
Abstract
We review areas of overlap between nucleoside diphosphate kinase (NDPK; nm23) and two proteins manifesting an equivalent diversity of action, each with many thousands of publications. The first is a constitutively active protein kinase, CK2 (formerly casein kinase 2), that includes NDPK amongst its hundreds of targets. The second is an enigmatic member of the ATP-binding cassette (ABC) family of membrane pumps that normally hydrolyse ATP to transport substrates. Yet our unusual family member (ABCC7) is not a pump but, uniquely, acts as a regulated anion channel. ABCC7 is the cystic fibrosis transmembrane conductance regulator (CFTR), and we discuss the highly prevalent CFTR mutation (F508del CFTR) in terms of the uncertainties surrounding the molecular basis of cystic fibrosis that cloud approaches to corrective therapy. Using lysates from cells stably expressing either wild-type or F508del CFTR, incubated with the CK2 substrate GTP, we show that the phosphoproteome of F508del CFTR-expressing cells both differs from wild-type CFTR-expressing cells and is significantly enhanced in intensity by ∼1.5-fold (p < 0.05, paired t test with Bonferroni correction, n = 4). Phosphorylation is about 50% attenuated with a specific CK2 inhibitor. We propose that a new function may exist for the CFTR region that is commonly mutated, noting that its sequence (PGTIKENIIF508GVSYDEYRYR) is not only highly conserved within the C sub-family of ABC proteins but also a related sequence is found in NDPK. We conclude that a latent path may exist between mutation of this conserved sequence, CK2 hyperactivity and disease pathogenesis that might also explain the heterozygote advantage for the common F508del CFTR mutant .
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The common autosomal recessive disease cystic fibrosis (CF) is found mostly in populations of European descent and remains an enigma despite over four decades of research with over 30,000 references in the literature. Its molecular origin is universally agreed in only one respect, namely after mutation of both alleles in the cystic fibrosis transmembrane conductance regulator (CFTR; ATP-binding cassette (ABC)C7) on human chromosome seven (Chen et al. 2010; Aleksandrov et al. 2007), the normally sterile lung epithelium acquires unusual bacterial flora shortly after birth and an inflammatory destructive lung disease ensues. After CFTR was discovered in 1989 (Riordan et al. 1989), early investigators showed that unlike other ABC proteins, CFTR was not a pump and therefore dissected the pathways controlling CFTR channel function proving beyond doubt that membrane-anchored, cyclic AMP-dependent protein kinase (PKA) opens a transmembrane pore within CFTR for the transport of chloride and other anions across epithelial cell membranes (Sheppard and Welsh 1999). In the epithelial cell, the prevailing electrochemical driving forces across the apical membrane facing the lumen (Fig. 1, bottom of right panel) are such that the direction of anion flow through CFTR is passive, i.e. dependent on the prevailing activities of other channels and transporters. Because anion flow may be into or out of the epithelial cell, this makes the CFTR channel useful both for secretion and absorption of electrolytes (and hence water). But, irrespective of a sufficient electrochemical driving force, anion flow through the pore only occurs provided enough hydrolysable ATP is sandwiched between the closely opposed faces of the two nucleotide binding domains (NBD1 and NBD2) that give the ABC family their leitmotif. This is why this channel is sometimes described as being ATP-gated, but the spatial and temporal relationship between ATP hydrolysis and pore opening is not agreed. For example, despite there being abundant cellular ATP in bulk cytosol, this CFTR pore is apparently inaccessible to external or internal anions in the quiescent epithelial cell because the channel rarely opens without prior cell stimulation through protein kinases such as the cAMP-regulated PKA, and indeed, at baseline, even when CFTR spontaneously but rarely flickers into life, it closes again promptly (Sheppard and Welsh 1999). So what keeps the open probability of CFTR so low in a quiescent epithelial cell? Feedback loops are one possible solution. One agreed inhibitory factor is the substantial, AMP-independent activity of a CFTR-binding protein kinase (AMP-activated kinase, AMPK) that acts on the regulatory domain in CFTR, largely through serine-768 (King et al. 2009; Kongsuphol et al. 2009a, b). It is thought that until this basal, inhibitory AMPK-mediated drive is overcome by a stimulus that (hyper)activates PKA (for simplicity, this is not shown in the model in Fig. 1); CFTR does not open fully (King et al. 2009; Kongsuphol et al. 2009a, b). These data also demonstrated that membrane-local destruction of cAMP, which produces AMP through membrane-bound phosphodiesterases (Fig. 1) local to CFTR, may also play a role in AMPK-dependent inhibition of CFTR.

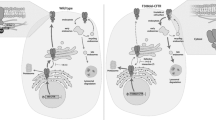
In bottom left panel, it is shown how membrane-bound NDPK, when ‘auto’ charged with phosphohistidine-118 acts as a high-energy phosphate distributor to other proteins such as to the beta subunit of G proteins which enables basal levels of cAMP synthesis without classical receptor-mediated activation. In bottom right panel, phosphoserine at S120 inhibits phosphohistidine-118, and in vitro, CK2 is thought to be the kinase. Should CK2 be overactive, NDPK is predicted to be dysfunctional because no phospho-His distribution can take place. One consequence is that cAMP falls, its product AMP falls thus relieving AMPK-mediated inhibition of CFTR. NDPK function is reviewed in Mehta and Orchard (2009), and the mechanisms linking NDPK with AMPK and CFTR are described in the Treharne et al. references in the manuscript
So how might the multifunctional nucleoside diphosphate kinase (NDPK) fit into this picture? Zollo and colleagues in this series of papers will focus on the role of a class of ‘DHH’ phosphodiesterases in NDPK binding and NDPK function, but this will not be discussed further here. The most direct evidence lies in cytosol-free epithelial membranes from a cell line bearing CFTR, where we reported that NDPK co-precipitates with AMPK despite high stringency salt washes (Treharne et al. 2009a). NDPK is present in the purified apical membrane of many airway species, but the exact interactions and consequences for CFTR regulation are unknown. Our work in progress (Fig. 1) suggests that CFTR, AMPK and NDPK form a complex at the epithelial membrane. For example, we observe that the phosphoserine and phosphohistidine content of NDPK changes in an AMP-dependent and GTP-selective manner in vitro (Treharne et al. 2009a). The interactions are under investigation, but it is interesting to note that, at a clinical level, independent confirmation of our proposed links between NDPK and CF has recently been published (Charro et al. 2009).
The ancient origins of the commonest form of cystic fibrosis, F508del CFTR
It is a curious fact that about 50% of CF patients across the globe are homozygous for only one type of CFTR mutation despite the existence of over 1,500 other CFTR mutations (Bobadilla et al. 2002). This commonest (by far) CFTR mutant deletes only three adjacent base pairs in exon 10 to produce an in-frame point deletion of a single phenylalanine-508 (the correct genetic name is p.phe508del CFTR, mostly written as F508del, Phe508del or ΔF508 in legacy nomenclature). Crystal structures of the isolated NBD1 domain in which this deleted phenylalanine residue resides suggest that the amino acids surrounding this F508 site are located in an accessible intracellular position on the roof of NBD1, just under the cell membrane, but too far removed for any direct interaction with the large hydrophobic NBD1 interface forming the ATP sandwich with NBD2 (Lewis et al. 2005). Furthermore, it is observed that the loss of F508 only induces local changes in the structure of NBD1 in the immediate vicinity of the missing phenylalanine. Thus, a key challenge is to explain how a very local change in CFTR structure leads first, to a heterozygote advantage and second to disease, a topic to which we will return when we consider the functional effects of 20-mer peptides corresponding to this mutated region.
A second curious fact emerges from analysis of the haplotype background of the chromosome bearing this single highly prevalent F508del CFTR mutant which suggests that this point mutation arose only once around 50,000 years ago (Morral et al. 1994). Thus, today, between apparently unrelated Europeans, there exists a commonality of structure of this F508-deleted chromosome which implies that over many hundreds of generations, this very same chromosome increased its prevalence from a vanishingly rare first CF carrier baby (the eponymous Fibrogenicum, born around the time one wave of early humans left Africa) to today’s carriage by every 50th–80th European (with a notable northwest to southeast decreasing gradient). The remarkable conclusion arises that, as this chromosome 7 was handed down between successive generations, a repeated survival advantage accrued to each heterozygote ‘fibrogenic’ carrier relative to non-carriers of F508del CFTR. It is interesting to note that up to about 50 years ago, almost all F508del CFTR homozygotes would have died in very early childhood because there was no therapy to correct their malabsorption of foodstuffs because the disease destroys the pancreas in foetal life. We will return to this concept of healthy European ‘super-survivors’ carrying F508del CFTR in single copy when we consider how the deleted F508 in NBD1 could create dominant control over protein kinase CK2 (formerly casein kinase 2), even in a heterozygote setting, thus gaining a survival advantage through recently understood effects of CK2 in inflammation and host defence (Singh and Ramji 2008).
The gap in understanding between CFTR dysfunction and cystic fibrosis disease
For decades, disputes have occurred on the molecular origins of CF disease that arises when a mutation is present in each cftr allele (typically, 50% are homozygous for F508del, 40% have a rare or very rare ‘private’ familial mutation with F508del and the remainder either have thousands of combinations of different rare mutations or have no detectable CFTR mutation, see below). We have discussed the spectrum of these mutations elsewhere (McCormick et al. 2002). Even today, there remain many unresolved molecular issues on the path from CFTR mutation to clinical disease. CFTR is undoubtedly an anion channel in epithelia lining the lung, pancreas, small bowel and sweat duct, but the exact in vivo nature of the negatively charged species that traverse the CFTR pore across the apical membrane is disputed (Muchekehu and Quinton 2010; Conner et al. 2007) and may even differ between tissues (evidence exists for chloride, bicarbonate and anions with epithelial surface-sterilising properties used to attack microbes). Other disagreements centre on the ‘chicken and egg’ scenario where a myriad of cellular compensatory mechanisms may masquerade as primary manifestations of CF disease (Chen et al. 2010). For example, although the epithelia lining the lungs are sterile at birth, inflammation is a rapidly age-increasing disease characteristic that drives childhood lung destruction leading to premature death, mostly in mid-adult life, but even today in parts of Europe, good outcomes to middle age only occur provided adequate resources for therapy are available (McCormick et al. 2010). Yet it remains disputed as to which comes first, ion channel dysfunction producing abnormal mucus and hence inflammation and premature death or whether excessive pre-inflammation is primed awaiting bacterial infection that precedes the destructive lung disease (Chen et al. 2010). We have reviewed this controversy recently adding our own corroborative data to the controversial views of another group that wild-type CFTR is an inflammation suppressor (Hunter et al. 2010) and have recently linked NDPK to inflammation (Treharne et al. 2009c). In contrast, Paul Quinton and colleagues from North America have persuasively argued that loss of CFTR-transported bicarbonate, through its damaging effects on mucus (release and quality), is the main driver of lung damage (Quinton 2010). A further complication is that the relative importance of the different origins of the fluid that hydrates the apical surface of epithelia is not agreed (the debate centres on the different roles of CFTR in submucosal glands versus the surface epithelium itself) making the correct targeting of potential therapies problematic.
Another confounding issue is that a small minority of patients have no detectable CFTR mutations and a tiny minority of these rare ‘phenotypically CF-like’ patients (Azad et al. 2009) have mutations in the epithelial sodium channel (ENaC). This may be relevant to CF because wild-type ENaC is reported to co-localise with wild-type CFTR (Gentzsch et al. 2009), and this interaction is thought to attenuate ENaC channel function, thus limiting sodium absorption and thereby preventing excessive surface drying that drags luminal water into the submucosal space along with the sodium. This is very controversial (Chen et al. 2010) as Michael Welsh and colleagues in Iowa find that, in young animals, sodium absorption from lung lumen to the blood is not enhanced in a pig model of CF where CFTR is deleted (Chen et al. 2010). This recent paper is an example of the overall mechanistic uncertainty into the relative importance of each ion transport process in CF pathogenesis at different ages (Joo et al. 2010; Quinton 2010; Sheppard and Welsh 1999). Uncertainty has not prevented development of therapies targeted to both secretory and absorptive disease aspects, but only clinical trial will resolve the relative importance of hyposecretion from glands versus hyperreabsorption of fluid by the surface epithelium. However, measuring drug-induced benefit based on either approach is not that easy because in CF pathogenesis, for a given patient’s CFTR genotype, when measured clinically against the resultant patient phenotype, CF disease severity is dramatically variable, even between CF siblings inheriting the same CFTR mutation (Mekus et al. 2000). Thus, even if general agreement existed on the optimum rank order of therapies, measuring clinical response is still a challenge more than 20 years after the discovery of CFTR.
CFTR is part of a network of proteins near the membrane
CF pathogenesis in the common form of CF is intrinsically complex because a given patient’s phenotypic ‘CF’ signs on clinical examination may be ‘secondary to or consequential of’ an attempt by the CF-affected cell bearing two defective CFTR copies to compensate for the absence of wild-type CFTR protein. Normally, wild-type CFTR lies within a membrane-delimited network containing many proteins where it acts as a regulatory hub (Wang et al. 2006) as befits the term regulator in its name. Here, we define the consequences of the absence of wild-type CFTR as loss of intra-membrane organisation (LIMBO). Should failure to compensate for LIMBO be the correct pathogenic notion, then CF signs and symptoms occur when LIMBO has no ‘wild-type signal input’ in CF patient cells. It is this approach that underpins much of current strategy to repair the CF defect by pharmacological correction of CFTR (function/stability) or its genetic replacement. Because the latter remains technologically problematic, probably because an airway epithelial cell has been designed by nature to restrict penetration by foreign DNA, it is thought by many that a drug-based approach is most likely to succeed. As proof of principle, a new small molecule has recently been reported to re-open the rare mutant G551D CFTR (<5% of alleles) which theoretically cannot transport anions because its nucleotide sandwich is defective in ATP hydrolysis (Accurso et al. 2010). In CF, the affected sweat gland duct produces too much sodium and chloride because defective G551D CFTR cannot open its pore to re-absorb chloride which consequently, together with luminal sodium, passes on by the chloride-impermeant apical membrane of the sweat duct onwards to the skin surface. The proof of principle arises because this new small molecule is the first to partially reverse the sweat duct LIMBO abnormality, partially reducing surface sweat chloride concentrations. That the observed LIMBO reversal is not complete suggests that the pathway from gene to disease is not fully understood and is consistent with this drug’s unexpected effects on CFTR mutants without this G551D-induced ATP hydrolytic defect. Further mechanistic confusion on the path between gene defect and disease arises when the effects of another new small molecule (Sermet-Gaudelus et al. 2010) are analysed. This drug is claimed to restore full-length CFTR bearing stop mutations that normally induce nonsense-mediated CFTR decay (for example, the G542X stop mutation in CFTR and others like it that together account for about 10% of CFTR mutants). At the very least, this drug, which was developed to create read-through at single stop codons such as G542X, should reverse the above ‘gold standard’ sweat test, but it absolutely fails to do so, thus adding further doubt to our ability to fully understand the path from CFTR defect to disease.
An alternative therapeutic approach posits that CF disease manifestations are ‘secondary’ and could result from some new, induced effect generated by the mutated CFTR itself, i.e. the common F508del mutant of CFTR, even as it degrades, in cellular transit, sends out a false signal that induces (dys)function through the miscreant signal’s ability to create a different sort of attack on the already damaged LIMBO network hub (as explained above, LIMBO is induced by a missing wild-type CFTR hub signal). A secondary, induced excess sodium absorption would be only one example, and its advocates suggest that pharmacological inhibition of ENaC, perhaps coupled to prevention of excessive ENaC activation by channel-activating proteases, is approached most likely to benefit the CF patient (Gentzsch et al. 2009). The literature is driven by champions of each hypothesis (Chen et al. 2010; Gentzsch et al. 2009). But it remains to be seen whether it is the near complete proteasomal destruction of the common F508del mutant of CFTR that generates disease by LIMBO that deprives the CF-affected cell of its wild-type CFTR network hub signal, or whether the process of destroying the mutant CFTR itself causes some unknown but secondary cellular network disruption signal that attacks a weakened LIMBO-affected cell. In the former model, we have a loss of signal and in the latter we have a gain of signal.
Channels are by no means the whole therapeutic story, even in the LIMBO scenario (Pier et al. 1997). Shortly after birth, CF epithelial surfaces acquire a strangely mixed population of both gram-negative and gram-positive species of bacteria such as pseudomonal and staphylococcal species and another major dispute in disease pathogenesis centres on the contribution of CFTR towards the innate immune system (Pier et al. 1997). Interestingly, NDPK is involved in both the cellular responses to viral invasion (as is CK2, see below), and others in this series will describe its role in immunological synapse function through a calcium-sensitive potassium channel which implies that the idea of immune dysregulation may have direct relevance to CF pathogenesis (we will consider the interface between cell calcium through its modulator calmodulin as a target for CK2 below). Some consider CFTR to be central to the bacterial invasion process by acting as a glycosylated pattern sensing molecule reporting on the external environment when epithelial cells encounter, bind and engulf bacteria such as salmonella species in the gut for example. This ‘non-channel’ receptor role for CFTR is further complicated by emerging functions attributed to the form of CFTR that is found in very low abundance in macrophages, lymphocytes, neutrophils and even in the brain. Although this nascent field lies outside the scope of the current paper, it is reasonable to consider the idea that not only does the CF epithelium fail to network normally through a missing wild-type CFTR but the incoming immune cells attracted to the epithelial surface bacteria may also be harbouring their very own dysfunctional CFTR, doubling the potential impact of any defect. This also is a very controversial area (Deriy et al. 2009), and it may be that NDPK function is playing a latent role (Punj et al. 2000; Yilmaz et al. 2008) because a decade of host–pathogen interactive data suggest that NDPK can be secreted by pathogenic bacteria such as CF-characteristic species of mucoid pseudomonas, invasive salmonella, atypical mycobacteria and others such as species that cause cholera. This ‘non-CF’ literature suggests that NDPK may be a key part of the process by which the host immune system is regulated during bacterial invasion by either eliminating, neutering, frustrating or subverting the first wave of professional phagocytes that traverse the epithelium to counteract the invaders (Punj et al. 2000). Here, it is very important to note the disparate nature of the literature because many decades of CFTR-centric research have stressed the role of ATP and its metabolites (Boucher 2004) in the control of ion transport through G-protein coupled purinergic (P) receptors on epithelia (Sheppard and Welsh 1999). Thus far, from the ‘epithelial-centred and channel-centred’ CF field, NDPK researchers focussing on bacteria report that certain members of the purinergic P2X-family are also functional on phagocytes and react to high concentrations of ATP, probably released from damaged epithelial cells, to provoke phagocyte suicide, thus killing any already engulfed bacteria. Through secreted bacterial NDPK, these P2X receptors are targeted by the above pathogenic species when they subvert macrophage function for example, either to promote extra-epithelial bacterial survival by killing incoming macrophages (prior to bacterial engulfment) or, paradoxically, by enhancing macrophage survival through apoptotic inhibition should the pathogen need to multiply intracellularly, if that is its preferred niche.
Many believe that augmenting channel function in a poorly functioning mutant CFTR will make the patient better. This idea is challenged by a recent study (Comer et al. 2009) examining lung function in CF patients with the above ‘severe’ G551D CFTR mutant that is supposedly devoid of any channel activity (Briel et al. 1998) in vitro. According to dogma, these classical, albeit rare, CF patients with ‘channel-dead’ CFTR should have very poor lung function as they should secrete very little chloride through CFTR. Yet the lungs of these patients, although moderately diseased to a severity comparable to that found in severe asthma say (75% of their predicted function is preserved), nevertheless exhibit significantly better lung function than age- and sex-matched F508del CFTR homozygous CF patients who only have 50% preserved function. This disparity occurs despite the fact that the latter mutant is supposedly absent because almost all of it is degraded in the ER and is therefore unavailable for ion transport. Thus, we have yet another theory-practice paradox of a dead but present G551D CFTR channel that is normally processed to the apical membrane creating better functioning lungs than those with a F508del CFTR missing channel that is degraded in the ER and probably never reaches the apical membrane. At the very least, these patient data challenge the widely held notion that G551D is always a ‘severe’ CF mutation. Those proposing the alternative ‘knock-in’ network dysfunction model would say that after G551D mutation, the sodium channel may not be as hyperactive as found in F508del CFTR, so we might expect better lungs. Yet others suggest that G551D CFTR also fails to inhibit the sodium channel in model systems (Briel et al. 1998), and the latest animal data suggest that the very idea of augmented sodium absorption needs to be treated with caution rather than drugs (Chen et al. 2010). On the other hand, those who say that channels are a distraction would say that the very presence of a channel-dead CFTR merely confirms that bacterial pattern recognition is intact as a glycosylated species of CFTR is present at the apical membrane (Pier et al. 1996) and that a better lung function compared to F508del CFTR is exactly what they would have predicted. It should be no surprise that over the years, various commentators have said that cystic fibrosis defies analysis (Kunzelmann 2001), for every argument there is a plausible counter argument.
Here, we conjecture that it is not only the understanding of the processes controlling CFTR, ENaC or bacterial pattern recognition that is likely to unlock the secrets of CF but in addition thought should be directed at the hidden route by which the most highly prevalent F508del CFTR induces its myriad of altered processes ‘in cell’, which number in their hundreds. We are not referring to ‘omic’ analyses of gene-based or protein-based arrays that are altered by losing F508 in CFTR expressed in different model systems such as cell lines, biopsies from patients at different disease stages or data from animal models. Our view is that these approaches generate large volumes of data (Collawn et al. 2010; Ollero et al. 2006; Pollard et al. 2006) that are useful in the LIMBO model in that they show which groups of proteins are likely to network with one another and with CFTR. Indeed one such approach has recently suggested that the isoform of NDPK involved in immune dysfunction is significantly different in the serum of CF patients (Charro et al. 2009).
We took a different approach, grounded on the accepted selective heterozygote advantage for F508del CFTR by first demonstrating common ground between the ability of the apparently constitutively active protein kinase CK2 to control both CFTR (Treharne et al. 2009d) and ENaC (Bachhuber et al. 2008; Kongsuphol et al. 2009a) noting in parallel the roles of CK2 in inflammation (Singh and Ramji 2008). We began by looking for evidence to support a secondary miscreant signal hypothesis given that CK2 is involved in apoptosis, inflammation, viral invasion and the correct sub-membrane anchoring of ion channels as described elsewhere (Pagano et al. 2008, 2010). Our novel conjecture lies at the heart of the dilemma that the heterozygote advantage driving the high prevalence of F508del CFTR must have repeatedly accrued to each healthy CF carrier (see above) who not only has both wild-type and mutant CFTR in the same cell but also must be a super-survivor of the high childhood mortality of around 75% that has characterised European populations, probably for thousands of years. Thus, at the very least, our delineation of a latent pathway from F508del CFTR to CK2 as described below might explain how this degraded common CFTR mutant creates a heterozygote advantage given that, pari passu, the wild-type channel works normally moving thousands of ions per second seemingly oblivious of the transit of the mutant F508del CFTR as it degrades from the ER before it ever matures at the plasma membrane. Our rather different approach also tackles head on the problem that a F508del CFTR heterozygote, whose remaining 50% of wild-type CFTR channels work normally, cannot accrue an obvious advantage from a defective ion channel hypothesis because no channel defect exists in heterozygote F508del CFTR carriers as far as we are aware. Our second argument is that wild-type CFTR is not a very abundant protein and the F508-deleted mutant must therefore constitute a miniscule fraction of the total proteasomal load at any one time. Hence, we could not see how any such cell, even if it expressed two defective CFTR copies, could be damaged by an ER stress response unless an amplifier from F508del CFTR was present. We reasoned that this degraded protein must be signalling separately from the LIMBO idea as a non-channel, signal amplifier and looked for a signal source with the widest reach, namely the pleiotropic protein kinase CK2.
Protein kinase CK2, a regulator of many proteins
CK2 (Meggio and Pinna 2003) is itself an enigma due to its constitutive activity that is unlike almost all other protein kinases which either need activating, either by another kinase, or through the binding of a regulatory small molecule such as cyclic AMP for PKA. In this respect, ‘master kinase’ CK2 shares common ground with NDPK, which is also constitutively active when ATP is added, albeit with its phosphotransfer potential towards targets uniquely realised through a conserved histidine residue, H118 (Fig. 1). Interestingly, in vitro data suggest that CK2 may target NDPK (Biondi et al. 1996) inhibiting NDPK function by phosphorylating S120 near the NDPK catalytic site at H118 (right panel in Fig 1). In addition, unlike the overwhelming majority of protein kinases, NDPK and CK2 can both use ATP or GTP as substrates whereas most kinases such as PKA and AMPK only use ATP.
CK2 is intrinsically complex as it may exist either as free catalytic alpha subunits or as a composite holoenzyme made from two alpha and two beta subunits (head and body, respectively). Indeed, the incredible versatility in CK2 targeting so many different substrates (Meggio and Pinna 2003) partly depends on its unusual assembly into two back-to-back catalytic subunit heads balanced on a beta body pair cemented into place only after subsequent beta subunit phosphorylation to create a stable heterotetramer. Thus, CK2 is aptly named with the potential to target two substrates simultaneously (see “Appendix”).
Although CK2 substrates number in their hundreds and possibly thousands, they can be classified to a degree, dependent on the structural form of CK2 they recognise. Some substrates such as heat shock proteins (such as HSP70 and HSP90, known CFTR interactors) are readily phosphorylated equally well by the alpha head (i.e. when no dimeric body is present) or the tetrameric holoenzyme. These are designated as type 1 substrates. Other substrates such as the calcium sensor calmodulin are subtly different because, when they encounter the CK2 holoenzyme, they do not immediately become phosphorylated. Instead, on binding, their phosphorylation is initially suppressed until another moiety (binding remotely on CK2) relieves this unusual substrate binding-induced catalytic inhibition (importantly, this ‘relief of inhibition’ phenomenon also occurs for the CFTR peptides corresponding to the F508 region, as described below). And yet these type 2 substrates are readily phosphorylated by the alpha subunits alone just like type one substrates. In contrast, type 3 substrates such as initiation factors for protein elongation during ribosomal synthesis are highly selective, never becoming phosphorylated by the catalytic head alone, only recognising the assembled heterodimeric CK2 structure because they need to bind to a site on the beta subunit to enable their access to one or other catalytic head. Finally, there exists a further complexity with respect to the fully assembled nature of the holoenzyme. The CK2 heterodimer itself becomes ‘auto’ phosphorylated at the N terminus of each beta subunit by a different CK2 molecule. This trans-phosphorylation is considered as a useful marker of completed holoenzyme assembly and was unexpectedly modulated when CK2 holoenzyme and a purified domain of CFTR-NBD1 were exposed to peptides derived from the F508del region of CFTR (PGTIKENIIF508GVSYDEYRYR). Because this beta subunit ‘auto’ phosphorylation changed so dramatically when we varied the sequence of this 20-mer peptide on either side of F508 for example, the data implied that CK2 might be controlled by peptides derived from that mutated domain within CFTR. This domain is conserved in the C group of ABC proteins. Our conjecture predicted that CK2 might control CFTR through the site of a CFTR mutation that generates an unexplained, multi-system CF disease.
What CK2 does to CFTR channel function
The alpha catalytic CK2 subunit has a smaller pocket near to its ATP/GTP binding site compared to most protein kinases because pocket size is reduced by two bulky hydrophobic amino acids. This structural difference has permitted the synthesis of relatively specific inhibitors. During ion transport studies, we exposed one of these cell-permeable inhibitors to cells expressing CFTR. First, with a patch clamp pipette in the ‘cell attached’ mode that does not perforate the cell membrane thereby minimally disturbing the (normal) intra-membrane organisation, David Sheppard and colleagues (Bristol, UK) opened CFTR pharmacologically with a cocktail of cell-permeant PKA activating stimuli that elevate cyclic AMP (Treharne et al. 2009d). When CK2 was subsequently inhibited, this nascently opened CFTR closed promptly within 80 s despite an ongoing PKA stimulus that normally keeps the channel open. This requirement for CK2 to be active to maintain an open PKA-activated CFTR was confirmed by Karl Kunzelmann (Regensburg, Germany) using CFTR over-expressed in oocytes (Treharne et al. 2009d) but complete CFTR closure post-CK2 inhibition was not seen in these frog eggs (∼20% residual wild-type channel activity was retained). Yet, in both cells and oocytes, pharmacological pre-inhibition of CK2 prevented the subsequent opening of CFTR, despite PKA activation. Control experiments, using oocytes over-expressing a mutant CK2 that was engineered by Lorenzo Pinna (Padua, Italy) to be kinase active but inhibitor insensitive (by mutating the two bulky amino acids in the pocket mentioned above), showed that PKA-activated CFTR was now insensitive to closure with the very same inhibitor of CK2. This proved that CFTR’s ability to conduct ions is largely CK2-dependent whilst confirming that the inhibitor had no effect on PKA or other unknown CFTR activating proteins in the normal CFTR-organised hub. Thus, the combined data suggest that when CFTR is stimulated by PKA, presumably within its intact membrane hub, active CK2 is not only required to maintain CFTR in its maximally open (PKA-driven) state but CK2 activity is also permissive for PKA action either to open (or keep open) CFTR.
Next, the requirement for CFTR to be nested within its hub of membrane-organised proteins was tested. The very same CFTR-containing patch bearing the recently CK2 regulated CFTR was re-studied in an excised inside out patch clamp configuration that cuts out the patched membrane for study by exposing its cytosolic face to the bath solution. Interestingly, this membrane fracture from the mother cell immediately closes CFTR requiring a re-activating cocktail of ATP and PKA to be added to the bath solution. Under these conditions, pharmacological CK2 inhibition that had recently shut CFTR in the intact cell could no longer close CFTR despite higher doses of CK2 inhibitor (Treharne et al. 2009d). This is consistent with the idea that either the normal intra-membrane organisation, fractured by patch excision (fracture-induced variant of the LIMBO idea but with wild-type CFTR present), was vital for CK2 to act on CFTR or that fracture-induced LIMBO displaced CK2 from wild-type CFTR.
The F508del CFTR point mutant is a temperature-sensitive allele that is not completely degraded at room temperature. Thus, oocytes have the advantage over mammalian cells that when F508del CFTR is over-expressed, it does not degrade due to the lower temperature of the amphibian egg. Curiously, F508del CFTR only manifests about 20% of wild-type channel activity in response to a maximal PKA stimulus. We wondered if this residual activity would be CK2 dependent but found that the PKA-activated F508del CFTR was completely CK2-inhibitor insensitive. Thus, we are left with the speculation that the missing 80% of PKA-driven wild-type CFTR opening that is absent in oocytes expressing the F508del mutant is the CK2-dependent part. However, we cannot exclude the possibility that this mutant form of CFTR has no bound CK2 in the vicinity of its different mutant hub of proteins as we had observed in human airway biopsies (Treharne et al. 2009d). Next, we switched our focus to the reciprocal actions of CFTR on CK2, i.e. we sought the aberrant signal generator.
What the F508 region of CFTR does to CK2 activity
Prior to these studies, we had noticed that in the immediate vicinity of the commonly mutated F508 in CFTR lay serine 511 in a potential CK2 consensus sequence just C-terminal to F508 (PGTIKENIIF508GVS 511 YDEYRYR). CK2 uses the proximity of negatively charged amino acids such as aspartate (D) and glutamate (E) in the example shown to locate its target serine (or threonine) residues for phosphorylation. To our surprise, in separate experiments, we had found that 20-mer peptides made from the region straddling this pathogenic F508 residue in CFTR were able to interfere with the ion channel activity of CFTR in a F508-dependent manner (Treharne et al. 2009d). We sought to explain how 20-mer peptides made from CFTR could inhibit CFTR itself given that these CFTR-derived peptides were also able to displace apically localised IgG in airway epithelial biopsies (Treharne et al. 2009b), consistent with a link to the immune system. Hence, we were fully expecting the above peptides to be phosphorylated by CK2 but, despite an excellent predicted consensus at S511YDE, we could never find any stoichiometric phosphate incorporation whether F508 was present or not. Paradoxically, we observed quite the reverse where this CFTR peptide appeared to be a robust inhibitor of the catalytic alpha subunit of CK2 in vitro, especially when F508 was deleted. These unexpected findings made us consider the idea that such CFTR peptide fragments, as might be generated when CFTR was degraded by the proteasome for example, might differ in composition and abundance post degradation of wild-type and F508del CFTR. In turn, this could explain how CFTR mutation disrupts so many apparently unrelated processes ranging from ion transport to immune dysfunction. We noted that CK2 controls a diverse array of substrates and looked for a new ‘knock-in’ CK2 signal that was generated by the F508del mutant given that the wild-type CFTR was controlled by such peptides and that the literature also contains many examples of the roles of CK2 in immunity and inflammation (Singh and Ramji 2008). We reasoned that for this peptide control to occur, they must first bind to CK2 and demonstrated a high affinity, F508-dependent interaction between the 20-mer CFTR peptides and both CK2 subunits using surface plasmon resonance (Pagano et al. 2008). Thus, both CK2 subunits could bind to CFTR peptides straddling the F508 site, but loss of F508 did not alter the affinity of binding to a significant degree. This made us consider the functional effects of these peptides towards CK2.
Our principal hypothesis was that degraded CFTR, whether wild-type or F508 mutant, generates peptide fragments that could feedback to differentially control CK2. Should this be correct, then these 20-mer peptides should modulate CK2 substrate phosphorylation in an F508-dependent manner. By applying 20-mer peptides from the F508 region of CFTR to CK2 incubated with its classical substrates in vitro, we demonstrated that CK2 activity against different class-specific substrates can be differentially controlled in a F508-dependent manner (Pagano et al. 2008, 2010). We found that these two peptides acted with significantly greater potency against CK2 when the F508 was deleted, regulating different classes of CK2 substrate in different directions. Thus, one peptide could create more than one signal just by losing F508. For example, the very same F508-deleted peptide that potently inhibited the in vitro catalytic activity of the isolated CK2 alpha catalytic subunit (in the absence of the beta subunit, see above) towards class 1 and 2 substrates (chaperones and the cell calcium binding protein, calmodulin as respective examples) nevertheless stimulated CK2 holoenzyme activity by 2–5-fold towards the very same substrates in vitro. We interpreted this bipartite peptide effect to suggest that the activity of any free CK2 catalytic subunit is downregulated whilst the holoenzyme is stimulated, thus fine tuning the balance of CK2 control. The potent inhibitory effect of this CFTR-derived peptide on the free alpha subunit also involved certain key residues on either side of the F508 site near S511, such that the combination of F508, V510 and the double isoleucine motif at I506/I507 were all critical determinants of efficacy, creating a rank order of inhibitory potential towards the alpha subunit. Interestingly, these are the very amino acids that are thought to lie differently oriented in the NBD1 domain structure when F508 is deleted. Next, we modelled the site of the alpha CK2 subunit–CFTR peptide interaction by kinetic analysis and demonstrated that CFTR peptide induced alpha CK2 inhibition was non-competitive against classical alpha CK2 substrates suggestive of allosteric inhibition. Yet this was complex allosteric control involving ATP concentration as co-substrate because we observed sharply bent slopes of the double reciprocal plots only when the ATP co-substrate was present at high micromolar to millimolar concentrations (as might be found in cell or, perhaps, locally enhanced by NDPK for example). In sharp contrast, when we investigated the stimulatory effect of the CFTR peptides on the CK2 holoenzyme, we found that the very same CFTR-derived peptides were now behaving like certain intensely positively charged small molecules known to relieve the inhibition normally observed on initial contact between the holoenzyme and type 2 substrates. This unexpected finding predicted that the small molecules normally used to relieve class 2 substrate inhibition in vitro such as polylysine would have their potency significantly reduced by direct competition with the CFTR peptides and indeed this was observed as predicted (Pagano et al. 2010). Thus, we had the unexpected conclusion that a constitutively active kinase, CK2 that is generally regarded as not being controlled by endogenous substrates, could nevertheless be regulated, in vitro by a potential naturally occurring peptide. Next, we demonstrated that when the whole domain of NBD1 was added to holoenzyme, NBD1 itself behaved like a type 2 substrate with inhibited phosphorylation relieved by either polylysine or the above F508del CFTR peptides.
To demonstrate that our conjecture of a single amino acid deletion in a channel that controls class-specific functions of a pleiotropic kinase can have meaning in a cellular environment, we now show some examples that take our current in vitro observations into an established cystic fibrosis model system.
Materials
All analytical grade reagents were from Sigma-Aldrich (St. Louis, MO, USA) and from Bio-Rad (Hercules, CA, USA). Monoclonal antibodies against CFTR were from the CFTR Folding Consortium (www.cftrfolding.org). Anti-CK2 beta subunit antibody was from Epitomics (Burlingame, CA, USA) whereas CK2 alpha subunit antiserum was raised in rabbit against the sequence of the human protein at C terminus (a.a. 376–391). All other antibodies were from Sigma and Perkin Elmer (Waltham, MA, USA). [γ-33P]GTP (3,000 Ci/mmol) was purchased from Hartmann Analytic GmbH (Braunschweig, Germany). Quinalizarin (>99% analytically pure) was provided by Produits Chimiques ACP Chemicals (Montreal, QC, Canada).
Cell cultures
C127 breast cancer cells engineerized to express either wild-type or F508del CFTR were grown in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% foetal bovine serum (FBS), 2 mM l-glutamine, penicillin (100 U/ml), streptomycin (0.1 mg/ml) and geneticin (200 μg/ml). Baby hamster kidney (BHK) cell lines transfected with either the empty vector or with CFTR, either wild-type or the F508 deletion mutant, in addition to glutamine and antibiotics at the concentrations reported for C127, required methotrexate (500 μM) to select stable transfectants in DMEM/F12 medium plus 5% FBS. All cells were grown at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Phosphorylation assays
BHK cells were cultured to reach 70–80% confluency and then lyzed in a buffer containing 50 mM Tris, 150 mM NaCl, 0.5% (v/v) Triton X-100, 2 mM DTT, 1 mM EDTA, 1 mM EGTA with a cocktail of anti-proteases (Roche, Mannheim, Germany) and anti-phosphatases (Sigma). After protein quantitation, lysates corresponding to 130 μg were incubated with 0.1 mM GTP/[γ-33P]GTP (1,000 cpm/pmol), 12 mM MgCl2, 0.1 M NaCl, 1 mM DTT with anti-protease and anti-phosphatase cocktails and in the absence or presence of 2 μM quinalizarin, a specific ATP-site-directed CK2 inhibitor (Cozza et al. 2009). After 20 min incubation at 37°C, the reaction was stopped with 20 mM EDTA.
Two-dimensional gel electrophoresis
All the samples were ultracentrifuged (Beckman MLA-130 rotor) at 100,000×g at 4°C for 30 min. The supernatants were collected and precipitated with 80% ice-cold acidic acetone (1 mM HCl) and kept overnight at −80°C. The precipitate was spun down at 13,000×g, 4°C for 30 min, the supernatant was removed and the pellet was air dried. The pellet was then resuspended in a buffer containing 7 M urea, 2 M thiourea, 3% (v/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 1% (v/v) ampholyte (3–10), 0.5% ASB-14 and 40 mM Tris. Samples were reduced with 65 mM DTT for 1 h and then alkylated by 1 h’s treatment with 60 mM acrylamide. Isoelectrofocusing was performed on ReadyStrip IPG strips, 3-10NL (Bio-Rad). For the second dimension, the strips were equilibrated in 375 mM Tris (pH 8.8), 6 M urea, 20% glycerol (v/v) and 2% sodium dodecyl sulphate (SDS; v/v) for 30 min. The strips were then run on 11.5% Tris–glycine gels. Radioactive spots were imaged by a Storm PhosphorImager system (Molecular Dynamics) and analysed by OptiQuant software (Perkin Elmer).
Western blotting
Thirty micrograms of proteins from cell lysates were resolved by SDS–polyacrylamide gel electrophoresis and transferred onto Immobilon-P membranes (Millipore, Billerica, MA, USA). Membranes were probed with the indicated antibody and developed by enhanced chemiluminescence (Amersham Biosciences). The signal was quantitated by chemiluminescence on Kodak Image Station 440cf (Eastman Kodak Co., New Haven, CT, USA) and analysed by the Kodak 1D image software. Spot intensities were compared by analysis of variance using data from three independent sets of cell samples.
Results
In Fig. 2, we show that in lysates from BHK cells there does not appear to be a significant change in CK2 subunit(s) abundance when these cells are stably transfected with either wild-type or F508del CFTR (BHK cells, gift of C. Farinha and M. Amaral, Lisbon). Nevertheless, when an equal protein concentration from each lysate is radiolabelled in the presence of [γ-33P]GTP, a phosphodonor substrate specific for CK2, and the radiolabelled proteins are resolved by 2D gel electrophoresis, significant differences in the phosphorylation of many proteins become evident when the phosphoproteomes of wild-type and F508del cells are compared (Fig. 3a, compare panels a and b). Notably, when compared to wild-type lysates, the relative excess phosphorylation of all spots detectable in panel b (F508del cell lysate) is drastically reduced by the CK2 specific inhibitor quinalizarin (compare panels b and c), thus providing an additional argument, besides usage of GTP as phosphate donor, to ascribe to endogenous CK2 most of the phosphorylating activity detected.

Protein level of CFTR and CK2 in different wild-type (wt) and F508del (ΔF) CFTR transfected cell lines. a Western blot analysis performed with anti-CFTR antibody on BHK and C127 cells transfected with wild-type and F508del CFTR. For BHK cells, the pattern of transfection with the empty vector (Mock) is also present. b Western blot analysis on BHK cells transfected with wt and F508del CFTR performed with anti-CFTR, anti-alpha CK2 (CK2α) subunit and anti-beta CK2 (CK2β) subunit antibodies. The protein levels of CK2 in wild-type and F508del CFTR transfected BHK cells are comparable. Actin was used as a loading control in all panels

a GTP-dependent phosphorylation pattern differs in lysates from BHK cells transfected with wild-type (wt) and F508del (ΔF508) CFTR. Representative autoradiographies of three independent experiments are shown. All experiments were conducted in the presence of GTP/[γ-33P]GTP as a phosphate donor and, where indicated, 2 μM quinalizarin, a specific CK2 inhibitor (Cozza et al. 2009), was applied to establish the contribution of CK2 to the phosphorylation pattern of BHK cells. Similar results were obtained with lysates of C127 cells transfected with wild-type and F508del CFTR (not shown). To quantitate the radioactivity incorporated, 18 radiolabelled areas as indicated in b were defined and analysed by OptiQuant software. Quantitative analysis is presented in Table 1
To quantify differences in phosphorylation, 18 radiolabelled areas, as indicated in Fig. 3b, were circumscribed and analysed by OptiQuant software. From four independent experiments, these preliminary data show that the overwhelming majority of radioactive spots were significantly more intense in the F508del cell lysates when compared to wild type (p < 0.05, paired t test with Bonferroni correction, n = 4), whilst a small minority were significantly less intense (typically by ∼30%, not shown). The phosphorylation of all areas was markedly reduced, albeit to variable extent per spot (Table 1), by the CK2 inhibitor quinalizarin, thus confirming in a quantitative manner the major implication of dysfunctional endogenous CK2 in the generation of most radiolabelled proteins in an F508del CFTR context. Thus, our preliminary analysis suggests that spots whose phosphorylation is dramatically enhanced by F508del CFTR expression are largely suppressed by quinalizarin, as might be expected for class II CK2 targets. The excess phosphorylation of class II targets such as calmodulin by CK2 holoenzyme has previously been shown to be selectively enhanced by F508del CFTR fragments in vitro (Pagano et al. 2010). Also remarkable is the ladder of spots included in area 4, whose behaviour is reminiscent of variably multi-phosphorylated proteins. Once again phosphoradiolabelling in this area is enhanced in F508del cells whilst becoming drastically inhibited by quinalizarin. In sharp contrast, a minority of spots appear to be significantly less enhanced in the F508del lysates and yet are likely to be targets of CK2 as judged from the inhibitory efficacy of quinalizarin. We are currently improving the resolution of the 2D gels to determine whether these spots, when better separated from their neighbours, represent CK2 substrates belonging to class III, whose phosphorylation is exceptionally inhibited rather than stimulated by F508del CFTR peptides (Pagano et al. 2010). In summary, the scenario disclosed by these experiments exploiting BHK cells is consistent with our earlier notion that in vitro, CK2 holoenzyme is susceptible to a dual control by synthetic fragments of F508del CFTR, resulting in variable and in some cases opposite effects on the phosphorylation of its targets (Pagano et al. 2008, 2010).
Discussion
Here, we have attempted to account for the heterozygote advantage from the commonest (by far) CFTR mutant by suggesting that the deletion of F508 alters the regulation of protein kinase CK2 in more than one direction. Our model shown in Fig. 4 suggests that altered CK2 regulation induced by F508del CFTR could occur irrespective of the presence of a wild-type copy of CFTR (compare the top and bottom of the model shown in Fig. 4). Thus, combining Figs. 1, 3 and 4, it might be imagined that both types of spot patterns may be present in a heterozygote F508del CFTR carrier. The higher intensity of F508del CFTR-induced labelling could act as an amplifier signal because CK2 has hundreds of targets belonging to three different classes—respectively, chaperones, signal transducers and regulators of growth in corresponding classes 1, 2 and 3. In this respect, a 20-mer peptide with the F508 deleted that was itself not a bona fide CK2 target nevertheless either downregulated or upregulated each of these different target classes in vitro depending on the structural form of CK2 they encounter. Because we find that the effect of F508del 20-mer peptide on a given substrate in vitro is between 2- and 20-fold stronger than on the wild type, it is our view that we have found a different path to CF disease. To set our findings into a normal intra-membrane network context for CFTR, in Fig. 1 we have also highlighted the potential overlap between four proteins, CFTR, CK2, NDPK and its membrane-bound binding partner, AMPK, each of which induces multiply deranged cellular defects when mutated ranging from multi-system disease (CFTR and NDPK) through cancer (all four proteins) to defects in inflammation (AMPK, CK2), viral invasion (CK2, NDPK) and the host–pathogen interaction with bacteria involving purinergic receptors (NDPK, CFTR). We note that an independent consortium (Charro et al. 2009) recently confirmed our ideas, first reported more than a decade ago, that NDPK is involved in CF pathogenesis by their invoking NDPK as a CF biomarker. Finally, we have taken our model into the cell where we observe significant F508del-induced differences in CK2-dependent phosphorylation. Furthermore, our unpublished data show that when the two-dimensional phosphoproteome equivalent to that shown in Fig. 3 is compared in another wild-type and mutant CFTR system using C127 cells (Fig. 2), then we once again observe the differences in CK2 signalling (unpublished data, cells were a gift of G. Cabrini, Verona, Italy). This provides cell-independent validation of our ideas.

A model for the effects of CFTR peptides on CK2. The proteasome does not completely degrade proteins to amino acids but instead produces short peptides. The model suggests that normal intra-membrane organisation (NIMBO; see text) is promoted by wild-type (wt) CFTR peptides (products of the upper part of figure). When CFTR is deleted, this signal is missing (LIMBO, see text). When F508del (ΔF508) CFTR is present in a heterozygote, the upper NIMBO signal is present but the signal shown in the lower figure is newly ‘knocked-in’. In F508del CFTR homozygotes, the NIMBO signal does not exist and the text describes those substrates for CK2 that might be mis-regulated
In conclusion, given that firstly, CK2 is thought to phosphorylate and inhibit NDPK in vitro at or near serine-120, adjacent to the catalytic site on NDPK at histidine-118; secondly, that membrane-bound NDPK and AMPK interact; thirdly, that AMPK binds and controls CFTR and fourthly, that NDPK both contains a similar motif to that surrounding F508 in CFTR, near to its own catalytic site and may be a biomarker in CF serum, we now have a model of the normal CFTR intra-membrane organisation (NIMBO) hub (left panel in Fig. 1) that is amenable to dissection and refinement. The predicted effects of this model (summarised in Fig. 4) will help us to bridge the current gap in understanding in CF pathophysiology by no longer exclusively focussing on what kinases do to CFTR, but in parallel asking what CFTR does to key CFTR-local kinases, thus rendering meaningful the final R (‘regulator’) of this CFTR acronym which to date makes little mechanistic sense.
Abbreviations
- ABC:
-
ATP-binding cassette
- AMPK:
-
AMP-activated kinase
- BHK:
-
Baby hamster kidney
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- CK2:
-
Protein kinase CK2 formerly casein kinase 2
- DMEM:
-
Dulbecco’s modified eagle’s medium
- ENaC:
-
Epithelial sodium (Na) channel
- FBS:
-
Foetal bovine serum
- LIMBO:
-
Loss of intra-membrane organisation
- NBD(1 and 2):
-
Nucleotide binding domain
- NDPK:
-
Nucleoside diphosphate kinase
- NIMBO:
-
Normal intra-membrane organisation
- PKA:
-
Cyclic AMP-activated protein kinase A
References
Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordonez CL, Campbell PW, Ashlock MA, Ramsey BW (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363:1991–2003
Aleksandrov AA, Aleksandrov LA, Riordan JR (2007) CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflugers Arch 453:693–702
Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, des Georges M, Stanke F, De Boeck K, Dupont L, Balascakova M, Hjelte L, Lebecque P, Radojkovic D, Castellani C, Schwartz M, Stuhrmann M, Schwarz M, Skalicka V, de Monestrol I, Girodon E, Ferec C, Claustres M, Tummler B, Cassiman JJ, Korbmacher C, Cuppens H (2009) Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum Mutat 30:1093–1103
Bachhuber T, Almaca J, Aldehni F, Mehta A, Amaral MD, Schreiber R, Kunzelmann K (2008) Regulation of the epithelial Na+ channel by the protein kinase CK2. J Biol Chem 283:13225–13232
Biondi RM, Engel M, Sauane M, Welter C, Issinger OG, Jimenez de Asua L, Passeron S (1996) Inhibition of nucleoside diphosphate kinase activity by in vitro phosphorylation by protein kinase CK2. Differential phosphorylation of NDP kinases in HeLa cells in culture. FEBS Lett 399:183–187
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat 19:575–606
Boucher RC (2004) New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23:146–158
Briel M, Greger R, Kunzelmann K (1998) Cl− transport by cystic fibrosis transmembrane conductance regulator (CFTR) contributes to the inhibition of epithelial Na+ channels (ENaCs) in Xenopus oocytes co-expressing CFTR and ENaC. J Physiol 508(Pt 3):825–836
Charro N, Hood BL, Faria D, Pacheco P, Azevedo P, Lopes C, de Almeida AB, Couto FM, Conrads TP, Penque D (2009) Serum proteomics signature of cystic fibrosis patients: a complementary 2-DE and LC-MS/MS approach. J Proteomics 74:110–126
Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ (2010) Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143:911–923
Collawn JF, Fu L, Bebok Z (2010) Targets for cystic fibrosis therapy: proteomic analysis and correction of mutant cystic fibrosis transmembrane conductance regulator. Expert Rev Proteomics 7:495–506
Comer DM, Ennis M, McDowell C, Beattie D, Rendall J, Hall V, Elborn JS (2009) Clinical phenotype of cystic fibrosis patients with the G551D mutation. QJM 102:793–798
Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M (2007) The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett 581:271–278
Cozza G, Mazzorana M, Papinutto E, Bain J, Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene M, Battistutta R, Meggio F, Moro S, Zagotto G, Pinna LA (2009) Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem J 421:387–395
Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, Gallan AJ, Shevchenko PD, Bindokas VP, Nelson DJ (2009) Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem 284:35926–35938
Gentzsch M, Dang H, Dang Y, Garcia-Caballero A, Suchindran H, Boucher RC, Stutts MJ (2009) The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J Biol Chem 285:32227–32232
Hunter MJ, Treharne KJ, Winter AK, Cassidy DM, Land S, Mehta A (2010) Expression of wild type CFTR suppresses NF-kappaB-driven inflammatory signalling. PLoS ONE 5:e11598
Joo NS, Cho HJ, Khansaheb M, Wine JJ (2010) Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest 120:3161–3166
King JD Jr, Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, Hallows KR (2009) AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol 297:C94–C101
Kongsuphol P, Cassidy D, Hieke B, Treharne KJ, Schreiber R, Mehta A, Kunzelmann K (2009a) Mechanistic insight into control of CFTR by AMPK. J Biol Chem 284:5645–5653
Kongsuphol P, Hieke B, Ousingsawat J, Almaca J, Viollet B, Schreiber R, Kunzelmann K (2009b) Regulation of Cl(−) secretion by AMPK in vivo. Pflugers Arch 457:1071–1078
Kunzelmann K (2001) CFTR: interacting with everything? News Physiol Sci 16:167–170
Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF, Emtage S (2005) Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem 280:1346–1353
McCormick J, Green MW, Mehta G, Culross F, Mehta A (2002) Demographics of the UK cystic fibrosis population: implications for neonatal screening. Eur J Hum Genet 10:583–590
McCormick J, Mehta G, Olesen HV, Viviani L, Macek M Jr, Mehta A (2010) Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet 375:1007–1013
Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17:349–368
Mehta A, Orchard S (2009) Nucleoside diphosphate kinase (NDPK, NM23, AWD): recent regulatory advances in endocytosis, metastasis, psoriasis, insulin release, fetal erythroid lineage and heart failure; translational medicine exemplified. Mol Cell Biochem 329:3–15
Mekus F, Ballmann M, Bronsveld I, Bijman J, Veeze H, Tummler B (2000) Categories of deltaF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res 3:277–293
Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Gimenez J, Reis A, Varon-Mateeva R, Macek M Jr, Kalaydjieva L et al (1994) The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat Genet 7:169–175
Muchekehu RW, Quinton PM (2010) A new role for bicarbonate secretion in cervico-uterine mucus release. J Physiol 588:2329–2342
Ollero M, Brouillard F, Edelman A (2006) Cystic fibrosis enters the proteomics scene: new answers to old questions. Proteomics 6:4084–4099
Pagano MA, Arrigoni G, Marin O, Sarno S, Meggio F, Treharne KJ, Mehta A, Pinna LA (2008) Modulation of protein kinase CK2 activity by fragments of CFTR encompassing F508 may reflect functional links with cystic fibrosis pathogenesis. Biochemistry 47:7925–7936
Pagano MA, Marin O, Cozza G, Sarno S, Meggio F, Treharne KJ, Mehta A, Pinna LA (2010) Cystic fibrosis transmembrane regulator fragments with the Phe508 deletion exert a dual allosteric control over the master kinase CK2. Biochem J 426:19–29
Pier GB, Grout M, Zaidi TS, Goldberg JB (1996) How mutant CFTR may contribute to Pseudomonas aeruginosa infection in cystic fibrosis. Am J Respir Crit Care Med 154:S175–S182
Pier GB, Grout M, Zaidi TS (1997) Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci U S A 94:12088–12093
Pollard HB, Eidelman O, Jozwik C, Huang W, Srivastava M, Ji XD, McGowan B, Norris CF, Todo T, Darling T, Mogayzel PJ, Zeitlin PL, Wright J, Guggino WB, Metcalf E, Driscoll WJ, Mueller G, Paweletz C, Jacobowitz DM (2006) De novo biosynthetic profiling of high abundance proteins in cystic fibrosis lung epithelial cells. Mol Cell Proteomics 5:1628–1637
Punj V, Zaborina O, Dhiman N, Falzari K, Bagdasarian M, Chakrabarty AM (2000) Phagocytic cell killing mediated by secreted cytotoxic factors of Vibrio cholerae. Infect Immun 68:4930–4937
Quinton PM (2010) Birth of mucus. Am J Physiol Lung Cell Mol Physiol 298:L13–L14
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL et al (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073
Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL (2010) Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 182:1262–1272
Sheppard DN, Welsh MJ (1999) Structure and function of the CFTR chloride channel. Physiol Rev 79:S23–S45
Singh NN, Ramji DP (2008) Protein kinase CK2, an important regulator of the inflammatory response? J Mol Med 86:887–897
Treharne KJ, Marshall LJ, Mehta A (1994) A novel chloride-dependent GTP-utilizing protein kinase in plasma membranes from human respiratory epithelium. Am J Physiol 267:L592–L601
Treharne KJ, Best OG, Mehta A (2009a) The phosphorylation status of membrane-bound nucleoside diphosphate kinase in epithelia and the role of AMP. Mol Cell Biochem 329:107–114
Treharne KJ, Cassidy D, Goddard C, Colledge WH, Cassidy A, Mehta A (2009b) Epithelial IgG and its relationship to the loss of F508 in the common mutant form of the cystic fibrosis transmembrane conductance regulator. FEBS Lett 583:2493–2499
Treharne KJ, Giles Best O, Mehta A (2009c) Transglutaminase 2 and nucleoside diphosphate kinase activity are correlated in epithelial membranes and are abnormal in cystic fibrosis. FEBS Lett 593:2789–2792
Treharne KJ, Xu Z, Chen JH, Best OG, Cassidy DM, Gruenert DC, Hegyi P, Gray MA, Sheppard DN, Kunzelmann K, Mehta A (2009d) Inhibition of protein kinase CK2 closes the CFTR Cl channel, but has no effect on the cystic fibrosis mutant deltaF508-CFTR. Cell Physiol Biochem 24:347–360
Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR 3rd, Balch WE (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127:803–815
Yilmaz O, Yao L, Maeda K, Rose TM, Lewis EL, Duman M, Lamont RJ, Ojcius DM (2008) ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol 10:863–875
Acknowledgements
We apologise to the many authors we could not site due to space limitations. We thank the Wellcome Trust (grant numbers 088929 to L.A.P. with A. M. and 069150 to A.M.) and the Fondazione per la Ricerca sulla Fibrosi Cistica (grant #4/2009 adopted by Loifur S.r.l. e Amici per la ricerca 09) and Associazione Italiana per la Ricerca sul Cancro. This work would not have been possible without the generosity of cell line and reagent provision from Margarida Amaral, Carlos Farinha and Giulio Cabrini.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
The problem of too much or too little airway surface fluid
Space limitations preclude a comprehensive discussion of the synthesis, trafficking, post-translational modification, residence time and destruction rates of apical membrane proteins that act as channels in apical membranes of epithelia. Here, we focus on the regulation of CFTR and ENaC that act in concert in the apical membrane facing the outside of epithelia as follows: Like the colon, the airway epithelium is set up to suck liquid and electrolytes from the lumen to avoid surface flooding. In reverse, in order to first wet the outer surface of the airway epithelium, kinase-stimulated CFTR lies amongst those anion channels that secrete negatively charged species such as bicarbonate and chloride out into the apical lining fluid, but can only act (outwardly) vectorially provided ENaC’s concurrent and inwardly directed sodium movement that drives fluid suction is simultaneously turned off. Otherwise, the inward (membrane-depolarising) driving forces produced by (unrestrained) sodium ion entry into the epithelial cell will not permit anion secretion out of the very same cell membrane where sodium is entering. Let us assume sodium entry towards the very low intracellular sodium concentration environment (∼10 mM) is somehow regulated, but in that instance, this CFTR-regulated secretory process must retain enough dynamic flexibility to turn ENaC on again should CFTR-driven secretion overshoot, i.e. feedback is essential in order to re-absorb any excess secreted fluid, thereby preventing surface flooding and consequent impairment of airway function (or in the colon, diarrhoea). Once flooding is threatened, this in turn now requires CFTR and other anion channels to act quickly in reverse mode to help re-absorb newly secreted surface anions. To control this loop locally, we thought that a constitutively active but regulated controller of ENaC and CFTR would be an essential first step.
CK2 controls ENaC (Bachhuber et al. 2008). Briefly, Karl Kunzelmann and colleagues (Regensburg, Germany) found that CK2 trafficked to the oocyte cell membrane only when both phosphorylation sites for CK2 on the beta and gamma subunits of ENaC were intact that is the normal situation when wild-type ENaC is expressed. When both CK2 sites were mutated to non-phosphorylatable alanines and despite there being abundant CK2 in the bulk of the oocyte, no CK2 fluorescence of the catalytic subunit could be found in the oocyte membrane, which contrasted with findings after wild-type ENaC expression, where abundant CK2 membrane fluorescence was observed. Under these doubly CK2 target mutated conditions, ENaC channel activity was almost nil and yet the channel itself trafficked normally to the membrane, just like wild type, but now without the CK2 ‘cargo’ found associated with wild-type ENaC. This suggests that as for CFTR, CK2 appears to be essential for channel function. Thus, we find a pleiotropic kinase is essential for both ENaC and CFTR to function as channels. Since ENaC and CFTR are thought to lie in close proximity to one another, our new conjecture is that the recognised physiological requirement to switch off ENaC when the cell is secreting and then rapidly switch it back on again when the cell is absorbing fluid make the different cellular forms of CK2 (doubly catalytic, heterotetrameric or singly catalytic CK2 alone) that target substrates differentially an attractive proposition to study given that CK2 can also use both ATP and GTP thus adding signal pleiotropy to the model. Separately, we found that in airway cells expressing F508del CFTR, CK2 was missing from the apical membrane (Treharne et al. 2009d) and previously we have speculated on the chloride and sodium sensitivity of membrane-localised NDPK that was lost upon NDPK purification (see references therein). We imagine a normal apical cell membrane: one head of CK2 targeting CFTR (NBD1 as a class 2 target, say) whilst the other targets ENaC (different substrate class) or even NDPK, such that one target might be on and the other off, depending on the nature of the CK2 signal. Note also the possibility of NDPK acting to flux-balance ATP, GTP, ADP and GDP near the membrane through substrate channelling. We then imagine a F508del CFTR bearing heterozygote cell where a new knock-in differential signal might arise from a F508del CFTR-derived peptide(s) creating an enhanced potency of CK2 signals to chaperones, calcium signalling through calmodulin and the protein synthetic machinery for example, a new signal that is many fold above that generated by the wild-type CFTR protein as it degrades (Fig. 4). But of course, there are many other possible ‘wiring diagrams’ that might include bi-directional effects of CFTR on AMPK for example, and the final model will have to incorporate NDPK as a CK2 target and AMPK as an NDPK binding protein, together acting as complex interacting elements within the NIMBO hub (see main text for definition). Note that the product of cAMP destruction is AMP, the very molecule that further controls the baseline activity of AMPK. At the very least, Fig. 3 demonstrates for the first time that such a scenario is at least theoretically possible in cell. Indeed, this project now comes back full circle to the apical airway of the CF patient. These human studies originated in 1991 when Mehta and colleagues began to analyse the constitutive phosphorylation patterns they found in lysates made from apically enriched human airway epithelial biopsies (Treharne et al. 1994). At that time, differences in phosphorylation pattern were repeatedly observed between CF patient biopsies and controls (Treharne and Mehta, unpublished), but the presence of excessive inflammation in CF patient airways made the data difficult to interpret. In control apical membranes from uninflamed volunteers, constitutive, ATP- and GTP-dependent membrane-delimited phosphorylation involving NDPK, sodium and anion concentrations was observed. In conclusion, we believe that our CK2 hub hypothesis now has a strong evidence base for further study given that we have evidence that ranges from human data to model systems lying on the path from CFTR mutation to disease pathogenesis.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Venerando, A., Pagano, M.A., Tosoni, K. et al. Understanding protein kinase CK2 mis-regulation upon F508del CFTR expression. Naunyn-Schmiedeberg's Arch Pharmacol 384, 473–488 (2011). https://doi.org/10.1007/s00210-011-0650-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-011-0650-x