
Abstract
Therapy of depression is difficult and still insufficient despite the presence of many antidepressants on the market. Therefore, there is a constant need to search for new, safer, and more effective drugs that could be used in the treatment of depression. Among many methods, chemical modification is an important strategy for new drug development. This study evaluates antidepressant-like effects and possible mechanism of action of two new arylpiperazine derivatives with isonicotinic and picolinic nuclei, compounds 4pN-(3-(4-(piperonyl)piperazin-1-yl)propyl) isonicotinamide and 3oN-(2-(4-(pyrimidin-2-yl)piperazin-1-yl)ethyl) picolinamide. The forced swim test (FST) and tail suspension test (TST), as two predictive tests for antidepressant effect in mice, were used. The possible involvement of serotonergic system in the effects of the new compounds in the FST through pharmacological antagonists/modulators of serotonergic transmission was also investigated. Compounds 4p and 3o were shown to possess antidepressant activity in both tests, FST and TST. The antidepressant-like effects of the new compounds in the FST were prevented by pretreatment of mice with pCPA (serotonin depletor), (−)pindolol (mixed 5-HT1A/1B and β-adrenergic antagonist), and WAY 100635 (selective 5-HT1A antagonist). Additionally, in drug interaction studies, the 5-HT2A/2C antagonist, ketanserin, and the classic antidepressant, imipramine, potentiated antidepressant-like effect of both new compounds. The obtained results demonstrate that the new compounds 4p and 3o produce an antidepressant-like effect in mice which seems to be mediated by interaction with the serotonin 5-HT1A receptors and in the case of 4p, also with the 5-HT2C receptors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Depression hinders emotionally and physically the everyday lives of the afflicted persons and thus is a vast public health problem leading to heavy socioeconomic burden. According to the World Health Organization (WHO), it will be the second most prevalent cause of illness-induced disability by the year 2020 (Manji et al. 2001; Nestler et al. 2002). The disease is characterized by a number of various symptoms. These include depressed mood, lack of interest, and recurrent thoughts of death and suicide (Aldous and Mann 1963; Nestler et al. 2002). It can also exacerbate or initiate other health problems, such as cardiovascular (Musselman et al. 1998; Fava and Kendler 2000; Neu 2009) and endocrine disorders (Peyrot 2003), as well as cancer (Lazure et al. 2009).
There are many drugs that are potent antidepressants. Treating depression, however, is difficult because of drug resistance, or drugs being poorly tolerated by patients due to many collateral undesirable side effects. The extent of these, in some cases, leads to the arrest of therapy and the worsening of morbidity. Additionally, a long latency (usually several weeks) is required for improvement of depressive symptoms. Such effect complicates the treatment due to the possibility of suicide, especially during the first period of therapy (Nemeroff and Owens 2002; Montgomery 2006; Loubinoux et al. 2012). Therefore, there is a constant need to search for new, safer, and more effective antidepressants (Montgomery 2006; Kędzierska and Wach 2016).
Serotonin (5-hydroxytryptamine, 5-HT) plays a key role in the pathophysiology of depression, and different groups of serotonin receptors are targets for antidepressant drugs (Petit-Demouliere et al. 2005; Adell et al. 2005; Brüning et al. 2011; Yohn et al. 2017). These receptors are divided into seven groups, of which only the 5-HT3 receptor belongs to the group of ionotropic receptors, as the others are all G protein–coupled receptors (GPCR). Serotonin receptors modulate the release of other neurotransmitters, such as glutamate, γ-aminobutyric acid (GABA), dopamine, noradrenaline, or acetylcholine (Hoyer et al. 1994; Chilmończyk et al. 2015). For the treatment of depression, the 5-HT1A and the 5-HT2C receptors are particularly important. The 5-HT1A receptors are present in the frontal cortex, hippocampus, amygdala, and raphe nuclei, and their distribution is similar in rodent and human brains. These receptors function both as presynaptic somatodendritic autoreceptors and as postsynaptic heteroreceptors. Of note, these two types act oppositely (Artigas 2013; Celada et al. 2013; Yohn et al. 2017). The 5-HT2 receptors are also widely distributed throughout the brain, in a pattern that suggests that their activation may be implicated in the regulation of mood disorders (Celada et al. 2004). Many ligands of serotonin 5-HT1A receptors produce beneficial effects in animal models of depression and anxiety (Kostowski et al. 1992; Schreiber and De Vry 1993; Singh and Lucki 1993; Deakin 1993; De Vry 1995; Redrobe et al. 1996), and some are already approved as drugs. For example, vilazodone and vortioxetine are marketed as antidepressants, while buspirone has found to be clinically effective in the treatment of either disorder (Taylor 1988; Faludi 1994; Marangell 2000; Appelberg et al. 2001; Mork et al. 2012; Artigas 2013; McIntyre 2017). Drugs acting at the 5-HT1A receptors offer a better safety profile because these receptors modulate rather than mediate neurotransmission in brain regions involved in development of anxiety and depression (Artigas 2013; Nautiyal and Hen 2017). They do not cause sedation, memory disturbances, or negative interactions with alcohol and do not have addictive potential (Taylor 1988; Faludi 1994). Therefore, it seems that the search for activity among the ligands of these receptors gives a good chance of obtaining effective drugs in the treatment of depression and anxiety.
In accordance with the aforementioned results and in aiming to develop novel pharmacological tools that could improve our knowledge of the signal transduction mechanism and lead creating compounds with high affinity and selectivity, a novel class of arylpiperazine derivatives containing isonicotinic (Fiorino et al. 2016) and picolinic nuclei (Fiorino et al. 2017) was designed and synthesized. All the new compounds were tested in vitro for their affinity, primarily for serotonin 5-HT1A, 5-HT2A, and 5-HT2C receptors. However, in view of the potential multireceptor profile of the derivatives, their ability to bind to α1 and α2 adrenoceptors, as well as to dopaminergic receptors (D1 and D2), was also examined. Subsequently, substances with the highest affinity and selectivity for 5-HT1A receptors were tested in vivo. Hence, we utilized a battery of preliminary behavioral tests to investigate the possible impact of new compounds on the central nervous system (Fiorino et al. 2016, 2017). On this basis, we chose compounds 4pN-(3-(4-(piperonyl)piperazin-1-yl)propyl) isonicotinamide (Fiorino et al. 2016) and 3oN-(2-(4-(pyrimidin-2-yl)piperazin-1-yl)ethyl) picolinamide (Fiorino et al. 2017) (Scheme 1) as the most promising agents for further pharmacological studies in vivo.

Chemical structures of compounds 4p and 3o
In such studies, compound 4p showed high affinity for serotonin 5-HT1A receptors with Ki = 0.0113 nM, but less affinity for 5-HT2C subtypes: Ki = 2.19 nM. In the behavioral studies performed up to now, it demonstrated the characteristics of presynaptic 5-HT1A receptor agonist; however, it was found to not act as an agonist or antagonist of postsynaptic receptors (Fiorino et al. 2016). Instead, compound 3o was characterized by having high affinity and selectivity for 5-HT1A serotonin receptors, with Ki = 0.046 nM. Moreover, it displayed the features of a postsynaptic partial agonist and presynaptic 5-HT1A receptor antagonist (Fiorino et al. 2017). The interesting affinity and selectivity profiles showed by piperonyl (4p) and 2-pyrimidinyl (3o) moieties are in line with those described in literature. In particular, it was already reported that the pyrimidinyl group, originally present in buspirone and later employed in many 2-pyrimidinylpiperazine analogues, produces a greater affinity for 5-HT1A receptors even if the plane of the 2-pyrimidinyl ring is parallel to that of piperazine due to delocalization of the sp2/sp3 nitrogen into the aromatic system, hence producing only two low-energy conformations which are essentially isoenergetic (Fiorino et al. 2017).
Furthermore, both compounds produced an antidepressant-like effect in the forced swim test (FST) and anxiolytic activity in the elevated plus-maze (EPM) test in mice (Fiorino et al. 2016, 2017). To confirm this antidepressant-like action, in the current study we conducted an additional predictive testing for antidepressants, the tail suspension test (TST). Based on literature reports concerning the involvement of 5-HT1A and 5-HT2C receptors in the pathomechanism of depression, we, thus, investigated the possible mechanism of action of the new derivatives for their antidepressant-like activity. The experiments were designed specifically to assess the putative engagement of the serotonergic neurotransmitter system. For this purpose, we used the FST, which is a behavioral paradigm that predicts the efficacy of various class antidepressants (Porsolt et al. 1977a, b; Borsini and Meli 1988; Millan et al. 2001; Willner and Mitchell 2002). In this context, we investigated the possible involvement of this system in the anti-immobility effect of the new compounds in the FST through pharmacological antagonists/modulators of serotonergic transmission. Firstly, we used p-chlorophenylalanine (pCPA, serotonin depletor) to ascertain if the antidepressant-like effects of the compounds occur after the inhibition of serotonin synthesis, and then, we analyzed the participation of 5-HT1A, 5-HT1B, and 5-HT2A/2C receptors in the observed effects of these compounds. Hence, in conducting the FST, we blocked the action of tested compounds using the following antagonists: the mixed 5-HT1A/1B and β-adrenergic antagonist (−)pindolol (Lejeune and Millan 2000) and the selective 5-HT1A antagonist WAY 100635 (Forster et al. 1995). Additionally, drug interaction studies were performed with the 5-HT2A/2C antagonist ketanserin (Kleven et al. 1997) and imipramine, a classic antidepressant (Glowinski and Axelrod 1964; Corrodi and Fuxe 1968). Doses and administration schedules of the tested substances and tool agents used here were selected on the basis of our previous results (Fiorino et al. 2016, 2017), as well as on the basis of literature data, and were reported not to increase locomotor activity (Redrobe and Bourin 1999; Zomkowski et al. 2004; Yalcin et al. 2005; Kaster et al. 2005; Guilloux et al. 2006).
Materials and methods
Experimental animals
The studies were conducted on 6-week-old male Albino Swiss mice (18–24 g). The mice were housed in cages, 38 × 18.5 × 13 cm, five individuals per cage. The bedding was corncob granules and it was changed once a week. The ambient temperature was 22 ± 1 °C, and the relative humidity was 50–60%. The mice were maintained on a 12-h light-dark cycle (lights on at 6 a.m.). Tap water and food pellets (LSM Agropol S.J., Motycz, Poland) were available ad libitum except for the short time when the mice were removed from their cages for testing. All experiments were carried out between 9:00 and 16:00. The study was performed under experimental protocols approved by the Local Ethics Committee (License No. 35/2017). Housing and experimental procedures were conducted in accordance with the European Union Directive of 22 September 2010 (2010/63/EU) and Polish legislation concerning animal experimentation. All efforts were made to minimize animal suffering as well as the number of animals used in the study.
Drug administration
After arrival from the breeding facility, the mice were allowed to acclimatize to the experimental room for at least 1 h before testing. After this time, the compounds were administered. The volume of all administrated solutions/suspension was 10 ml/kg. Animals were weighed immediately before injection. Each study group consisted of eight to ten individuals (eight in groups receiving antagonists/modulators of the serotonergic system and ten in other groups). The control groups received respective vehicles at respective time points before testing. It was one injection of tylose in the TST and two injections (tylose and physiological saline) in all other studies with co-administration of drugs. Between the injections, mice were provided with stable living conditions and unrestricted access to food and water.
The new arylpiperazine derivatives 4p and 3o were synthesized in the Department of Pharmacy of Federico II University in Naples, Italy, and the synthesis was described earlier (Fiorino et al. 2016, 2017). For behavioral studies, they were dissolved in DMSO (at final concentration of 0.1%) and then diluted by aqueous solution of 0.5% methylcellulose (tylose) and injected intraperitoneally (i.p.) 60 min before the tests.
To establish the involvement of the serotonin-mediated mechanism in the anti-immobility effect of tested compounds (4p 30 mg/kg i.p., 3o 15 mg/kg i.p.) in the FST, the animals were pretreated with p-chlorophenylalanine methyl ester hydrochloride (pCPA, an inhibitor of serotonin synthesis) dissolved in saline, at a dose of 100 mg/kg once a day, for four consecutive days (Wang et al. 2008; Girish et al. 2012). After the last pCPA injection, the experimental mice were treated with the test compounds and tested in the FST 60 min later.
In another set of experiments, to investigate the involvement of particular subtypes of serotonin receptors in the antidepressant-like effect of the new compounds in the FST, the mice were pretreated with either a mixed 5-HT1A/1B and β-adrenergic antagonist (−)pindolol at dose 10 mg/kg i.p., or the selective 5-HT1A antagonist WAY 100635, 0.1 mg/kg subcutaneously (s.c.) each of them 15 min before the tested compounds (Savegnago et al. 2007; Wang et al. 2008; Jesse et al. 2010). Furthermore, ketanserin, a 5-HT2A/2C antagonist, at a dose of 1 mg/kg was given i.p. 15 min before tested compounds (Brüning et al. 2011), and imipramine at the threshold dose of 15 mg/kg, diluted in physiological saline, was concomitantly administered with the new derivatives, 60 min before the test (Poleszak et al. 2014). All of the tool substances used were purchased from Sigma-Aldrich company, and the used doses and pretreatment times were selected from the mentioned referential literature and from previous experiments conducted within our laboratory (Fiorino et al. 2016, 2017).
Behavioral tests
Forced swim test (Porsolt’s test)
The study was carried out using the test proposed by Porsolt et al. (1977a, b). The method is based on the observation of an animal forced to swim in a situation without possibility of escape. After the initial period of vigorous movements, the animals give up further attempts, which reflects a human sense of hopelessness (Porsolt et al. 1977a, b; Kędzierska and Wach 2016). The test involved putting the mouse into a beaker (diameter 10 cm, height 25 cm) filled with water at 23–25 °C for a period of 6 min. The time of immobility was then recorded in the last 4 min of the test session (between the second and sixth minutes), in real time, by a blinded observer using the summing stoppers. Immobility was assumed when the animal floated passively, performing only movements necessary to keep its head above water (a semi-horizontal position).
Tail suspension test
The procedure was carried out according to the method of Steru et al. (1985). The mice were suspended 50 cm above the floor by adhesive tape placed approximately 1 cm from the tip of the tail for 6 min. The total duration of immobility was then recorded by a blinded observer in real time, between the second and sixth minute (as in the FST) using the summing stoppers. Mice were judged to be immobile when they hung passively, making only the small movements necessary to breathe (Steru et al. 1985; Poleszak et al. 2016).
Locomotor activity test
The spontaneous motility of mice was measured using a photocell apparatus (Multiserv, Lublin, Poland). The apparatus consisted of round cages (diameter 25 cm) made of plastic (PP, PVC) with the measuring element being made of infrared motion sensors. The results were presented on an LCD display. Motility was interpreted as the number of light beams crossed by the freely moving mouse. The animals were placed in the cage individually, 50 min after the administration of the tested compounds, for a period of 10 min for acclimatization. After this time, their activity was noted after 2 and 6 min (corresponded to the observation period in the FST and TST) and after 10 min to observe the dynamics of changes.
Statistical analysis
The data obtained from the experiments were subjected to statistical evaluation. One-way analysis of variance (ANOVA) was used, with Bonferroni’s test applied as a post hoc test. The results are presented as the means ± SEM (standard error of the mean); p < 0.05 was considered statistically significant. All analyses were performed using the Prism software ver. 5.0 (GraphPad Software, San Diego, CA, USA).
Results
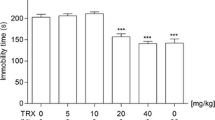
The effect of new compounds 4p and 3o (15 and 30 mg/kg) on the duration of mouse immobility in the tail suspension test
Statistical analysis of the results obtained in the TST revealed that compounds 4p and 3o at doses of 15 and 30 mg/kg exerted a statistically significant antidepressant effect: 4p at p < 0.001 and p < 0.05 and 3o at p < 0.01 and p < 0.05, respectively. This manifested in the reduction of total immobility time, in comparison with the control group (Fig. 1; ANOVA: F(4,30) = 5.293; p < 0.01; Bonferroni’s test).

The effect of new compounds 4p and 3o (15 and 30 mg/kg) on the duration of immobility in the tail suspension test (TST) in mice. The tested compounds 4p and 3o were injected i.p. 60 min before the test. Data are expressed as mean ± SEM values. *p < 0.05; **p < 0.01; ***p < 0.001 vs. control vehicle-treated group (Bonferroni’s test; n = 10)
Effect of pretreatment with pCPA on the antidepressant-like effect of new compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test and on mice locomotor activity
Statistical analysis of the results showed that the inhibitor of serotonin synthesis, pCPA alone (100 mg/kg, once a day, for four consecutive days), did not modify the immobility time, while pretreatment of mice with pCPA significantly prevented the reduction of immobility elicited by both new compounds 4p and 3o, p < 0.01 vs. respective compound (Fig. 2a; ANOVA: F(5,46) = 9461; p < 0.0001).

Effect of pretreatment with pCPA on the antidepressant-like effect of new compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test (FST) (a) and on locomotor activity of mice (b). Mice were pretreated by pCPA, 5-HT synthesis inhibitor, 100 mg/kg i.p. for 4 consecutive days before administration of the tested compounds 4p and 3o, injected i.p. 60 min before the test. Data are expressed as mean ± SEM values of immobility time in the FST and the mobility count of mice after 2, 6, and 10 min in the locomotor activity test. ***p < 0.001 vs. control vehicle-treated group, ##p < 0.01 vs. respective compound (Bonferroni’s test; n = 8 for pCPA group, n = 10 for other groups)
Figure 2b shows that the administration of the substances at the same doses did not change the spontaneous locomotor activity in mice, neither after 2 min (ANOVA: F(5,41) = 0.7980; p = 0.5575) nor after 6 min (ANOVA: F(5,38) = 2.192; p = 0.0753) nor after 10 min (ANOVA: F(5,41) = 0.7235; p = 0.6096; Bonferroni’s test).
The role of serotonin receptors in the antidepressant-like effect of new compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test and on mice locomotor activity
The results in Fig. 3a show that pindolol alone (a mixed 5-HT1A/1B and β-adrenergic antagonist, 10 mg/kg i.p.) did not modify the immobility time, while pretreatment of mice with pindolol significantly prevented the reduction of immobility elicited by both new compounds 4p and 3o (p < 0.01 and p < 0.05) vs. respective compound (ANOVA: F(5,40) = 9.261; p < 0.0001).

Effect of pretreatment with pindolol (10 mg/kg i.p.) on the antidepressant-like effect of new compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test (FST) (a) and on locomotor activity of mice (b). Mice were pretreated by pindolol, the mixed 5-HT1A/5-HT1B and β-adrenergic antagonist, 10 mg/kg i.p., 15 min before administration of the tested compounds 4p and 3o. Data are expressed as mean ± SEM values of immobility time in the FST and the mobility count of mice after 2, 6, and 10 min in the locomotor activity test. ***p < 0.001 vs. control vehicle-treated group, #p < 0.05, ##p < 0.01 vs. respective compound (Bonferroni’s test; n = 8 for pindolol group, n = 10 for other groups)
Figure 3b reveals that the administration of the substances at the same doses did not change the spontaneous locomotor activity in mice, neither after 2 min (ANOVA: F(5,43) = 0.5841; p = 0.7120) nor after 6 min (ANOVA: F(5,40) = 3.380; p < 0.05) nor after 10 min (ANOVA: F(5,42) = 1.491; p = 0.2134; Bonferroni’s test).
The results in Fig. 4a show that WAY 100635 alone (the selective 5-HT1A receptor antagonist, 0.1 mg/kg s.c.) did not modify the immobility time, while pretreatment of mice with WAY 100635 significantly prevented the reduction of immobility elicited by both new compounds 4p and 3o (p < 0.01) vs. respective compound (ANOVA: F(5,44) = 10.30; p < 0.0001).

Effect of pretreatment with WAY 100635 (0.1 mg/kg s.c.) on the antidepressant-like effect of new compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test (FST) (a) and on locomotor activity of mice (b). Mice were pretreated by WAY 100635, the selective 5-HT1A receptor antagonist, 0.1 mg/kg s.c., 15 min before administration of the tested compounds 4p and 3o. Data are expressed as mean ± SEM values of immobility time in the FST and the mobility count of mice after 2, 6, and 10 min in the locomotor activity test. **p < 0.01, ***p < 0.001 vs. control vehicle-treated group, ##p < 0.01 vs. respective compound (Bonferroni’s test; n = 8 for WAY 100635 group, n = 10 for other groups)
Figure 4b indicates that the administration of the substances at the same doses did not change the spontaneous locomotor activity in mice, neither after 2 min (ANOVA: F(5,40) = 0.7134; p = 0.6170) nor after 6 min (ANOVA: F(5,41) = 1.081; p = 0.3850) nor after 10 min (ANOVA: F(5,42) = 0.5608; p = 0.7294; Bonferroni’s test)
Effect of combined administration of ketanserin (1 mg/kg i.p.) and tested compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test and on mice locomotor activity
The results in Fig. 5a show that both ketanserin (1 mg/kg) alone and tested compounds 4p and 3o, administered at ineffective doses of 7.5 and 3.75 mg/kg alone, respectively, had no effect on the immobility time in the FST. However, simultaneous administration of ketanserin and the tested compounds 4p and 3o at the abovementioned doses resulted in a statistically significant reduction of the immobility time as compared with that of the ketanserin group (p < 0.05), but not as compared with that of the control group (ANOVA: F(5,32) = 2.299; p = 0.0681; Bonferroni’s test).

Effect of combined administration of ketanserin (1 mg/kg i.p.) and tested compounds 4p and 3o (7.5 and 3.75 mg/kg) in the forced swim test (FST) (a) and on locomotor activity of mice (b). Mice were pretreated by ketanserin 1 mg/kg i.p. 15 min before administration of the tested compounds 4p and 3o. Data are expressed as mean ± SEM values of immobility time in the FST and the mobility count of mice after 2, 6, and 10 min in the locomotor activity test. *p < 0.01 vs. control vehicle-treated group, ^p < 0.05 vs. ketanserin group (Bonferroni’s test; n = 8 for ketanserin group, n = 10 for other groups)
Figure 5b reveals that the administration of the substances at the same doses did not change the spontaneous locomotor activity in mice, neither after 2 min (ANOVA: F(5,29) = 0.9901; p = 0.4408) nor after 6 min (ANOVA: F(5,27) = 0.9441; p = 0.4688), but concomitant administration of ketanserin and tested compound 3o resulted in a statistically significant reduction of motility count after 10 min (ANOVA: F(5,30) = 2.482; p = 0.0538; Bonferroni’s test). Of note, there is a general visible trend to decrease locomotor activity after administration of ketanserin alone, and also concomitantly with tested compounds.
Effect of combined administration of imipramine (15 mg/kg i.p.) and tested compounds 4p and 3o (15 and 30 mg/kg) in the forced swim test and on mice locomotor activity
The results in Fig. 6a demonstrate that both imipramine (IMI, 15 mg/kg) and tested compounds 4p and 3o, administered alone at ineffective doses of 7.5 and 3.75 mg/kg, respectively, had no effect on the immobility time in the FST. However, simultaneous administration of IMI and tested compounds 4p and 3o at the abovementioned doses resulted in a statistically significant reduction of the immobility time as compared with that of the IMI group (p < 0.05), as well as with that of the control group (p < 0.01 and p < 0.05) and also vs. respective compound (p < 0.05; ANOVA: F(5,34) = 4.613; p < 0.001; Bonferroni’s test).

Effect of combined administration of imipramine (IMI, 15 mg/kg i.p.) and tested compounds 4p and 3o (7.5 and 3.75 mg/kg) in the forced swim test (FST) (a) and on locomotor activity of mice (b). Mice were treated with imipramine 1 mg/kg i.p. concomitantly with the tested compounds 4p and 3o. Data are expressed as mean ± SEM values of immobility time in the FST and the mobility count of mice after 2, 6, and 10 min in the locomotor activity test. *p < 0.05, **p < 0.01 vs. control vehicle-treated group, ^p < 0.05 vs. IMI 15 mg/kg, #p < 0.05 vs. respective compound (Bonferroni’s test; n = 8 for IMI group, n = 10 for other groups)
Figure 6b shows that the administration of the substances at the same doses did not change the spontaneous locomotor activity in mice, neither after 2 min (ANOVA: F(5,34) = 0.3038; p = 0.9072) nor after 6 min (ANOVA: F(5,39) = 0.7998; p = 0.5566) nor after 10 min (ANOVA: F(5,40) = 0.5620; p = 0.7284; Bonferroni’s test).
Discussion
Preclinical and clinical findings suggest the involvement of the serotonergic system in the neurobiology of depression, as well as in the action of many antidepressants (Hensler 2002; Yohn et al. 2017). Herein, alterations in mood and emotion are mainly associated with the 5-HT1A and 5-TH2A/2C receptors, with the 5-HT1A receptors being known to function as presynaptic autoreceptors and postsynaptic heteroreceptors (Yohn et al. 2017); the first-type receptors are located somatodendrically on the serotonin neurons in the raphe nuclei area, while the second-type receptors are found postsynaptically to serotonin axon terminals in the corticolimbic structures (Celada et al. 2013; Yohn et al. 2017). In depressed patients, in postmortem research, a reduced number and sensitivity of postsynaptic 5-HT1A receptors and hyperfunction of the 5-HT1A autoreceptors, as well as increased density of 5-HT2A/2C receptors, have been observed (Barnes and Sharp 1999; Savitz et al. 2009; McAllister-Williams et al. 2014). Newer imaging techniques also confirmed these observations (Sargent et al. 2000). Autoreceptors inhibit the activity of the system, causing suppression of endogenous 5-HT secretion into the synaptic cleft by negative feedback. By this mechanism, 5-HT1A receptors control the general tone of serotonin activity (Chilmończyk et al. 2015). The limited clinical efficacy of selective serotonin reuptake inhibitors (SSRIs) and their delayed action are partly due to this negative feedback mechanism (Celada et al. 2013). Upon chronic treatment with SSRI, 5-HT1A autoreceptors succumb to downregulation, leading to the recovery of serotonergic activity and enhanced 5-HT release (Blier and de Montigny 1994; Artigas et al. 1996; Stahl 1998).
Stimulation of postsynaptic 5-HT1A heteroreceptors in corticolimbic structures induces an increase in the activity of the monoamine system and, as a result, produces an antidepressant effect. In addition, these receptors have a modulatory action on other neurons (e.g., glutamatergic) in various brain regions, which can also contribute to this outcome (Artigas 2015). Research has shown that mice lacking 5-HT1A heteroreceptors do not respond to SSRI-based treatment. In contrast, chronic SSRI treatment tonically stimulates hippocampal 5-HT1A receptors, indicating their substantial role in mediating the behavioral response to antidepressants (Santarelli et al. 2003; Artigas 2015; Chilmończyk et al. 2015; Yohn et al. 2017).
Such distribution and function of pre- and postsynaptic receptors suggest that the blockade of 5-HT1A autoreceptors can increase serotonergic transmission by protecting against the self-inhibitory action of 5-HT without affecting the postsynaptic hippocampal 5-HT1A receptors that contribute to the antidepressant-like effect (Kinney et al. 2000; Ohno 2010). Hence, supporting the actions of heteroreceptors should ensure better antidepressant effects. That is why it is suggested that in searching for new compounds useful in the treatment of mental illness targeting 5-HT1A receptors, focus should be placed on specific modulation of either autoreceptors or heteroreceptors (but not both).
The compounds presented in this study are selective ligands with high affinity to 5-HT1A receptors: 4p with Ki = 0.0113 nM and 3o with Ki = 0.046 nM (Fiorino et al. 2016, 2017). Additionally, compound 4p exhibits high affinity toward serotonin 5-HT2C receptors (with Ki = 2.19 nM). In previous work, both compounds have demonstrated significant antidepressant-like activity reducing immobility time in the FST (Fiorino et al. 2016, 2017). To confirm the observed effects, in this study, we performed experiments using another test (TST) to predict the antidepressant activity. TST is considered to be more sensitive to serotonergic drugs such as SSRI (Lucki et al. 2001). In line with our earlier findings (Fiorino et al. 2016, 2017), the new compounds, at the same doses as in FST, i.e., 15 and 30 mg/kg, exerted significant antidepressant-like activity in the TST (Fig. 1), amplifying the assumption that these compounds might play a role in the modulation of depression. Such results prompted us to study the mechanisms engaged in the observed effects. Based on the in vitro study, we suspected that the antidepressant-like effect of the new compounds may involve the 5-HT receptors, especially 5-HT1A, and in the case of 4p, also 5-HT2C.
For further study, we chose to employ FST. This is a simple, fast, widely used, very reliable tool for studying depression and the mechanisms underlying the action of agents with antidepressant potency (Borsini 1995; Petit-Demouliere et al. 2005; Cryan and Slattery 2007). In this test, rodents are subjected to an unavoidable and inescapable situation. In such situations, after the time of vigorous attempts to escape, they abandon any activity, expressing a behavioral despair which is thought to reflect human depression (Dixon 1998). Via such testing, compounds that intensify active behavior and decrease immobility are evaluated as antidepressants. Indeed, a significant correlation exists between clinical potency and the effectiveness of the drugs in this test (Porsolt et al. 1977a, b; Steru et al. 1985; Dixon 1998; Cryan et al. 2005; Petit-Demouliere et al. 2005).
Motor impairment is known to influence animals’ behavior in the FST and TST. However, in the performed studies, the doses of tested compounds effective in the FST and TST did not modify the motility of mice, as this was measured in the locomotor activity cages during the time equal to the observational period in these tests (i.e., in a 6-min period (Fiorino et al. 2016, 2017)). Therefore, presumably, the antidepressant-like action produced by the new derivatives in these tests was not masked by other general pharmacological activities and is not a result of unspecific actions. The shortening of immobility time induced by antidepressant drugs in FST and TST, therefore, depends on the enhancement of the central 5-HT and catecholaminergic transmission (Porsolt et al. 1977a, b; Borsini and Meli 1988; Borsini 1995). In fact, most of antidepressant drugs in current clinical use are known to promote an increase in 5-HT availability. This directly affects serotonin turnover in the brain by inhibiting serotonin reuptake and also interacting with the 5-HT1A and 5-HT2 receptors (Wong and Licinio 2001; Millan 2004).
In the present study, antidepressant-like action is exerted by both the compounds: 3o, which behaves as a presynaptic 5-HT1A receptor antagonist and a postsynaptic 5-HT1A receptor partial agonist, and compound 4p, which acts as presynaptic 5-HT1A receptor agonist. In the case of 3o, blockade of 5-HT1A autoreceptors and stimulation of 5-HT1A heteroreceptors can result in antidepressant-like activity, while in the case of 4p, the antidepressant effect may result not only from the interaction with 5-HT1A, but also from that with 5-HT2C receptors.
In order to confirm or exclude the contribution of the serotonergic system in the antidepressant-like activity of the tested compounds, we conducted an experiment utilizing pCPA. Data already reported in literature have shown that the administration of pCPA (an inhibitor of serotonin synthesis that blocks tryptophan hydroxylase) for four consecutive days depletes the endogenous stores of 5-HT by about 60% in mice, without having influence on noradrenaline and dopamine levels (Redrobe and Bourin 1998). pCPA was also reported to block the antidepressant-like effect of selective 5-HT reuptake inhibitors (SSRIs such as fluoxetine and citalopram) in the TST and FST, but not of noradrenaline reuptake inhibitors (NRIs such as reboxetine) or tricyclic antidepressants (such as desipramine) (Page et al. 1999; O’Leary et al. 2007). This is consistent with the hypothesis that SSRI compounds elicit their acute behavioral effects by increasing extracellular 5-HT (Bymaster et al. 2002). In our experiments, in accordance with other reports (Kaster et al. 2005; Wang et al. 2008), pCPA alone did not affect the immobility time of mice in the FST, but the pCPA-induced reduction in brain 5-HT prevented the antidepressant-like effect of the tested compounds, indicating the important role played by this monoamine in their effects in the FST.
To analyze further the mechanism of action of the new compounds in the FST, we attempted to block the observed effects with antagonists of different serotonin receptor subtypes which had no effect in the FST per se. Taking into account the affinity studies of new compounds, we primarily wanted to assess the contribution of 5-HT1A receptors. We observed that (−)pindolol, being inactive at this dose in the FST, as reported by Wang et al. (2008), fully blocked the antidepressant-like action of both tested compounds. Similarly, WAY 100635 administered alone had no effect in the FST, as previously shown (Kaster et al. 2005; Wang et al. 2008), and reversed the effects of both compounds. (−)Pindolol is an unspecific serotonergic antagonist which binds to 5-HT1A, 5-HT1B, and β-adrenergic receptors (Lejeune and Millan 2000), while WAY 100635 is a specific 5-HT1A receptor antagonist with 100 times greater affinity to their sites than for other serotonergic antagonists, and which also has affinity for dopaminergic and noradrenergic sites (for review, see Forster et al. 1995). The results obtained from studies with these antagonists clearly demonstrate the participation of the serotonergic system, particularly the serotonin 5-HT1A receptor subtype, in the antidepressant-like action of the tested compounds. The findings are also in accordance with radioligand in vitro binding assays (Fiorino et al. 2016, 2017). This indicates, therefore, that the new compounds are selective ligands of the 5-HT1A receptors. Additional confirmation of our results is the studies of other researchers, who indicate the contribution of serotonin 5-HT1A receptors in the antidepressant-like effects of new derivatives from the arylpiperazine group (Zajdel et al. 2007; Partyka et al. 2015; Zagórska et al. 2015).
In contrast, in vitro studies have demonstrated that the new compound 4p also possesses affinity toward the 5-HT2C receptors which are widely distributed in the brain regions connected with regulation of mood (Celada et al. 2004). In fact, in suicide victims, an altered level of the m-RNA encoding 5-HT2C receptors has been reported in the prefrontal cortex (Gurevich et al. 2002). Moreover, desensitization of these receptors has been reported following chronic SSRI treatment. In addition, preclinical data suggest that the blockage of 5-HT2C can enhance the antidepressant effects of SSRI (Redrobe and Bourin 1997, 1998; Cremers et al. 2004). Therefore, in the present study, an additional experiment was conducted to explore the possible participation of these receptors in the effects of the tested compounds in the FST.
We found that administration of ketanserin at an ineffective dose of 1 mg/kg 15 min prior to tested compound 3o (used also at an ineffective dose, 3.75 mg/kg) did not produce statistically significant effects in the FST, whereas in the case of co-administration of 4p compound (7.5 mg/kg) with ketanserin, a certain decrease in immobility time was observed. Still, we cannot exclude that these results may be slightly affected by impaired locomotor activity after co-administration of ketanserin with the new compounds (Fig. 5b). Yet, with these results, after using serotonergic tool substances, we can confirm the participation of 5-HT1A receptors in the antidepressant-like effect of both compounds, but cannot exclude the involvement of 5-HT2C receptors in the effects of 4p, but not of the 3o compound. The aforementioned result is in line with our in vitro studies. Moreover, considering that pCPA is suggested to act presynaptically (Luscombe et al. 1993), the observed antidepressant-like effect of compound 3o may be mediated, at least in part, by the presynaptic part of the 5-HT system. This notion is basically compatible with our previous observations that this compound behaved as a presynaptic 5-HT1A receptor antagonist (Fiorino et al. 2017). Acting at this site, it can inhibit negative feedback on serotonergic neurons, hence increasing 5-HT release, which plays a role in reducing immobility. Since pCPA does not abrogate the anti-immobility effect of 8-OH-DPAT (a 5-HT1A receptor agonist) in the FST, the antidepressant activity of this compound is attributed to postsynaptic 5-HT1A receptor activation (Wieland and Lucki 1990; Luscombe et al. 1993; Kitamura and Nagatani 1996). In the case of the 3o compound, however, stimulation of postsynaptic receptors seems to be insufficient to produce antidepressant-like effects. Based on the results of our experiments, we can speculate that the antidepressant-like activity of 3o is related both to pre- and postsynaptic sites of the serotonergic synapse.
In contrast, compound 4p was shown to behave as a presynaptic 5-HT1A agonist in the earlier studies (Fiorino et al. 2016), wherein pCPA, acting presynaptically by blocking synthesis of 5-HT, reversed the antidepressant-like effect of 4p. It is difficult to explain these results. As the effect of 4p was potentiated by ketanserin, this suggests that 5-HT2A/2C receptors are involved in its antidepressant-like activity. In numerous rodent tests, 5-HT2C receptor antagonists have been shown to display strong, rapid, and sustained anxiolytic/antidepressant effects (Di Giovanni et al. 2002; Harada et al. 2006; Jensen et al. 2010). Agomelatine is one of these, and this compound has been recognized to be a 5-HT2C receptor antagonist and a melatonin MT1 and MT2 receptor agonist (Millan et al. 2011). Of note, most of the third-generation antipsychotics with antidepressant-like properties are blockers of these receptors (Van Oekelen et al. 2003; Reynolds 2011). In addition, it was shown that prior administration of ketanserin potentiated the effects of subactive doses of imipramine (Redrobe and Bourin 1997), which is a conventional antidepressant that has been available on the market for years for the treatment of major depression (Amsterdam et al. 1986; Nielsen et al. 1993). Imipramine is known to be a non-selective monoamine reuptake inhibitor (Glowinski and Axelrod 1964; Corrodi and Fuxe 1968). In our studies, the tested compounds injected at ineffective doses were able to potentiate the antidepressant action of imipramine (at a subeffective dose of 15 mg/kg) in FST by decreasing immobility time. This effect was not the result of increased motor activity, so it seems to be specific (Fig. 6a, b). Thus, we cannot exclude other presynaptic mechanisms involved in facilitation of serotonergic neurotransmission by 4p and 3o. Therefore, it is reasonable to speculate that the tested compounds can act on other non-investigated targets, such as the serotonin transport.
In order to exclude false positive results, we assessed the locomotor activity of the animals in an analogous schedule to the presented experiments. Such work showed that neither new compounds 4p and 3o alone (Fiorino et al. 2016, 2017) nor in combination with the serotonergic tool substances changed the locomotor activity of mice in this test (see Figs. 2B, 3B, 4B, and 6B). One exception is the concomitant treatment of 3o with ketanserin, wherein a significant decrease in locomotor activity was observed (Fig. 5b). Still, we hold that the results obtained in the FST can be considered genuine.
Additionally, our previous in vitro studies proved that both tested compounds had no affinity toward other receptors beyond the serotonin ones (Fiorino et al. 2016, 2017). This could represent a possible advantage in respect to other antidepressants, since interactions with few neurotransmitter systems are responsible for their side effects (Stahl 1998). However, further studies are needed to confirm this assumption.
Conclusion
This study demonstrates that the new arylpiperazine derivatives, compounds 4p (N-(3-(4-(piperonyl)piperazin-1-yl)propyl) isonicotinamide and 3oN-(2-(4-(pyrimidin-2-yl)piperazin-1-yl)ethyl) picolinamide, produce an antidepressant-like effect in FST and TST—two classical animal tests predictive of antidepressant properties. The action of tested compounds appears to be mediated at least in part by an interaction with the serotonin 5-HT1A receptors and in the case of 4p, also the 5-HT2C receptors.
References
Adell A, Castro E, Celada P, Bortolozzi A, Pazos A, Artigas F (2005) Strategies for producing faster acting antidepressants. Drug Discov Today 10:578–585. https://doi.org/10.1016/S1359-6446(05)03398-2
Aldous NR, Mann AM (1963) The pathophysiology of depression. Can Med Assoc J 89:937–943
Amsterdam JD, Kaplan M, Potter L, Bloom L, Rickels K (1986) Adinazolam, a new triazolobenzodiazepine, and imipramine in the treatment of major depressive disorder. Psychopharmacology 88:484–488
Appelberg BG, Syvälahti EK, Koskinen TE et al (2001) Patients with severe depression may benefit from buspirone augmentation of selective serotonin reuptake inhibitors: results from a placebo-controlled, randomized, double-blind, placebo wash-in study. J Clin Psychiat 62:448–452
Artigas F (2013) Serotonin receptors involved in antidepressant effects. Pharmacol Ther 137:119–131. https://doi.org/10.1016/j.pharmthera.2012.09.006
Artigas F (2015) Developments in the field of antidepressants, where do we go now? Eur Neuropsychopharmacol 25:657–670. https://doi.org/10.1016/j.euroneuro.2013.04.013
Artigas F, Romero L, de Montigny C, Blier P (1996) Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci 19:378–383. https://doi.org/10.1016/S0166-2236(96)10037-0
Barnes NM, Sharp T (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38:1083–1152
Blier P, de Montigny C (1994) Current advances and trends in the treatment of depression. Trends Pharmacol Sci 15:220–226
Borsini F (1995) Role of the serotonergic system in the forced swimming test. Neurosci Biobehav Rev 19:377–395
Borsini F, Meli A (1988) Is the forced swimming test a suitable model for revealing antidepressant activity? Psychopharmacology 94:147–160. https://doi.org/10.1007/BF00176837
Brüning CA, Souza ACG, Gai BM, Zeni G, Nogueira CW (2011) Antidepressant-like effect of m-trifluoromethyl-diphenyl diselenide in the mouse forced swimming test involves opioid and serotonergic systems. Eur J Pharmacol 658:145–149. https://doi.org/10.1016/j.ejphar.2011.02.039
Bymaster FP, Zhang W, Carter PA, Shaw J, Chernet E, Phebus L, Wong DT, Perry KW (2002) Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology 160:353–361. https://doi.org/10.1007/s00213-001-0986-x
Celada P, Puig M, Amargós-Bosch M et al (2004) The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci 29:252–265
Celada P, Bortolozzi A, Artigas F (2013) Serotonin 5-HT1A receptors as targets for agents to treat psychiatric disorders: rationale and current status of research. CNS Drugs 27:703–716. https://doi.org/10.1007/s40263-013-0071-0
Chilmończyk Z, Bojarski A, Pilc A, Sylte I (2015) Functional selectivity and antidepressant activity of serotonin 1A receptor ligands. Int J Mol Sci 16:18474–18506. https://doi.org/10.3390/ijms160818474
Corrodi H, Fuxe K (1968) The effect of imipramine on central monoamine neurons. J Pharm Pharmacol 20:230–231
Cremers TIFH, Giorgetti M, Bosker FJ, Hogg S, Arnt J, Mørk A, Honig G, Bøgesø KP, Westerink BHC, den Boer H, Wikstrom HV, Tecott LH (2004) Inactivation of 5-HT2C receptors potentiates consequences of serotonin reuptake blockade. Neuropsychopharmacology 29:1782–1789. https://doi.org/10.1038/sj.npp.1300474
Cryan JF, Slattery DA (2007) Animal models of mood disorders: recent developments. Curr Opin Psychiatr 20:1–7. https://doi.org/10.1097/YCO.0b013e3280117733
Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29:571–625. https://doi.org/10.1016/j.neubiorev.2005.03.009
De Vry J (1995) 5-HT1A receptor agonists: recent developments and controversial issues. Psychopharmacology 121:1–26
Deakin JFW (1993) A review of clinical efficacy of 5-HT 1A agonists in anxiety and depression. J Psychopharmacol 7:283–289. https://doi.org/10.1177/026988119300700308
Di Giovanni G, Di Matteo V, Esposito E (2002) Serotonin/dopamine interaction--focus on 5-HT2C receptor, a new target of psychotropic drugs. Indian J Exp Biol 40:1344–1352
Zomkowski ADE, Rosa AO, Lin J, Santos ARS, Calixto JB, Rodrigues ALS (2004) Evidence for serotonin receptor subtypes involvement in agmatine antidepressant like-effect in the mouse forced swimming test. Brain Res 1023:253–263. https://doi.org/10.1016/j.brainres.2004.07.041
Dixon AK (1998) Ethological strategies for defence in animals and humans: their role in some psychiatric disorders. Br J Med Psychol 71(Pt 4):417–445
Faludi G (1994) Buspirone: a new possibility in the treatment of anxiety. Orv Hetil 135:1807–1813
Fava M, Kendler KS (2000) Major depressive disorder. Neuron 28:335–341
Fiorino F, Ciano A, Magli E, Severino B, Corvino A, Perissutti E, Frecentese F, di Vaio P, Izzo AA, Capasso R, Massarelli P, Nencini C, Rossi I, Kędzierska E, Orzelska-Górka J, Bielenica A, Santagada V, Caliendo G (2016) Synthesis, in vitro and in vivo pharmacological evaluation of serotoninergic ligands containing an isonicotinic nucleus. Eur J Med Chem 110:133–150. https://doi.org/10.1016/j.ejmech.2016.01.021
Fiorino F, Magli E, Kędzierska E, Ciano A, Corvino A, Severino B, Perissutti E, Frecentese F, di Vaio P, Saccone I, Izzo AA, Capasso R, Massarelli P, Rossi I, Orzelska-Górka J, Kotlińska JH, Santagada V, Caliendo G (2017) New 5-HT 1A, 5HT 2A and 5HT 2C receptor ligands containing a picolinic nucleus: synthesis, in vitro and in vivo pharmacological evaluation. Bioorg Med Chem 25:5820–5837. https://doi.org/10.1016/j.bmc.2017.09.018
Forster EA, Cliffe IA, Bill DJ, Dover GM, Jones D, Reilly Y, Fletcher A (1995) A pharmacological profile of the selective silent 5-HT1A receptor antagonist, WAY-100635. Eur J Pharmacol 281:81–88
Girish C, Raj V, Arya J, Balakrishnan S (2012) Evidence for the involvement of the monoaminergic system, but not the opioid system in the antidepressant-like activity of ellagic acid in mice. Eur J Pharmacol 682:118–125. https://doi.org/10.1016/j.ejphar.2012.02.034
Glowinski J, Axelrod J (1964) Inhibition of uptake of tritiated-noradrenaline in the intact rat brain by imipramine and structurally related compounds. Nature 204:1318–1319
Guilloux J-P, David DJP, Guiard BP, Chenu F, Repérant C, Toth M, Bourin M, Gardier AM (2006) Blockade of 5-HT1A receptors by (±)-pindolol potentiates cortical 5-HT outflow, but not antidepressant-like activity of paroxetine: microdialysis and behavioral approaches in 5-HT1A receptor knockout mice. Neuropsychopharmacology 31:2162–2172. https://doi.org/10.1038/sj.npp.1301019
Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C (2002) Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 34:349–356
Harada K, Aota M, Inoue T, Matsuda R, Mihara T, Yamaji T, Ishibashi K, Matsuoka N (2006) Anxiolytic activity of a novel potent serotonin 5-HT2C receptor antagonist FR260010: a comparison with diazepam and buspirone. Eur J Pharmacol 553:171–184. https://doi.org/10.1016/j.ejphar.2006.09.042
Hensler J (2002) Differential regulation of 5-HT1A receptor-G protein interactions in brain following chronic antidepressant administration. Neuropsychopharmacology 26:565–573. https://doi.org/10.1016/S0893-133X(01)00395-5
Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP (1994) International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol Rev 46:157–203
Jensen NH, Cremers TI, Sotty F (2010) Therapeutic potential of 5-HT 2C receptor ligands. Sci World J 10:1870–1885. https://doi.org/10.1100/tsw.2010.180
Jesse CR, Wilhelm EA, Bortolatto CF, Nogueira CW (2010) Evidence for the involvement of the serotonergic 5-HT2A/C and 5-HT3 receptors in the antidepressant-like effect caused by oral administration of bis selenide in mice. Prog Neuro-Psychopharmacol Biol Psychiatry 34:294–302. https://doi.org/10.1016/j.pnpbp.2009.11.023
Kaster MP, Santos ARS, Rodrigues ALS (2005) Involvement of 5-HT1A receptors in the antidepressant-like effect of adenosine in the mouse forced swimming test. Brain Res Bull 67:53–61. https://doi.org/10.1016/j.brainresbull.2005.05.025
Kędzierska E, Wach I (2016) Using tests and models to assess antidepressant-like activity in rodents. Curr Issues Pharm Med Sci 29:61–65. https://doi.org/10.1515/cipms-2016-0013
Kinney GG, Taber MT, Gribkoff VK (2000) The augmentation hypothesis for improvement of antidepressant therapy: is pindolol a suitable candidate for testing the ability of 5HT1A receptor antagonists to enhance SSRI efficacy and onset latency? Mol Neurobiol 21:137–152
Kitamura Y, Nagatani T (1996) Buspirone enhances immobility in the forced swim test in mice. Pharmacol Biochem Behav 55:445–451
Kleven MS, Assié MB, Koek W (1997) Pharmacological characterization of in vivo properties of putative mixed 5-HT1A agonist/5-HT(2A/2C) antagonist anxiolytics. II. Drug discrimination and behavioral observation studies in rats. J Pharmacol Exp Ther 282:747–759
Kostowski W, Dyr W, Krząścik P, Järbe T, Archer T (1992) 5-Hydroxytryptamine 1A receptor agonists in animal models of depression and anxiety. Pharmacol Toxicol 71:24–30. https://doi.org/10.1111/j.1600-0773.1992.tb00515.x
Lazure KE, Lydiatt WM, Denman D, Burke WJ (2009) Association between depression and survival or disease recurrence in patients with head and neck cancer enrolled in a depression prevention trial. Head Neck 31:888–892. https://doi.org/10.1002/hed.21046
Lejeune F, Millan MJ (2000) Pindolol excites dopaminergic and adrenergic neurons, and inhibits serotonergic neurons, by activation of 5-HT1A receptors. Eur J Neurosci 12:3265–3275
Loubinoux I, Kronenberg G, Endres M, Schumann-Bard P, Freret T, Filipkowski RK, Kaczmarek L, Popa-Wagner A (2012) Post-stroke depression: mechanisms, translation and therapy. J Cell Mol Med 16:1961–1969. https://doi.org/10.1111/j.1582-4934.2012.01555.x
Lucki I, Dalvi A, Mayorga AJ (2001) Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology 155:315–322
Luscombe GP, Martin KF, Hutchins LJ, Gosden J, Heal DJ (1993) Mediation of the antidepressant-like effect of 8-OH-DPAT in mice by postsynaptic 5-HT1A receptors. Br J Pharmacol 108:669–677
Manji HK, Drevets WC, Charney DS (2001) The cellular neurobiology of depression. Nat Med 7:541–547. https://doi.org/10.1038/87865
Marangell LB (2000) Augmentation of standard depression therapy. Clin Ther 22(Suppl A):A25–A38 discussion A39–41
McAllister-Williams RH, Alhaj HA, Massey A et al (2014) Somatodendritic 5-hydroxytryptamine1A (5-HT1A) autoreceptor function in major depression as assessed using the shift in electroencephalographic frequency spectrum with buspirone. Psychol Med 44:767–777. https://doi.org/10.1017/S0033291713001475
McIntyre RS (2017) The role of new antidepressants in clinical practice in Canada: a brief review of vortioxetine, levomilnacipran ER, and vilazodone. Neuropsychiatr Dis Treat Volume 13:2913–2919. https://doi.org/10.2147/NDT.S150589
Millan MJ (2004) The role of monoamines in the actions of established and “novel” antidepressant agents: a critical review. Eur J Pharmacol 500:371–384. https://doi.org/10.1016/j.ejphar.2004.07.038
Millan MJ, Dekeyne A, Papp M, la Rochelle CD, MacSweeny C, Peglion JL, Brocco M (2001) S33005, a novel ligand at both serotonin and norepinephrine transporters: II. Behavioral profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther 298:581–591
Millan MJ, Marin P, Kamal M, Jockers R, Chanrion B, Labasque M, Bockaert J, Mannoury la Cour C (2011) The melatonergic agonist and clinically active antidepressant, agomelatine, is a neutral antagonist at 5-HT2C receptors. Int J Neuropsychopharmacol 14:768–783. https://doi.org/10.1017/S1461145710001045
Montgomery SA (2006) Why do we need new and better antidepressants? Int Clin Psychopharmacol 21:S1–S10. https://doi.org/10.1097/01.yic.0000199455.39552.1c
Mork A, Pehrson A, Brennum LT, Nielsen SM, Zhong H, Lassen AB, Miller S, Westrich L, Boyle NJ, Sanchez C, Fischer CW, Liebenberg N, Wegener G, Bundgaard C, Hogg S, Bang-Andersen B, Stensbol TB (2012) Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther 340:666–675. https://doi.org/10.1124/jpet.111.189068
Musselman DL, Evans DL, Nemeroff CB (1998) The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry 55:580–592
Nautiyal KM, Hen R (2017) Serotonin receptors in depression: from A to B. F1000Research 6:123. https://doi.org/10.12688/f1000research.9736.1
Nemeroff CB, Owens MJ (2002) Treatment of mood disorders. Nat Neurosci 5:1068–1070. https://doi.org/10.1038/nn943
Nestler EJ, Barrot M, DiLeone RJ et al (2002) Neurobiology of depression. Neuron 34:13–25
Neu P (2009) Wechselwirkungen zwischen Depression und Schlaganfall. Nervenarzt 80:772–780. https://doi.org/10.1007/s00115-009-2720-6
Nielsen BM, Behnke K, Arup P, Christiansen PE, Geisler A, Ipsen E, Maach-Møller B, Øhrberg SC (1993) A comparison of fluoxetine and imipramine in the treatment of outpatients with major depressive disorder. Acta Psychiatr Scand 87:269–272
O’Leary OF, Bechtholt AJ, Crowley JJ et al (2007) Depletion of serotonin and catecholamines block the acute behavioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology 192:357–371. https://doi.org/10.1007/s00213-007-0728-9
Ohno Y (2010) New insight into the therapeutic role of 5-HT1A receptors in central nervous system disorders. Cent Nerv Syst Agents Med Chem 10:148–157
Page ME, Detke MJ, Dalvi A, Kirby LG, Lucki I (1999) Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology 147:162–167
Partyka A, Chłoń-Rzepa G, Wasik A, Jastrzębska-Więsek M, Bucki A, Kołaczkowski M, Satała G, Bojarski AJ, Wesołowska A (2015) Antidepressant- and anxiolytic-like activity of 7-phenylpiperazinylalkyl-1,3-dimethyl-purine-2,6-dione derivatives with diversified 5-HT1A receptor functional profile. Bioorg Med Chem 23:212–221. https://doi.org/10.1016/j.bmc.2014.11.008
Petit-Demouliere B, Chenu F, Bourin M (2005) Forced swimming test in mice: a review of antidepressant activity. Psychopharmacology 177:245–255. https://doi.org/10.1007/s00213-004-2048-7
Peyrot M (2003) Depression: a quiet killer by any name. Diabetes Care 26:2952–2953. https://doi.org/10.2337/DIACARE.26.10.2952
Poleszak E, Wośko S, Serefko A, Wlaź A, Kasperek R, Dudka J, Wróbel A, Nowak G, Wlaź P (2014) The effects of ifenprodil on the activity of antidepressant drugs in the forced swim test in mice. Pharmacol Rep 66:1031–1036. https://doi.org/10.1016/j.pharep.2014.06.016
Poleszak E, Szopa A, Wyska E, Kukuła-Koch W, Serefko A, Wośko S, Bogatko K, Wróbel A, Wlaź P (2016) Caffeine augments the antidepressant-like activity of mianserin and agomelatine in forced swim and tail suspension tests in mice. Pharmacol Rep 68:56–61. https://doi.org/10.1016/j.pharep.2015.06.138
Porsolt RD, Bertin A, Jalfre M (1977a) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229:327–336
Porsolt RD, Le Pichon M, Jalfre M (1977b) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732. https://doi.org/10.1038/266730a0
Redrobe JP, Bourin M (1997) Partial role of 5-HT2 and 5-HT3 receptors in the activity of antidepressants in the mouse forced swimming test. Eur J Pharmacol 325:129–135
Redrobe JP, Bourin M (1998) Clonidine potentiates the effects of 5-HT1A, 5-HT1B and 5-HT2A/2C antagonists and 8-OH-DPAT in the mouse forced swimming test. Eur Neuropsychopharmacol 8:169–173
Redrobe JP, Bourin M (1999) Evidence of the activity of lithium on 5-HT1B receptors in the mouse forced swimming test: comparison with carbamazepine and sodium valproate. Psychopharmacology 141:370–377
Redrobe JP, MacSweeney CP, Bourin M (1996) The role of 5-HT1A and 5-HT1B receptors in antidepressant drug actions in the mouse forced swimming test. Eur J Pharmacol 318:213–220
Reynolds GP (2011) Receptor mechanisms of antipsychotic drug action in bipolar disorder – focus on asenapine. Ther Adv Psychopharmacol 1:197–204. https://doi.org/10.1177/2045125311430112
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301:805–809. https://doi.org/10.1126/science.1083328
Sargent PA, Kjaer KH, Bench CJ, Rabiner EA, Messa C, Meyer J, Gunn RN, Grasby PM, Cowen PJ (2000) Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry 57:174–180
Savegnago L, Jesse CR, Pinto LG, Rocha JBT, Nogueira CW, Zeni G (2007) Monoaminergic agents modulate antidepressant-like effect caused by diphenyl diselenide in rats. Prog Neuro-Psychopharmacol Biol Psychiatry 31:1261–1269. https://doi.org/10.1016/j.pnpbp.2007.05.006
Savitz J, Lucki I, Drevets WC (2009) 5-HT1A receptor function in major depressive disorder. Prog Neurobiol 88:17–31. https://doi.org/10.1016/j.pneurobio.2009.01.009
Schreiber R, De Vry J (1993) 5-HT1A receptor ligands in animal models of anxiety, impulsivity and depression: multiple mechanisms of action? Prog Neuro-Psychopharmacol Biol Psychiatry 17:87–104
Singh A, Lucki I (1993) Antidepressant-like activity of compounds with varying efficacy at 5-HT1A receptors. Neuropharmacology 32:331–340
Stahl SM (1998) Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord 51:215–235
Steru L, Chermat R, Thierry B, Simon P (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85:367–370
Taylor DP (1988) Buspirone, a new approach to the treatment of anxiety. FASEB J 2:2445–2452
Van Oekelen D, Luyten WHML, Leysen JE (2003) 5-HT2A and 5-HT2C receptors and their atypical regulation properties. Life Sci 72:2429–2449
Wang R, Xu Y, Wu H-L, Li YB, Li YH, Guo JB, Li XJ (2008) The antidepressant effects of curcumin in the forced swimming test involve 5-HT1 and 5-HT2 receptors. Eur J Pharmacol 578:43–50. https://doi.org/10.1016/j.ejphar.2007.08.045
Wieland S, Lucki I (1990) Antidepressant-like activity of 5-HT1A agonists measured with the forced swim test. Psychopharmacology 101:497–504
Willner P, Mitchell PJ (2002) The validity of animal models of predisposition to depression. Behav Pharmacol 13:169–188
Wong M-L, Licinio J (2001) Research and treatment approaches to depression. Nat Rev Neurosci 2:343–351. https://doi.org/10.1038/35072566
Yalcin I, Aksu F, Belzung C (2005) Effects of desipramine and tramadol in a chronic mild stress model in mice are altered by yohimbine but not by pindolol. Eur J Pharmacol 514:165–174. https://doi.org/10.1016/j.ejphar.2005.03.029
Yohn CN, Gergues MM, Samuels BA (2017) The role of 5-HT receptors in depression. Mol Brain 10:28. https://doi.org/10.1186/s13041-017-0306-y
Zagórska A, Kołaczkowski M, Bucki A, Siwek A, Kazek G, Satała G, Bojarski AJ, Partyka A, Wesołowska A, Pawłowski M (2015) Structure–activity relationships and molecular studies of novel arylpiperazinylalkyl purine-2,4-diones and purine-2,4,8-triones with antidepressant and anxiolytic-like activity. Eur J Med Chem 97:142–154. https://doi.org/10.1016/j.ejmech.2015.04.046
Zajdel P, Subra G, Bojarski AJ, Duszyńska B, Tatarczyńska E, Nikiforuk A, Chojnacka-Wójcik E, Pawłowski M, Martinez J (2007) Novel class of arylpiperazines containing N-acylated amino acids: their synthesis, 5-HT1A, 5-HT2A receptor affinity, and in vivo pharmacological evaluation. Bioorg Med Chem 15:2907–2919. https://doi.org/10.1016/j.bmc.2007.02.018
Funding
This work was supported by Funds for the Statutory Activity of the Medical University of Lublin.
Author information
Authors and Affiliations
Contributions
EK and EP conceived and designed the research.
EK, JO-G, and BK conducted behavioral experiments.
FF and EM contributed new compounds tested in the study.
EK analyzed data and wrote the manuscript.
FF, PW, and JHK revised the manuscript.
All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kędzierska, E., Fiorino, F., Magli, E. et al. New arylpiperazine derivatives with antidepressant-like activity containing isonicotinic and picolinic nuclei: evidence for serotonergic system involvement. Naunyn-Schmiedeberg's Arch Pharmacol 392, 743–754 (2019). https://doi.org/10.1007/s00210-019-01620-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-019-01620-7