 mg/kg were required to reverse deficits in PPI induced by 0.63 mg/kg apomorphine. In addition, it is important to note that the reversal of Apo-PPI produced by asenapine occurred at doses that did not consistently impact overall startle magnitude, suggesting that nonspecific motor effects did not influence these results.
Overall, these LMA and PPI data provide additional preclinical support for the antipsychotic profile of asenapine. In a previous report, doses of asenapine ranging from 0.05 to 0.2 mg/kg s.c. demonstrated dose-dependent antipsychotic effects, as measured by suppression of the conditioned avoidance response (Franberg et al. 2008). The interpretation of these effects in animal models is further bolstered by the results of a clinical study in patients with schizophrenia in which asenapine was found to be effective in the treatment of positive and negative symptoms (Potkin et al. 2007).
Neither asenapine nor the SGAs tested demonstrated cognitive-enhancing properties in the DNMTP or 5-CSR tasks under the test conditions and doses here in normal rats. Furthermore, high-dose treatment with asenapine, risperidone, or olanzapine was generally associated with impaired performance (e.g., reduced accuracy, increased omissions, and increased response latencies) that may be associated with impaired motor performance or sedation. However, it should be noted that there is a somewhat different pattern of effects between the compounds. Asenapine significantly reduced spontaneous activity from the lowest dose tested 0.01 (mg/kg). Although numerically the effects of 0.01 mg/kg asenapine (2,268 counts) compared well with those of the other agents tested (2,334, 2,461, and 2,554 counts for haloperidol, olanzapine, and risperidone, respectively) at the lowest doses tested, only intermediate doses of those agents proved to be effective. However, a different pattern emerges from the parameters associated with sedation and motivation in the cognitive tasks (e.g., the percentage of omissions in the 5-CSR task or correct latency in the DNMTP task). Asenapine impairs both measures at a dose of 0.1 mg/kg, a dose that is a log unit higher than the lowest effective dose for spontaneous locomotion. Olanzapine increases omissions and correct response latency at 0.1 and 0.5 mg/kg, respectively. These doses are lower than the dose, which reduces spontaneous activity (3.0 mg/kg). Similarly, risperidone has deleterious effects on these 5-CSR and DNMTP performance measures at doses that are lower than the dose, which impairs spontaneous activity (0.03 and 0.3 mg/kg, respectively, vs 1.0 mg/kg). Thus, these effects are generally consistent with previous reports of the effects of antipsychotics in tasks of cognitive function in normal adult rats (Didriksen 1995; Levin and Christopher 2006; Rosengarten and Quartermain 2002; Smith et al. 2000; Terry et al. 2003). However, the different patterns of effect may suggest that high doses of asenapine begin to impair cognitive task performance due to motor retardation or sedation. By contrast, olanzapine and risperidone induce motor and/or motivational impairment in cognitive tasks at doses that do not cause locomotor effects, possibly indicating a bradyphrenic profile at non-sedative doses.
The data from the cognitive tasks suggest that asenapine, olanzapine, and risperidone each impaired short-term spatial memory and attention, as measured by DNMTP and 5-CSR performance, respectively. However, the effects of asenapine on performance were less pronounced than those of olanzapine and risperidone. In contrast to olanzapine, asenapine did not induce any cognitive impairment in the standard version of the 5-CSR task. When the demand of the task was increased, cognitive impairment was observed most consistently after administration of higher asenapine doses (0.1 and 0.3 mg/kg), whereas impairment was observed after olanzapine and risperidone doses as low as 0.03 mg/kg. Although the exact mechanism underlying the difference between asenapine and the other agents requires further investigation, it is hypothesized that it may be related to differences in their overall receptor-binding profile and in particular to antagonism at 5-HT6 and histamine H1 receptors. Asenapine (k i, 0.25 nM) has much higher 5-HT6 receptor affinity than risperidone (k i, 2,187 nM; Shahid et al. 2009). This difference could be of relevance as antagonism at the 5-HT6 receptor, with selective ligands such as SB-399885 and SB-271046, has cognitive-enhancing effects in animal models (Hatcher et al. 2005; Hirst et al. 2006). Although olanzapine, similar to asenapine, has a 6-fold higher affinity for 5-HT6 (k i, 3.2 nM) compared to D2 receptors (k i, 20 nM), the relative affinity of olanzapine for the H1 receptor over the D2 is higher than that of asenapine (6- vs 1.3-fold; Shahid et al. 2009). Given that strong H1 receptor antagonism is associated with sedation (Miller 2004), it could be this aspect in the differential pharmacologies that contributes to the difference observed between asenapine and olanzapine in the cognition tests used in the current study. In short, all three of the drugs examined in this study have complex and varied pharmacologies. The subtle interaction between these various components in each different scenario makes it difficult to attribute these effects on the affinity at any one given receptor. Similarly, caution must be exercised in making an a priori prediction on performance in a particular task based purely on knowledge of individual receptor affinities.
Previous reports have demonstrated that atypical antipsychotics routinely reverse cognitive deficits induced by different experimental manipulations (Abdul-Monim et al. 2003, 2006; Idris et al. 2005; Marcus et al. 2005; Terranova et al. 2005) but rarely improve cognitive performance in healthy rats (Wolff and Leander 2003). A number of reports have demonstrated the reversal of cognitive deficits in reversal learning in rats using experimental procedures (i.e., phencyclidine- or amphetamine-induced deficits) relevant to psychiatric disease (Abdul-Monim et al. 2003, 2006; Idris et al. 2005; Neill et al. 2008). Therefore, it would appear that, as in humans, these agents are either neutral or deleterious to cognitive function in healthy individuals, whereas improvements may be observed in impaired individuals. In this regard, recent pre-clinical data has indicated that asenapine can attenuate experimentally induced cognitive deficits in rodents (Neill et al. 2006, 2008; Tait et al. 2009), and primates (Jentsch et al. 2006). These pre-clinical data mirrored the preliminary findings of a clinical study performed in schizophrenic patients (Fleming et al. 2007).
In conclusion, the reversal of amphetamine-induced hyperactivity and apomorphine-induced impairments in PPI by asenapine provides support for antipsychotic properties and utility in the treatment of schizophrenia (Potkin et al. 2007). In contrast, the lack of improvement in DNMTP and 5-CSR performance following treatment with asenapine, olanzapine, or risperidone suggests that these agents do not have cognitive-enhancing effects in healthy adult rats. This finding may indicate the need to move to studies based upon experimentally induced impairments (Abdul-Monim et al. 2003, 2006; Birrell and Brown 2000; Idris et al. 2005; Marcus et al. 2005; Terranova et al. 2005) in order to determine if there is potential for clinical utility in the cognitive domain.
References
Abdul-Monim Z, Reynolds GP, Neill JC (2003) The atypical antipsychotic ziprasidone, but not haloperidol, improves phencyclidine-induced cognitive deficits in a reversal learning task in the rat. J Psychopharmacol 17:57–65
Abdul-Monim Z, Reynolds GP, Neill JC (2006) The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav Brain Res 169:263–273
Addington J, Addington D, Gasbarre L (2001) Neurocognitive and social functioning in schizophrenia and other diagnoses. Schizophr Res 48:367–368
Amitai N, Markou A (2009) Chronic nicotine improves cognitive performance in a test of attention but does not attenuate cognitive disruption induced by repeated phencyclidine administration. Psychopharmacology (Berl) 202:275–286
Amitai N, Semenova S, Markou A (2007) Cognitive-disruptive effects of the psychotomimetic phencyclidine and attenuation by atypical antipsychotic medications in rats. Psychopharmacology (Berl) 193:521–537
Arnt J (1995) Differential effects of classical and newer antipsychotics on the hypermotility induced by two dose levels of D-amphetamine. Eur J Pharmacol 283:55–62
Arnt J, Skarsfeldt T (1998) Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology 18:63–101
Auclair AL, Kleven MS, Besnard J, Depoortere R, Newman-Tancredi A (2006) Actions of novel antipsychotic agents on apomorphine-induced PPI disruption: influence of combined serotonin 5-HT1A receptor activation and dopamine D2 receptor blockade. Neuropsychopharmacology 31:1900–1909
Birrell JM, Brown VJ (2000) Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci 20:4320–4324
Cuesta MJ, Peralta V, Zarzuela A (2001) Effects of olanzapine and other antipsychotics on cognitive function in chronic schizophrenia: a longitudinal study. Schizophr Res 48:17–28
Didriksen M (1995) Effects of antipsychotics on cognitive behaviour in rats using the delayed non-match to position paradigm. Eur J Pharmacol 281:241–250
Didriksen M, Skarsfeldt T, Arnt J (2007) Reversal of PCP-induced learning and memory deficits in the Morris' water maze by sertindole and other antipsychotics. Psychopharmacology (Berl) 193:225–233
Feldman DJ, Frank RA, Kehne JH, Flannery R, Brown D, Soni S, Byrd G, Shah S (1997) Mixed D2/5-HT2 antagonism differentially affects apomorphine- and amphetamine-induced stereotyped behavior. Pharmacol Biochem Behav 58:565–572
Fleming K, Potkin SG, Binneman B, Keller DS, Alphs L, Panagides J (2007) Asenapine improves cognitive function in acute schizophrenia: a placebo- and risperidone-controlled trial. Paper presented at: 160th Annual Meeting of the American Psychiatric Association, May 19–24, 2007; San Diego, CA
Franberg O, Wiker C, Marcus MM, Konradsson A, Jardemark K, Schilstrom B, Shahid M, Wong EH, Svensson TH (2008) Asenapine, a novel psychopharmacologic agent: preclinical evidence for clinical effects in schizophrenia. Psychopharmacology (Berl) 196:417–429
Geyer MA, Ellenbroek B (2003) Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry 27:1071–1079
Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR (2001) Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 156:117–154
Grayson B, Idris NF, Neill JC (2007) Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav Brain Res 184:31–38
Hagan JJ, Jones DN (2005) Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull 31:830–853
Hatcher PD, Brown VJ, Tait DS, Bate S, Overend P, Hagan JJ, Jones DN (2005) 5-HT6 receptor antagonists improve performance in an attentional set shifting task in rats. Psychopharmacology (Berl) 181:253–259
Hirst WD, Stean TO, Rogers DC, Sunter D, Pugh P, Moss SF, Bromidge SM, Riley G, Smith DR, Bartlett S, Heidbreder CA, Atkins AR, Lacroix LP, Dawson LA, Foley AG, Regan CM, Upton N (2006) SB-399885 is a potent, selective 5-HT6 receptor antagonist with cognitive enhancing properties in aged rat water maze and novel object recognition models. Eur J Pharmacol 553:109–119
Huang M, Li Z, Dai J, Shahid M, Wong EH, Meltzer HY (2008) Asenapine increases dopamine, norepinephrine, and acetylcholine efflux in the rat medial prefrontal cortex and hippocampus. Neuropsychopharmacology 33:2934–2945
Idris NF, Repeto P, Neill JC, Large CH (2005) Investigation of the effects of lamotrigine and clozapine in improving reversal-learning impairments induced by acute phencyclidine and D-amphetamine in the rat. Psychopharmacology (Berl) 179:336–348
Institute of Laboratory Animal Resources (1996) Guide for the care and use of laboratory animals—ILAR, National Research Council. National Academy Press, Washington, DC
Jentsch JD, Shahid M, Wong EHF, Roth RH (2006) Asenapine improves cognitve function in monkeys repeatedly exposed to the psychotomimetic drug phencyclidine. Biol Psychiatry 59:471
Kapur S, Mamo D (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry 27:1081–1090
Levin ED, Christopher NC (2006) Effects of clozapine on memory function in the rat neonatal hippocampal lesion model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 30:223–229
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK, Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators (2005) Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353:1209–1223
Mansbach RS, Geyer MA, Braff DL (1988) Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 94:507–514
Marcus MM, Jardemark KE, Wadenberg ML, Langlois X, Hertel P, Svensson TH (2005) Combined alpha2 and D2/3 receptor blockade enhances cortical glutamatergic transmission and reverses cognitive impairment in the rat. Int J Neuropsychopharmacol 8:315–327
McIntyre R, Hirschfeld R, Calabrese J, Szegedi A (2008) Asenapine in bipolar disorder: an overview of the clinical trials in the Olympia program. Int J Neuropsychopharmacol 11(suppl 1):183
Miller DD (2004) Atypical antipsychotics: sleep, sedation, and efficacy. Prim Care Companion J Clin Psychiat 6:3–7
Nakai S, Hirose T, Mori T, Stark A, Araki H, Kikuchi T (2008) The effect of aripiprazole on prepulse inhibition of the startle response in normal and hyperdopaminergic states in rats. Int J Neurosci 118:39–57
Neill JC, Shahid M, Wong EHF, Idris NF (2006) Comparison of the efficacy of asenapine, risperidone, and olanzapine to improve a reversal learning deficit in the rat. Schizophr Res 81:s105–s106
Neill JC, Shahid M, Grayson B, Marston HM, Snigdha S (2008) Asenapine improves a subchronic phencyclidine-induced deficit in object recognition memory in the rat. Biol Psychol 63:75S–76S
Nordquist RE, Risterucci C, Moreau JL, von Kienlin M, Kunnecke B, Maco M, Freichel C, Riemer C, Spooren W (2008) Effects of aripiprazole/OPC-14597 on motor activity, pharmacological models of psychosis, and brain activity in rats. Neuropharmacology 54:405–416
Potkin SG, Cohen M, Panagides J (2007) Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry 68:1492–1500
Quarta D, Naylor CG, Morris HV, Patel S, Genn RF, Stolerman IP (2007) Different effects of ionotropic and metabotropic glutamate receptor antagonists on attention and the attentional properties of nicotine. Neuropharmacology 53:421–430
Remington G (2003) Understanding antipsychotic “atypicality”: a clinical and pharmacological moving target. J Psychiatry Neurosci 28:275–284
Rosengarten H, Quartermain D (2002) The effect of chronic treatment with typical and atypical antipsychotics on working memory and jaw movements in three- and eighteen-month-old rats. Prog Neuropsychopharmacol Biol Psychiatry 26:1047–1054
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, De Loore K, Leysen JE (1996) Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 124:57–73
Shahid M, Walker G, Zorn S, Wong E (2009) Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol 23:65–73
Sharma T, Antonova L (2003) Cognitive function in schizophrenia. Deficits, functional consequences, and future treatment. Psychiatr Clin North Am 26:25–40
Smith AD, Smith DL, Zigmond MJ, Amalric M, Koob GF (2000) Differential effects of dopamine receptor subtype blockade on performance of rats in a reaction-time paradigm. Psychopharmacology (Berl) 148:355–360
Sukhotina IA, Dravolina OA, Novitskaya Y, Zvartau EE, Danysz W, Bespalov AY (2008) Effects of mGlu1 receptor blockade on working memory, time estimation, and impulsivity in rats. Psychopharmacology (Berl) 196:211–220
Swerdlow NR, Varty GB, Geyer MA (1998) Discrepant findings of clozapine effects on prepulse inhibition of startle: is it the route or the rat? Neuropsychopharmacology 18:50–56
Tait DS, Marston HM, Shahid M, Brown VJ (2009) Asenapine restores cognitive flexibility in rats with medial prefrontal cortex lesions. Psychopharmacology (Berl) 202:295–306
Tarazi FI, Moran-Gates T, Wong EH, Henry B, Shahid M (2008) Differential regional and dose-related effects of asenapine on dopamine receptor subtypes. Psychopharmacology (Berl) 198:103–111
Terranova JP, Chabot C, Barnouin MC, Perrault G, Depoortere R, Griebel G, Scatton B (2005) SSR181507, a dopamine D(2) receptor antagonist and 5-HT(1A) receptor agonist, alleviates disturbances of novelty discrimination in a social context in rats, a putative model of selective attention deficit. Psychopharmacology (Berl) 181:134–144
Terry AV Jr, Hill WD, Parikh V, Waller JL, Evans DR, Mahadik SP (2003) Differential effects of haloperidol, risperidone, and clozapine exposure on cholinergic markers and spatial learning performance in rats. Neuropsychopharmacology 28:300–309
Varty GB, Geyer MA (1998) Effects of isolation rearing on startle reactivity, habituation, and prepulse inhibition in male Lewis, Sprague–Dawley, and Fischer F344 rats. Behav Neurosci 112:1450–1457
Varty GB, Higgins GA (1995) Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology (Berl) 122:15–26
Ward CP, Harsh JR, York KM, Stewart KL, McCoy JG (2004) Modafinil facilitates performance on a delayed nonmatching to position swim task in rats. Pharmacol Biochem Behav 78:735–741
Wolff MC, Leander JD (2003) Comparison of the effects of antipsychotics on a delayed radial maze task in the rat. Psychopharmacology (Berl) 168:410–416
Acknowledgments
This research was supported by Schering-Plough and Pfizer Inc. Editorial support was provided by Complete Healthcare Communications, Inc., and was funded by Schering-Plough. H.M. Marston and M. Shahid are employees of Schering-Plough. K. Serpa and C. Moore are employees of Pfizer Inc. E.H.F. Wong, L. Gold, and L. Meltzer were employees of Pfizer Inc. at the time this research was conducted. M.A. Geyer holds an equity interest in San Diego Instruments. M. R. Azar is an employee of Behavioral Pharma, Inc.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Marston, H.M., Young, J.W., Martin, F.D.C. et al. Asenapine effects in animal models of psychosis and cognitive function. Psychopharmacology 206, 699–714 (2009). https://doi.org/10.1007/s00213-009-1570-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1570-z