
Abstract
HMG-CoA-Reductase inhibitors (HMGRIs) are currently the most widely used group of drugs in patients with coronary artery disease (CAD) and are given preemptively to patients with high levels of cholesterol, including those with diabetes mellitus (DM). However, intake of HMGRIs also increases the progression of coronary artery calcification (CAC) and the risk of developing DM. This study aimed to investigate whether HMGRI intake interacts with the diabetes-associated genetic risk score (GRS) to affect CAC progression using data from the population-based Heinz Nixdorf Recall (HNR) study. CAC was measured in 3157 participants using electron-beam computed tomography twice, at baseline (CACb) and 5 years later (CAC5y). CAC progression was classified as slow, expected, or rapid based on predicted values. Weighted DM GRS was constructed using 100 diabetes mellitus–associated single nucleotide polymorphisms (SNPs). We used log-linear regression to evaluate the interaction of HMGRI intake with diabetes-associated GRS and individual SNPs on CAC progression (rapid vs. expected/slow), adjusting for age, sex, and log(CACb + 1). The prevalence of rapid CAC progression in the HNR study was 19.6%. We did not observe any association of the weighted diabetes mellitus GRS with the rapid progression of CAC (relative risk (RR) [95% confidence interval (95% CI)]: 1.01 [0.94; 1.10]). Furthermore, no indication of an interaction between GRS and HMGRI intake was observed (1.08 [0.83; 1.41]). Our analyses showed no indication that the impact of HMGRIs on CAC progression is significantly more severe in patients with a high genetic risk of developing DM than in those with a low GRS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is the primary cause of coronary artery disease (CAD) and precedes the onset of coronary heart disease (CHD) by decades (Erbel & Budoff 2012; McClelland et al. 2015; Möhlenkamp et al. 2011). Coronary artery calcification (CAC) is one of the most sensitive and specific markers of coronary atherosclerosis, and the quantification of CAC has been shown to improve the ability to predict future CHD events (Budoff et al. 2010; Elias-Smale et al. 2010; Erbel & Budoff 2012; Lehmann et al. 2018; McClelland et al. 2015; Möhlenkamp et al. 2011; Taylor et al. 2008).
HMG-CoA-Reductase inhibitors (HMGRIs; also commonly referred to as statins) are the most widely used lipid-lowering medication for the majority of people with CHD risk, including those with diabetes mellitus type 2 (DM) (Knuuti et al. 2020). They are strongly recommended in many primary prevention guidelines from the European Association for Cardiovascular Prevention and Rehabilitation (EACPR) and the American College of Cardiology/American Heart Association (ACC/AHA) ("JBS 2: Joint British Societies' guidelines on prevention of cardiovascular disease in clinical practice," 2005; Piepoli et al. 2016; Robson 2008). However, recent results from an observational study and a clinical trial indicate that HMGRIs promote the progression of CAC (Dykun et al. 2016; Henein et al. 2015). Likewise, earlier studies concluded that HMGRI intake does not halt progression of CAC and enhances it in type 2 diabetes patients (Anand et al. 2007; Houslay et al. 2006; Saremi et al. 2012; Terry et al. 2007). Other studies have shown that HMGRIs promote coronary atheroma calcification and are implicated in the calcification of vascular smooth muscle cells as well as mesenchymal cells (Kupcsik et al. 2009; Puri et al. 2015; Trionet al. 2008).
HMGRIs bind directly to 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase within the mevalonate pathway to impair endogenous cholesterol synthesis, thus lowering LDL-cholesterol levels (Fig. 1). Problems may arise from the “pleiotropic effects” of HMGRIs, a collective term describing the multitude of (sometimes unintentional and often less well understood) HMGRI effects independent of cholesterol synthesis. They are mostly mediated through altered levels of the mevalonate pathway’s isoprenoid intermediates farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) (Fig. 1). For instance, heme A and ubiquinone, both of which are involved in oxidative phosphorylation, are derived from isoprenoid precursors. The available data suggest that HMGRIs modulate redox systems that are implicated in the development of atherosclerosis (Okuyama et al. 2015; Rhee et al. 2015).

HMG-CoA-Reductase inhibitors (HMGRIs; formerly known as statins) lead to reduced FPP levels, which is the reason for reduced cholesterol synthesis and the rationale for their use in the prevention of coronary heart disease (CHD). Additionally, HMGRIs exhibit pleiotropic (i.e., cholesterol synthesis independent) effects, some of which may counteract their desired LDL-cholesterol lowering effect. Red arrows indicate up- and downregulation upon HMGRI treatment
Other pleiotropic effects originate from reduced protein prenylation, a posttranslational modification involving the direct transfer of FPP or GGPP, which is crucial for the proper function of hundreds of proteins. There is evidence that prenylated proteins play an important role in the pathogenesis of diabetes and the regulation of glucose levels (Kowluru & Kowluru 2015). At the same time, several observational studies have suggested an association between HMGRIs and elevated risks of developing DM (Carter et al. 2013; Cederberg et al. 2015; Waters et al. 2011). Furthermore, several studies indicated an association of DM and, more specifically, poor glycemic control with CAC progression (Koulaouzidis et al. 2013; Snell-Bergeon et al. 2003).
Recent large-scale genome-wide association studies (GWASs) and meta-analyses have identified 100 genetic variants that are associated with DM (Locke et al. 2015; Mahajan et al. 2014; Scott et al. 2017; Zhao et al. 2017). Moreover, it is not known whether genetic variation at most of these loci exacerbates the effects of HMGRIs on the risk of progression of CAC. A desirable clinical goal is to incorporate genetic information in the form of a genetic risk score (GRS) for DM. This would improve the predictive power of a model when compared with a model consisting of only known (lifestyle) risk factors in asymptomatic individuals so that preventive measures, if available, can be taken.
It is important to preclude a disproportionately high risk of developing DM or intense CAC in particular patient groups treated with HMGRIs. The necessity of such investigations is highlighted by three facts: (i) the widespread use of HMGRIs, (ii) the fact that individuals under HMGRI therapy have been shown to have higher CAC as well as a higher risk of developing DM, and (iii) the evidence that DM is associated with CAC progression. To this end, we investigated the putative interaction between the DM GRS (constructed using GWAS-identified genetic variants) and HMGRI intake concerning its influence on the progression of coronary artery calcification.
Materials and methods
Study population
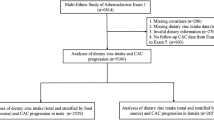
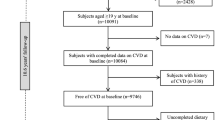
At baseline (b), 4814 participants aged between 45 and 75 years (50% women) from the Heinz Nixdorf Recall Study (Risk Factors, Evaluation of Coronary Calcium, and Lifestyle) were randomly selected from the registration lists of the densely populated Ruhr metropolitan cities in Germany (residents of Essen, Bochum, and Mülheim) between December 2000 and August 2003. The rationale and design of the study were previously described in detail (Schmermund et al. 2002). The Heinz Nixdorf Recall Study is an already well-described population-based cohort study (Stang et al. 2005). Data of the study participants have been repeatedly used to address research questions of different medical fields (Heilmann-Heimbach et al. 2017; Locke et al. 2015; Malhotra et al. 2019; Orban et al. 2017; Shungin et al. 2015; Stang et al. 2007; Thanassoulis et al. 2013; Tzivian et al. 2016). To avoid identification and profiling of the participants, strict data protection is applied. However, for the purpose of replication, other researchers are allowed to access data upon request, which is the same way the authors of the present paper obtained the data. Data requests can be addressed to recall@uk-essen.de. The first follow-up examination took place 5 years after the baseline examination, and the participants were re-invited to attend.
The study participants were selected based on several exclusion and inclusion criteria. First, we excluded participants with prior CAD (coronary artery bypass surgery and/or interventional revascularization and a history of prior myocardial infarction) (n = 327) at baseline. Of the remaining participants, only the participants with CAC measurements at baseline (CACb) and first follow-up (CAC5y) were included (approximately 5 years apart, 5.1 ± 0.3 years) (n = 3675). Additionally, the following groups of participants were excluded: (i) participants with stent implementation, bypass, balloon dilatation, or myocardial infarction during the 5-year follow-up (n = 154); (ii) participants outside the study age range (45–74 at baseline, 50–79 at the 5-year follow-up, n = 12); and (iii) participants with missing Framingham risk factors information (n = 28) (Erbel et al. 2014; Lehmann et al. 2016, 2018).
Lymphocyte DNA was isolated from EDTA anticoagulated venous blood using a Chemagic Magnetic Separation Module I (Chemagen, Baesweiler, Germany). Genotyping was performed using different Illumina microarrays (Metabochip, Omni1-Quad, OmniExpressv1.0, HumanCoreExome (v1.0 and v1.1); Illumina, San Diego, USA) according to the manufacturer’s protocols. Quality control was applied prior to the imputation, separately for each chip, and was first performed on the subject level including sex, ethnicity, and relatedness checks, excluding subjects with missing genotype data > 5%. Furthermore, single nucleotide polymorphisms (SNPs) with a minor allele frequency (MAF) < 1%, a missing genotype frequency > 5%, or a deviation from Hardy–Weinberg Equilibrium (HWE) (p < 10−5) were excluded. Imputation was carried out using IMPUTE v.2.3.1 with reference data from 1000 Genomes Phase 1, release March 2012, for the Metabochip and 1000 Genomes Phase 3, release October 2014, for all other microarray data (Frank et al. 2019; Geisel et al. 2016; Pechlivanis et al. 2020, 2013). The imputed data were then converted to the PLINK ped format using the threshold ≥ 0.8 in GTOOL v0.7.5.
For the present study, data from 3157 participants was used. The participants were included with CAC measurements at two points of time (CACb, CAC5y), and no missing information on age, sex, genetic risk score, and intake of HMGRIs. The study was approved by the ethical committees at the University Hospital of Essen, Germany, and was conducted in accordance with the principles expressed in the Declaration of Helsinki. The study was certified and recertified according to DIN EN ISO 9001:2000/2008. All study participants gave their written informed consent.
Genetic risk scores
The SNPs for the GRS were selected from the published diabetes mellitus GWASs (Gaulton et al. 2015; Mahajan et al. 2014; Scott et al. 2017; Zhao et al. 2017). The average weighted GRS for each individual was constructed by using the risk estimate (transformed by natural log; bn) from the published study and multiplying it by the number of risk alleles (xn: 0 (no risk allele), 1 (1 risk allele), 2 (2 risk alleles)) for each SNP as previously published (Pechlivanis et al. 2020). The products were then summed and divided by the number of SNPs (n = 100):
If the genotype in the score for a particular individual was missing, then the expected value was imputed based on the sample allele frequency. To calculate the GRS, the allelic scoring routine in PLINK was used (Purcell et al. 2007). The mean (0.0498) and standard deviation (SD 0.005) of the study population were used to standardize the GRS to have a mean of 0 and unit variance. Genetic risk was then analyzed per SD of the standardized GRS.
Assessment of coronary artery calcification at baseline and first follow-up
A nonenhanced electron-beam scan with a C-100 or C-150 scanner (GE Imatron, San Francisco, CA, USA) was used to assess the CACb. (Schmermund et al. 2002). The CAC5y computer tomography (CT) was performed at the Radiology Department of the Alfred Krupp-Hospital, Essen with a C-150 scanner (Erbel et al. 2014; Lehmann et al. 2014). Prospective ECG triggering was performed at 80% of the RR interval, and contiguous 3-mm-thick slices from the pulmonary bifurcation to the apex of the heart were obtained in both scans at an image acquisition time of 100 ms (Lehmann et al. 2016). The methods of Agatston et al. were used to determine the CAC score (Agatston et al. 1990). The total CAC score was computed, comprising all calcified lesions in the coronary artery system. Analyses were performed using a Virtuoso workstation (Siemens Medical Solutions, Forchheim, Germany). CT scan results were not disclosed to the participants or to the study center. As previously reported, a reassessment of CAC scoring was implemented when extreme progression or regression from baseline to the 5-year examination was found (CACb ≤ 10 to CAC5y > 50, CACb > 20 to CAC5y ≤ 10, or otherwise, > 30% or < − 7% annual change), accounting for the reproducibility by the given correction factors (R. C. Detrano et al. 2005; Lehmann et al. 2016). A reader with several years of experience in the evaluation of cardiac CT, who was blinded to the results of the initial reading as well as the risk factor profile of the participants, performed a second reading of the CAC score in two hundred forty-four cases. At the end, the images of both CT examinations were re-evaluated offline using the same workstation (Aquarius, TerraRecon, Foster City, CA, USA) (Lehmann et al. 2016). We used the loge transformation of the CAC score plus 1, as previously suggested by Detrano et al., to address the right-skewed distribution of the CAC (R. Detrano et al. 2008).
As described previously, CAC5y was predicted exponentially from the age-specific and sex-specific CAC percentiles at baseline (Erbel et al. 2014). The observed CAC5y was then compared with the predicted CAC5y. The values within the predefined acceptance range (20% of the observed CAC5y around the individually predicted CAC5y) were classified as “expected progression,” values above that range were classified as “rapid progression,” and values below that range as “slow progression.” For our analysis, we used a binary outcome, classifying CAC progression as either rapid or expected/slow.
Assessment of cardiovascular risk factors
The CVD risk factors were evaluated at baseline. Body mass index (BMI) was measured as weight divided by height squared (kg/m2). Smoking status (current, past, and non-smokers) was evaluated as described previously (Lehmann et al. 2014). All of the participants were queried about their regular use of cardiovascular medication. The current use of medication was then recorded by means of a computer-based system with a barcode scanner. The information regarding the use of antihypertensive and HMGRI medication (ATC code: C10AA) was recorded.
The resting blood pressure was measured using an automated oscillometric blood pressure device (Omron, HEM-705CP-E) with the participants seated. The mean of the second and third values of the three measurements was calculated (Stang et al. 2006). Standardized enzymatic methods were used to determine serum triglycerides, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol values (ADVIA 1650, Siemens Medical Solutions, Erlangen, Germany) (Lehmann et al. 2018). DM was defined as meeting any of the following four criteria: (1) the participants reported a history of clinically diagnosed diabetes, (2) the participants took glucose-lowering drugs (ATC code: A10), (3) the participants had fasting glucose levels of greater than 125 mg/dL, or (4) the participants had nonfasting glucose levels of 200 mg/dL or greater. Socioeconomic status was defined by combining school and vocational training as total years of formal education according to the International Standard Classification of Education (UNESCO 1997) and categorized into two groups (≤ 13 vs. > 13 years).
Statistical methods
The continuous data are presented as the mean ± SD or median (first quartile, Q1; third quartile, Q3) if the distributions of the data were substantially skewed. The count data are presented as frequencies and percentages. We first assessed the influence of DM GRS as well as each of the DM SNPs on the rapid progression of CAC adjusting for age, sex, and log(CACb + 1) (model 1: rapid progression of CAC = DM GRS/SNP + age + sex + log(CACb + 1)). Next, the influence of intake of HMGRIs on rapid progression of CAC adjusting for age, sex, and log(CACb + 1) was assessed (model 2: rapid progression of CAC = HMGRI intake + age + sex + log(CACb + 1)). Furthermore, an interaction between HMGRI intake and DM GRS/SNP on the progression of CAC (model 3: rapid progression of CAC = DM GRS/SNP + age + sex + log(CACb + 1) + HMGRI intake + HMGRI intake × DM GRS) was assessed. We further stratified the analyses by the use of HMGRI. Log-linear regression was used, adjusted for age, sex, and log(CACb + 1), to estimate the relative risks (RR) and 95% confidence interval (95% CI) (Spiegelman & Hertzmark 2005). For all the analyses, we have reported the main effect of the DM GRS/SNPs (model 1) as well as DM GRS × HMGRI intake interaction terms (model 3) on rapid progression of CAC.
Multiple testing at 5% was done for the two main questions regarding the interaction between HMGRI intake and DM GRS (HMGRI intake × DM GRS) as well as HMGRI intake × DM SNPs with progression of CAC adjusting of age, sex, and log(CACb + 1). We corrected consequently for 101 tests that translate into αBF = 0.0005 using the Bonferroni procedure.
Analyses were performed using Plink v.19 (https://www.cog-genomics.org/plink2) (Purcell et al. 2007) and SAS v.9.4 (SAS Institute, Cary, NC, USA).
Results
Characteristics of the study population
Table 1 shows the clinical and demographic characteristics of the Heinz Nixdorf Recall Study population. During the median follow-up time of 5.1 years, the prevalence of rapid CAC progression in the HNR Study was 19.6%. Participants with HMGRI intake (N (%): 229 (7.3)), compared with non-HMGRI users (N (%): 2928 (92.7%)), were older (mean ± SD: 61.9 ± 6.9 vs. 58.7 ± 7.5 years), had more often diabetes (17.9% vs. 10.9%), and reported greater use of antihypertensive medication (55.5% vs. 29.2%). Similarly, participants with HMGRI intake had lower levels of LDL cholesterol, and higher levels of CAC at baseline and at first follow-up (Table 1). Additionally, Table 2 shows the names and defined daily doses of the HMGRI drugs used. Almost 45% of the participants used atorvastatin followed by simvastatin (26.2%), fluvastatin (12.2%), pravastatin (6.6%), lovastatin (5.7%), and cerivastatin (4.4%). The weighted DM GRSs in the HMGRI users (0.0497 ± 0.0047) and non-HMGRI users (0.0499 ± 0.005) were similar (Table 1).
Table 3 describes all of the SNPs that are included in the GRS, indicating the chromosome number, the base pair position, nearby gene, risk allele/other allele, and risk allele frequency (RAF ≥ 3%), as well as the HWE. All the SNPs were in HWE (p > 0.01) except rs495828 (p = 0.003).
Looking at the main effect, in the age, sex, and log(CACb + 1)-adjusted model, no association of the weighted DM GRS with the rapid progression of CAC was observed (model 1: RR per SD increase in the DM GRS [95% CI]: 1.01 [0.94; 1.10]) (Table 3). Table 3 further shows the association of individual SNPs with the rapid progression of CAC. Although a genetic association at a nominal significance level with rapid CAC progression was observed for five SNPs (rs10203174 (THADA), rs7674212 (CISD2), rs1552224 (ARAP1 (CENTD2)), rs9940149 (ITFG3), and rs10401969 (CILP2)) (Table 3), after correction for multiple testing, none of the SNPs was significantly associated.
As almost twofold odds of HMGRI intake for CAC progression are already published for the HNR study population, we re-analyzed the current dataset (Dykun et al. 2016). The main effect of HMGRI intake on rapid progression of CAC in the model adjusting for age, sex, and log(CACb + 1) showed similar significant effect (model 2: 1.64 [1.27; 2.11], p = 0.0001) (data not shown).
Furthermore, looking at the interaction of DM GRS × HMGRI intake on rapid progression of CAC, no indication of interaction was found after multiple testing (model 3: 1.08 [0.83; 1.41]) (Table 3). Table 3 further shows the interaction of individual SNP × HMGRI intake on rapid progression of CAC. Indication of SNP × HMGRI intake interaction was not observed for any of the five SNPs that appeared to be associated with CAC progression at nominal significance level (rs10203174, rs7674212, rs1552224, rs9940149, and rs10401969) (Table 3). Apart from that, SNP × HMGRI intake interactions at the nominal significance level were observed for four other SNPs (rs9470794 (ZFAND3), rs10886471 (GRK5), rs10842994 (KLHDC5), and rs8068804 (ZZEF1)) (Table 3). However, none of the SNP × HMGRI intake interactions remained significant after correcting for multiple testing (p < 0.0005).
Looking in the HMGRI intake stratified groups, although the group taking HMGRIs showed higher effect size for the DM GRS on the rapid progression of CAC (1.09 [0.85; 1.40]), this effect was not statistically significant. The group without HMGRI intake showed no significant influence of DM GRS on the rapid progression of CAC either (1.01 [0.93; 1.10]) (Table 4).
Discussion and conclusion
Analyzing data from the population-based Heinz Nixdorf Recall study, we investigated for the first time the influence that results from the interplay of both a diabetes mellitus genetic risk score (DM GRS; constructed using GWAS-identified diabetes-associated genetic variants) and individual SNPs with HMGRI intake on the progression of CAC (Table 5). We did not find any evidence of an association of CAC progression with DM GRS alone nor with the interaction of DM GRS and HMGRI intake. The interaction of individual SNPs and HMGRI intake was not correlated with CAC progression either.
HMGRIs are typically used to reduce LDL cholesterol levels in order to prevent CHD. They are generally considered safe and have even been shown to enhance life expectancy (Jacobs et al. 2013), but due to their pleiotropic effects, their use can have unexpected consequences, such as increased progression of CAC or increased risk of developing DM. There is evidence from several studies that the use of HMGRIs (especially in diabetic patients) leads to a greater degree of progression of CAC (Anand et al., 2007; Houslay et al. 2006; Saremi et al. 2012; Terry et al. 2007). In accordance with those results, Dykun et al. already showed that the prevalence of rapid progression of CAC and cardiovascular risk factors in the HNR study was higher among HMGRI users than among non-HMGRI users (Dykun et al. 2016).
As our analyses used the data from the same cohort, we could also see that HMGRIs promote CAC progression irrespective of the DM GRS. However according to our research question, our study did not reveal any impact of the individual diabetes-associated genetic variants that had previously been identified via diabetes-associated GWAS on the progression of CAC. Neither was CAC progression affected by a DM GRS that included the entirety of these 100 SNPs. Furthermore, the interaction of DM GRS × HMGRI intake was also not significantly associated with CAC progression. These results suggest that the association of DM and CAC progression is mediated by factors other than a genetic disposition to diabetes. Further investigations will be needed to determine whether this association is due to poor glycemic control, as suggested by Snell-Bergeon et al., or other causes like DM-associated lifestyle factors.
Importantly, these negative results persisted upon additionally taking into account the participants’ HMGRI intake status, which shows that DM GRS does not statistically significantly affect CAC progression. This can be judged as a point in favor of the common practice of treating patients with increased CHD risk with HMGRIs—even those with an increased genetic disposition for developing DM and despite a growing body of evidence that HMGRIs can also promote the progression of CAC. Presently, we witness both widespread use of HMGRIs and a globally increasing prevalence of DM and CHD. Considering this, an acceleration of CAC progression due to the interaction of HMGRIs and DM-associated genetic variants could have represented a substantial hidden risk factor for developing CHD—and presumably a considerable burden for public health. In that case, reconsideration of HMGRI medication for DM patients would likely have become necessary. However, the results of our study do not show a statistical significant influence of such an interaction on CAC progression, therefore delivering an important confirmation of current guidelines regarding HMGRI prescription.
The strength of the present study is the long follow-up time of 5 years for the assessment of CAC progression. However, we must emphasize that due to the small cohort size, especially regarding the subgroup of HMGRI users, the statistical power of the present study is limited. The lack of an association of the DM GRS could be attributed to the sample size being relatively small relative to small effect size observed for any of the individual diabetes mellitus–related SNPs which were integrated together into a genetic risk score. The small sample size has further limited the statistical power for the analyses of the individual SNPs. It is also possible that several lifestyle factors such as physical activity, consumption of alcohol, or dietary factors can modify the effect of the diabetes mellitus–related genetic variants on the rapid progression of CAC, which could be investigated in larger studies. Nevertheless, our study provides first insights into the hitherto disregarded pharmacogenetic aspects of HMGRI medication in the context of diabetes and CAC progression.
In conclusion, our study showed no accelerated progression of CAC that could be attributed to the combination of HMGRI intake and genetic DM risk factors; and thus, it corroborates the current recommendations of the EACPR and ACC/AHA regarding the use of HMGRIs. While limited in scale, it supports the prescription of HMGRIs as the preferred preemptive CHD medication in the face of new insights into the various pleiotropic effects of HMGRIs, even in individuals exhibiting an increased genetic disposition for the development of diabetes mellitus.
Data availability
Due to data security reasons, i.e., the data contain potentially participant identifying information, the Heinz Nixdorf Recall Study does not allow sharing data as a public use file. However, for the purpose of replication, other authors/researchers are allowed to access data upon request, which is the same way the authors of the present paper obtained the data. Data requests can be addressed to recall@uk-essen.de.
References
Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr, Detrano R (1990) Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol 15(4):827–832. https://doi.org/10.1016/0735-1097(90)90282-t
Anand, D. V., Lim, E., Darko, D., Bassett, P., Hopkins, D., Lipkin, D., . . . Lahiri, A. (2007). Determinants of progression of coronary artery calcification in type 2 diabetes role of glycemic control and inflammatory/vascular calcification markers. J Am Coll Cardiol, 50(23), 2218-2225. https://doi.org/10.1016/j.jacc.2007.08.032
Budoff, M. J., Hokanson, J. E., Nasir, K., Shaw, L. J., Kinney, G. L., Chow, D., . . . Raggi, P. (2010). Progression of coronary artery calcium predicts all-cause mortality. JACC Cardiovasc Imaging, 3(12), 1229-1236. https://doi.org/10.1016/j.jcmg.2010.08.018
Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM (2013) Risk of incident diabetes among patients treated with statins: population based study. BMJ 346:f2610. https://doi.org/10.1136/bmj.f2610
Cederberg H, Stančáková A, Yaluri N, Modi S, Kuusisto J, Laakso M (2015) Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia 58(5):1109–1117. https://doi.org/10.1007/s00125-015-3528-5
Detrano, R., Guerci, A. D., Carr, J. J., Bild, D. E., Burke, G., Folsom, A. R., . . . Kronmal, R. A. (2008). Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med, 358 (13), 1336-1345. https://doi.org/10.1056/NEJMoa072100
Detrano RC, Anderson M, Nelson J, Wong ND, Carr JJ, McNitt-Gray M, Bild DE (2005) Coronary calcium measurements: effect of CT scanner type and calcium measure on rescan reproducibility–MESA study. Radiology 236(2):477–484. https://doi.org/10.1148/radiol.2362040513
Dykun, I., Lehmann, N., Kälsch, H., Möhlenkamp, S., Moebus, S., Budde, T., . . . Mahabadi, A. A. (2016). Statin medication enhances progression of coronary artery calcification: the Heinz Nixdorf Recall Study. J Am Coll Cardiol, 68 (19), 2123-2125. https://doi.org/10.1016/j.jacc.2016.08.040
Elias-Smale, S. E., Proença, R. V., Koller, M. T., Kavousi, M., van Rooij, F. J., Hunink, M. G., . . . Witteman, J. C. (2010). Coronary calcium score improves classification of coronary heart disease risk in the elderly: the Rotterdam study. J Am Coll Cardiol, 56 (17), 1407-1414. https://doi.org/10.1016/j.jacc.2010.06.029
Erbel R, Budoff M (2012) Improvement of cardiovascular risk prediction using coronary imaging: subclinical atherosclerosis: the memory of lifetime risk factor exposure. Eur Heart J 33(10):1201–1213. https://doi.org/10.1093/eurheartj/ehs076
Erbel, R., Lehmann, N., Churzidse, S., Rauwolf, M., Mahabadi, A. A., Möhlenkamp, S., . . . Jöckel, K. H. (2014). Progression of coronary artery calcification seems to be inevitable, but predictable - results of the Heinz Nixdorf Recall (HNR) study. Eur Heart J, 35 (42), 2960-2971. https://doi.org/10.1093/eurheartj/ehu288
Frank, M., Dragano, N., Arendt, M., Forstner, A. J., Nöthen, M. M., Moebus, S., . . . Schmidt, B. (2019). A genetic sum score of risk alleles associated with body mass index interacts with socioeconomic position in the Heinz Nixdorf Recall Study. PLoS One, 14 (8), e0221252. https://doi.org/10.1371/journal.pone.0221252
Gaulton, K. J., Ferreira, T., Lee, Y., Raimondo, A., Mägi, R., Reschen, M. E., . . . Morris, A. P. (2015). Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet, 47 (12), 1415-1425. https://doi.org/10.1038/ng.3437
Geisel, M. H., Coassin, S., Heßler, N., Bauer, M., Eisele, L., Erbel, R., . . . Kronenberg, F. (2016). Update of the effect estimates for common variants associated with carotid intima media thickness within four independent samples: The Bonn IMT Family Study, the Heinz Nixdorf Recall Study, the SAPHIR Study and the Bruneck Study. Atherosclerosis, 249, 83-87. https://doi.org/10.1016/j.atherosclerosis.2016.03.042
Heilmann-Heimbach, S., Herold, C., Hochfeld, L. M., Hillmer, A. M., Nyholt, D. R., Hecker, J., . . . Nöthen, M. M. (2017). Meta-analysis identifies novel risk loci and yields systematic insights into the biology of male-pattern baldness. Nat Commun, 8, 14694. https://doi.org/10.1038/ncomms14694
Henein M, Granåsen G, Wiklund U, Schmermund A, Guerci A, Erbel R, Raggi P (2015) High dose and long-term statin therapy accelerate coronary artery calcification. Int J Cardiol 184:581–586. https://doi.org/10.1016/j.ijcard.2015.02.072
Houslay, E. S., Cowell, S. J., Prescott, R. J., Reid, J., Burton, J., Northridge, D. B., . . . Newby, D. E. (2006). Progressive coronary calcification despite intensive lipid-lowering treatment: a randomised controlled trial. Heart, 92 (9), 1207-1212. https://doi.org/10.1136/hrt.2005.080929
Jacobs JM, Cohen A, Ein-Mor E, Stessman J (2013) Cholesterol, statins, and longevity from age 70 to 90 years. J Am Med Dir Assoc 14(12):883–888. https://doi.org/10.1016/j.jamda.2013.08.012
JBS 2: Joint British Societies' guidelines on prevention of cardiovascular disease in clinical practice (2005). Heart, 91 Suppl 5(Suppl 5), v1–52. https://doi.org/10.1136/hrt.2005.079988
Knuuti, J., Wijns, W., Saraste, A., Capodanno, D., Barbato, E., Funck-Brentano, C., . . . Bax, J. J. (2020). 2019 ESC guidelines for the diagnosis and management of chronic coronary syndromes. Eur Heart J, 41(3), 407-477. https://doi.org/10.1093/eurheartj/ehz425
Koulaouzidis G, Charisopoulou D, Maffrett S, Tighe M, Jenkins PJ, McArthur T (2013) Coronary artery calcification progression in asymptomatic individuals with initial score of zero. Angiology 64(7):494–497. https://doi.org/10.1177/0003319712459213
Kowluru A, Kowluru RA (2015) Protein prenylation in islet beta-cell function in health and diabetes: Putting the pieces of the puzzle together. Biochem Pharmacol 98(3):363–370. https://doi.org/10.1016/j.bcp.2015.07.004
Kupcsik L, Meurya T, Flury M, Stoddart M, Alini M (2009) Statin-induced calcification in human mesenchymal stem cells is cell death related. J Cell Mol Med 13(11–12):4465–4473. https://doi.org/10.1111/j.1582-4934.2008.00545.x
Lehmann, N., Erbel, R., Mahabadi, A. A., Kälsch, H., Möhlenkamp, S., Moebus, S., . . . Jöckel, K. H. (2016). Accelerated progression of coronary artery calcification in hypertension but also prehypertension. J Hypertens, 34(11), 2233-2242. https://doi.org/10.1097/hjh.0000000000001080
Lehmann, N., Erbel, R., Mahabadi, A. A., Rauwolf, M., Möhlenkamp, S., Moebus, S., . . . Jöckel, K. H. (2018). Value of progression of coronary artery calcification for risk prediction of coronary and cardiovascular events: result of the HNR Study (Heinz Nixdorf Recall). Circulation, 137(7), 665-679. https://doi.org/10.1161/circulationaha.116.027034
Lehmann, N., Möhlenkamp, S., Mahabadi, A. A., Schmermund, A., Roggenbuck, U., Seibel, R., . . . Jöckel, K. H. (2014). Effect of smoking and other traditional risk factors on the onset of coronary artery calcification: results of the Heinz Nixdorf Recall study. Atherosclerosis, 232(2), 339-345. https://doi.org/10.1016/j.atherosclerosis.2013.11.045
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E., Pers, T. H., Day, F. R., . . . Speliotes, E. K. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature, 518(7538), 197-206. https://doi.org/10.1038/nature14177
Mahajan, A., Go, M. J., Zhang, W., Below, J. E., Gaulton, K. J., Ferreira, T., . . . Morris, A. P. (2014). Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet, 46(3), 234-244. https://doi.org/10.1038/ng.2897
Malhotra, R., Mauer, A. C., Lino Cardenas, C. L., Guo, X., Yao, J., Zhang, X., . . . O'Donnell, C. J. (2019). HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet, 51(11), 1580-1587. https://doi.org/10.1038/s41588-019-0514-8
McClelland, R. L., Jorgensen, N. W., Budoff, M., Blaha, M. J., Post, W. S., Kronmal, R. A., . . . Burke, G. L. (2015). 10-Year coronary heart disease risk prediction using coronary artery calcium and traditional risk factors: derivation in the MESA (Multi-Ethnic Study of Atherosclerosis) with validation in the HNR (Heinz Nixdorf Recall) study and the DHS (Dallas Heart Study). J Am Coll Cardiol, 66(15), 1643-1653. https://doi.org/10.1016/j.jacc.2015.08.035
Möhlenkamp, S., Lehmann, N., Moebus, S., Schmermund, A., Dragano, N., Stang, A., . . . Erbel, R. (2011). Quantification of coronary atherosclerosis and inflammation to predict coronary events and all-cause mortality. J Am Coll Cardiol,57(13), 1455-1464. https://doi.org/10.1016/j.jacc.2010.10.043
Okuyama H, Langsjoen PH, Hamazaki T, Ogushi Y, Hama R, Kobayashi T, Uchino H (2015) Statins stimulate atherosclerosis and heart failure: pharmacological mechanisms. Expert Rev Clin Pharmacol 8(2):189–199. https://doi.org/10.1586/17512433.2015.1011125
Orban E, Sutcliffe R, Dragano N, Jöckel KH, Moebus S (2017) Residential surrounding greenness, self-rated health and interrelations with aspects of neighborhood environment and social relations. J Urban Health 94(2):158–169. https://doi.org/10.1007/s11524-016-0112-3
Pechlivanis, S., Moebus, S., Lehmann, N., Erbel, R., Mahabadi, A. A., Hoffmann, P., . . . Heinz Nixdorf Recall Study Investigative, G. (2020). Genetic risk scores for coronary artery disease and its traditional risk factors: their role in the progression of coronary artery calcification-results of the Heinz Nixdorf Recall study. PLoS One, 15(5), e0232735. https://doi.org/10.1371/journal.pone.0232735
Pechlivanis, S., Muhleisen, T. W., Mohlenkamp, S., Schadendorf, D., Erbel, R., Jockel, K. H., . . . Heinz Nixdorf Recall Study Investigative, G. (2013). Risk loci for coronary artery calcification replicated at 9p21 and 6q24 in the Heinz Nixdorf Recall Study. BMC Med Genet, 14, 23. https://doi.org/10.1186/1471-2350-14-23
Piepoli, M. F., Hoes, A. W., Agewall, S., Albus, C., Brotons, C., Catapano, A. L., . . . Verschuren, W. M. M. (2016). 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis, 252, 207-274. https://doi.org/10.1016/j.atherosclerosis.2016.05.037
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., . . . Sham, P. C. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 81(3), 559-575. https://doi.org/10.1086/519795
Puri, R., Nicholls, S. J., Shao, M., Kataoka, Y., Uno, K., Kapadia, S. R., . . . Nissen, S. E. (2015). Impact of statins on serial coronary calcification during atheroma progression and regression. J Am Coll Cardiol, 65(13), 1273-1282. https://doi.org/10.1016/j.jacc.2015.01.036
Rhee, E. J., Park, S. E., Oh, H. G., Park, C. Y., Oh, K. W., Park, S. W., . . . Lee, W. Y. (2015). Statin eligibility and cardiovascular risk burden assessed by coronary artery calcium score: comparing the two guidelines in a large Korean cohort. Atherosclerosis, 240(1), 242-249. https://doi.org/10.1016/j.atherosclerosis.2015.03.006
Robson J (2008) Lipid modification: cardiovascular risk assessment and the modification of blood lipids for the primary and secondary prevention of cardiovascular disease. Heart 94(10):1331–1332. https://doi.org/10.1136/hrt.2008.150979
Saremi A, Bahn G, Reaven PD (2012) Progression of vascular calcification is increased with statin use in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 35(11):2390–2392. https://doi.org/10.2337/dc12-0464
Schmermund, A., Möhlenkamp, S., Stang, A., Grönemeyer, D., Seibel, R., Hirche, H., . . . Erbel, R. (2002). Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: rationale and design of the Heinz Nixdorf RECALL Study. Risk Factors, Evaluation of Coronary Calcium and Lifestyle. Am Heart J, 144(2), 212-218. https://doi.org/10.1067/mhj.2002.123579
Scott, R. A., Scott, L. J., Mägi, R., Marullo, L., Gaulton, K. J., Kaakinen, M., . . . Prokopenko, I. (2017). An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes, 66(11), 2888-2902. https://doi.org/10.2337/db16-1253
Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, Mohlke KL (2015) New genetic loci link adipose and insulin biology to body fat distribution. Nature 518(7538):187–196. https://doi.org/10.1038/nature14132
Snell-Bergeon, J. K., Hokanson, J. E., Jensen, L., MacKenzie, T., Kinney, G., Dabelea, D., . . . Rewers, M. (2003). Progression of coronary artery calcification in type 1 diabetes: the importance of glycemic control. Diabetes Care, 26(10), 2923-2928. https://doi.org/10.2337/diacare.26.10.2923
Spiegelman D, Hertzmark E (2005) Easy SAS calculations for risk or prevalence ratios and differences. Am J Epidemiol 162(3):199–200. https://doi.org/10.1093/aje/kwi188
Stang, A., Dragano, N., Poole, C., Moebus, S., Möhlenkamp, S., Schmermund, A., . . . Jöckel, K. H. (2007). Daily siesta, cardiovascular risk factors, and measures of subclinical atherosclerosis: results of the Heinz Nixdorf Recall Study. Sleep, 30(9), 1111-1119. https://doi.org/10.1093/sleep/30.9.1111
Stang, A., Moebus, S., Dragano, N., Beck, E. M., Möhlenkamp, S., Schmermund, A., . . . Jöckel, K. H. (2005). Baseline recruitment and analyses of nonresponse of the Heinz Nixdorf Recall Study: identifiability of phone numbers as the major determinant of response. Eur J Epidemiol, 20(6), 489-496. https://doi.org/10.1007/s10654-005-5529-z
Stang, A., Moebus, S., Möhlenkamp, S., Dragano, N., Schmermund, A., Beck, E. M., . . . Jöckel, K. H. (2006). Algorithms for converting random-zero to automated oscillometric blood pressure values, and vice versa. Am J Epidemiol, 164(1), 85-94. https://doi.org/10.1093/aje/kwj160
Taylor, A. J., Bindeman, J., Le, T. P., Bauer, K., Byrd, C., Feuerstein, I. M., . . . O'Malley, P. G. (2008). Progression of calcified coronary atherosclerosis: relationship to coronary risk factors and carotid intima-media thickness. Atherosclerosis, 197(1), 339-345. https://doi.org/10.1016/j.atherosclerosis.2007.05.027
Terry, J. G., Carr, J. J., Kouba, E. O., Davis, D. H., Menon, L., Bender, K., . . . Crouse, J. R., 3rd. (2007). Effect of simvastatin (80 mg) on coronary and abdominal aortic arterial calcium (from the Coronary Artery Calcification Treatment with Zocor [CATZ] study). Am J Cardiol, 99(12), 1714-1717. https://doi.org/10.1016/j.amjcard.2007.01.060
Thanassoulis, G., Campbell, C. Y., Owens, D. S., Smith, J. G., Smith, A. V., Peloso, G. M., . . . Post, W. S. (2013). Genetic associations with valvular calcification and aortic stenosis. N Engl J Med, 368(6), 503-512. https://doi.org/10.1056/NEJMoa1109034
Trion A, Schutte-Bart C, Bax WH, Jukema JW, van der Laarse A (2008) Modulation of calcification of vascular smooth muscle cells in culture by calcium antagonists, statins, and their combination. Mol Cell Biochem 308(1–2):25–33. https://doi.org/10.1007/s11010-007-9608-1
Tzivian, L., Dlugaj, M., Winkler, A., Hennig, F., Fuks, K., Sugiri, D., . . . Weimar, C. (2016). Long-term air pollution and traffic noise exposures and cognitive function: a cross-sectional analysis of the Heinz Nixdorf Recall study. J Toxicol Environ Health A, 79(22-23), 1057-1069. https://doi.org/10.1080/15287394.2016.1219570
Waters, D. D., Ho, J. E., DeMicco, D. A., Breazna, A., Arsenault, B. J., Wun, C. C., . . . Barter, P. (2011). Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol, 57(14), 1535-1545. https://doi.org/10.1016/j.jacc.2010.10.047
Zhao, W., Rasheed, A., Tikkanen, E., Lee, J. J., Butterworth, A. S., Howson, J. M. M., . . . Saleheen, D. (2017). Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet, 49(10), 1450-1457. https://doi.org/10.1038/ng.3943
Acknowledgements
This study was supported by the German Heart Foundation/German Foundation of Heart Research. We are indebted to all of the study participants and to the dedicated personnel of both the study center of the Heinz Nixdorf Recall study and the EBT scanner facilities, including D. Grönemeyer, Bochum, and R. Seibel, Mülheim, as well as to the investigative group, in particular to U. Roggenbuck, U. Slomiany, E. M. Beck, M. Bauer, A. Öffner, S. Münkel, S. Schrader, R. Peter, H. Kälsch, and H. Hirche.
Scientific advisory board: T. Meinertz, Hamburg (Chair); M. Blettner, Mainz; C. Bode, Freiburg; P. J. de Feyter, Rotterdam, Zürich, Niederlande; B. Güntert, Hall i.T., Schweiz; F. Gutzwiller, Schweiz; H. Heinen, Bonn; O. Hess‡, Bern, Schweiz; B. Klein‡, Essen; H. Löwel, Neuherberg; M. Reiser, München; G. Schmidt‡, Essen; M. Schwaiger, München; C. Steinmüller, Bonn; T. Theorell, Stockholm, Schweden; S. N. Willich, Berlin.
Criteria and end point committee: C. Bode, Freiburg (Chair), K. Berger, Münster; H. R. Figulla, Jena; C. Hamm, Bad Nauheim; P. Hanrath, Aachen; W. Köpcke, Münster; C. Weimar, Essen; M. Dichgans. München, A. Zeiher, Frankfurt. Technical support for the imputation of the Heinz Nixdorf Recall study data on the Supercomputer Cray XT6m was provided by the Center for Information and Media Services, University of Duisburg-Essen.
Artwork was created using Inkscape (Version 0.92.3—https://www.inkscape.org).
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the German Heart Foundation/German Foundation of Heart Research.
Author information
Authors and Affiliations
Contributions
Conceptualization: SP, HSB. Project administration: SP. Investigation: RE, SM. Methodology: RE, SM, AAM, NL. Genotyping: PH, MMN. Imputation: PH, SP. Data analysis: SP. Visualization: SP, HSB, DJ. Writing original draft preparation: SP and DJ. Writing – review and editing: all authors. Supervision: HSB. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethics approval
The study has been approved by the ethical committee at the University Hospital Essen, Germany and is conducted in accordance with the principles expressed in the Declaration of Helsinki. The study was certified and recertified according to DIN EN ISO 9001:2000/2008. All study participants gave their written informed consent. In order to access the data for the present study, the authors have obtained permission from the internal committee of the Heinz Nixdorf Recall study.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pechlivanis, S., Jung, D., Moebus, S. et al. Pharmacogenetic association of diabetes-associated genetic risk score with rapid progression of coronary artery calcification following treatment with HMG-CoA-reductase inhibitors —results of the Heinz Nixdorf Recall Study. Naunyn-Schmiedeberg's Arch Pharmacol 394, 1713–1725 (2021). https://doi.org/10.1007/s00210-021-02100-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-021-02100-7