
Abstract
Reliable in vitro models to assess developmental toxicity of drugs and chemicals would lead to improvement in fetal safety and a reduced cost of drug development. The validated embryonic stem cell test (EST) uses cardiac differentiation of mouse embryonic stem cells (mESCs) to predict in vivo developmental toxicity, but does not take into account the stage-specific patterning of progenitor populations into anterior (ventricular) and posterior (atrial) compartments. In this study, we generated a novel dual reporter mESC line with fluorescent reporters under the control of anterior and posterior cardiac promoters. Reporter expression was observed in nascent compartments in transgenic mouse embryos, and mESCs were used to develop differentiation assays in which chemical modulators of Wnt (XAV939: 3, 10 µM), retinoic acid (all-trans retinoic acid: 0.1, 1, 10 µM; 9-cis retinoic acid: 0.1, 1, 10 µM; bexarotene 0.1, 1, 10 µM), and Tgf-β (SB431542: 3, 10 µM) pathways were tested for stage- and dose-dependent effects on in vitro anterior–posterior patterning. Our results suggest that with further development, the inclusion of anterior–posterior reporter expression could be part of a battery of high-throughput tests used to identify and characterize teratogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart diseases (CHDs) are the most common birth defects, with a prevalence in Western Europe of 8.3 cases per 1000 births (0.83%) (Moons et al. 2009). Among the risk factors for heart malformation are genetic predisposition, maternal health, and exposure to teratogens (Donofrio et al. 2014). These include drugs commonly taken by women of reproductive age, such as anticonvulsants, lithium, ACE inhibitors, and retinoids (Donofrio et al. 2014). After the retinoid isotretinoin was approved as an acne treatment in the early 1980s, it was taken by 120,000 women of childbearing age, and induced spontaneous abortions or birth defects in 83% of pregnancies (Stern et al. 1984). In line with these potentially severe outcomes, developmental toxicity studies in pregnant mice and rats are currently mandated before approval of novel drugs (DeSesso 2017). However, these in vivo tests are low throughput, difficult to stage, require specialized expertise for determination of adverse effects, and rely on costly animal models (DeSesso 2017). In Europe alone, the number of animals used for toxicological risk assessment is 1 million per year, and this will increase with new testing of industrial chemicals (Kugler et al. 2016a, b). The development of robust, high-throughput in vitro tests for developmental toxicity is thus an important step in the reduction of both industrial expenditure and the use of laboratory animals as mandated by 3R principles of animal experiments (reduce, refine, replace) (Beekhuijzen 2017; Russell and Burch 1959).
For in vitro developmental toxicity tests to be reliable, multiple mechanisms of drug-induced developmental perturbation should be taken into account. These are thought to include folate antagonism, neural crest (progenitor) cell disruption, endocrine disruption, oxidative stress, vascular disruption, and specific receptor- or enzyme-mediated effects (van Gelder et al. 2010). These mechanisms are not mutually exclusive, as in the case of progenitor cell perturbation via binding of toxic compounds to specific receptors (van Gelder et al. 2010). Additionally, neural crest cell disruption might be expanded to include disruption of the diverse progenitor populations which give rise to the fully formed heart (Jain and Epstein 2018). Cell-based assays intended to detect developmental toxicity should thus include cells similar to those encountered during embryogenesis, such as undifferentiated progenitors derived from pluripotent stem cells. The validated embryonic stem cell test (EST) is an in vitro assay which involves compound testing in mouse embryonic stem cells (mESCs), immortalized fibroblasts (3T3 cells), and the cardiac differentiation of mESCs (Seiler and Spielmann 2011). The cardiac differentiation protocol used in the EST might be improved by considering the diverse progenitor populations underlying embryogenesis, the heterogeneous cell types composing the fully developed heart, the temporal and spatial control of cell signaling, and cell-specific molecular markers underlying the differentiation process.
Recent studies have shown the tight temporal control of signaling pathways during cardiomyocyte differentiation. Increased canonical Wnt and bone morphogenetic protein (Bmp) signaling promotes the mesodermal specification of ESCs, whereas inhibition of these pathways underlies the differentiation of progenitor cells to cardiomyocytes (Kattman et al. 2011; Kwon et al. 2007; Lian et al. 2012). Additionally, there is significant spatial variation in deployment of signaling pathways which governs the patterning of the heart into anterior (ventricular) and posterior (atrial) compartments and is predominantly controlled by time- and dose-dependent retinoic acid signaling in the developing posterior compartment (Ryckebusch et al. 2008; Xavier-Neto et al. 1999). However, retinoic acid signaling has also been shown to govern heart size via the negative regulation of cardiac progenitors (Keegan et al. 2005). Thus, anterior–posterior patterning of organs is a complex process which itself encompasses multiple, stage-specific molecular programs.
Though it is not feasible to have an in vitro model which incorporates all aspects of development, testing for the perturbation of multiple, related lineages simultaneously might allow for improved detection of adverse events. Further optimization of protocols to include multiple, short compound treatment windows more closely following developmental stages and capable of detecting multiple lineages simultaneously will provide information on perinatal safety windows and mechanisms of developmental toxicity. This is especially important when attempting to use in vitro models to predict effects in clinical settings, as both the stage of pregnancy and dosage of exposure to toxic compounds affect the likelihood of inducing malformations (Bérard et al. 2007; Cooper et al. 2006; Diav-Citrin et al. 2008).
We here describe steps toward improving the existing EST using reporter lines for developmental markers of anterior–posterior patterning of the heart. To confirm suitability of markers for genetic modification, gene expression profiles were generated of embryonic atria and ventricles using a selection of markers of atrial cardiomyocytes, ventricular cardiomyocytes, cardiac progenitors, nonmyocyte (stromal cells), and signaling pathways. Genome editing was used to generate a novel dual reporter mESC line for detecting anterior and posterior cardiac lineages simultaneously, and gene expression analyses of differentiating reporter mESCs informed assay development. Chimeric embryos generated from these mESCs displayed fluorescent expression in relevant cardiac tissues, and the selected compounds were then tested in the spontaneous differentiation of reporter mESCs. Finally, a serum-free embryoid body (EB) differentiation protocol was conducted in a scalable suspension format, allowing the high-throughput testing of differentiation modulators in cells of a defined mesodermal population. Our results give insight into cardiac development and provide a basis for further improvements to in vitro toxicological assessment of teratogens.
Materials and methods
qRT-PCR analysis of embryonic atria and ventricles
Embryonic day 10.5 (E10.5) embryonic hearts were removed from wild-type pregnant mice (C57BL/6JOlaHsd). Atria and ventricles were collected into TRIzol reagent (ThermoFisher Scientific), and RNA was extracted according to the manufacturer’s instructions. RNA purity was improved using the RNeasy MinElute Cleanup kit combined with DNase treatment (Qiagen), and RNA quality was determined via both Qubit (ThermoFisher Scientific) and Bioanalyzer (Agilent). qRT-PCR reactions were performed using the Fluidigm Biomark HD and Taqman gene expression assays (Supplementary Table 1). A general list of abbreviations is shown in Table 1. Values were normalized to a reference gene (Actb). Each biological replicate consisted of four embryos. Samples from spontaneous differentiation of reporter mESCs were similarly processed.
TALEN design, assembly, and validation
A Tal effector nuclease (TALEN) pair targeting the Myl2 locus was designed using TAL effector-nucleotide targeter (Doyle et al. 2012). TALEN-encoding plasmids were assembled using Golden Gate Cloning as described (Cermak et al. 2011). Plasmids for Golden Gate Cloning were obtained from Addgene. The TAL1 repeat variable diresidue (RVD) sequence was ‘NI NN NI NI NN NN HD NN NN NN NI NN HD NG HD HD NI’ and the TAL 2 RVD sequence was ‘NN NN NI NG HD NG NN NN NN NG HD NG NN HD NG’. This TALEN pair recognizes the genomic target sequence 5′- AGAAGGCGGGAGCTCCAACGTGTTCTCCATGTTTGAGCAGACCCAGATCC-3′ within the Myl2 exon.
Generation of ventricle-specific targeting vector
The Myl2 targeting vector consisted of a promoterless eGFP-bGH-PGK-Puro cassette flanked by 5′ and 3′ homology arms. A large fragment encompassing the total homologous region was amplified from E14 wild-type mESC genomic DNA by PCR using 5′-AACCCCCTCCCAAGCCAAGCA-3′ and 5′-ATTCCCCCAAGGGCCAGGTGA-3′. The 5′ homology arm was amplified from this amplicon by PCR using 5′-TGGCGGCCGCTCTAGCCCTCCCAAGCCAAGCACCC-3′ and 5′-GCTCACCATGCTAGCCACGTTGGAGCTCCCGCCTTC-3′, whereas the 3′ homology arm was amplified from the homologous template by PCR using 5′ GCTCTATGGAGGCCTTCCATGTTTGAGCAGACCCAGA-3′ and 5′-ATCCACTAGTTCTAGATTCCCCCAAGGGCCAGGTG-3′. The eGFP-bGH-PGK-Puro cassette was constructed by first excising eGFP from PITX3-eGFP-PGK-Puro using EcoRI and XhoI restriction enzymes, and subcloning this fragment into the pCDNA3.1(−) expression vector upstream of the bGH polyA sequence. This eGFP-bGH cassette was amplified by PCR using 5′-GCTAGCATGGTGAGCAAGGGCG-3′ and 5′-AGGCCTCCATAGAGCCCACCGCATCCC-3′. Primer regions not in bold were added to facilitate HD fusion cloning. The 5′ homology arm, eGFP-bGH-PGK-Puro cassette, and 3′ homology arm were cloned into XbaI digested pBluescript II KS( +) using the HD fusion cloning kit (Clontech). A PGK-Puro fragment amplified by 5′-TGGGCTCTATGGAGGTCCGATCATATTCAATAACCCTTAAT-3′ and 5′-CTCAAACATGGAAGGGGCGCGCCTAGTGAACCTCT-3′ was ligated into a StuI restriction site in the resulting vector to generate the final Myl2-eGFP-PGK-Puro donor construct. The plasmid was sequenced to ensure fidelity.
Generation of atrial-specific transgene
The 840 bp sinoatrial-specific SMyHC3 promoter has been described previously (Xavier-Neto et al. 1999) and a construct containing the sequence was obtained from Dr. Jose Xavier-Neto. A plasmid was generated in which this promoter is used to drive the TdTomato fluorescent protein using infusion HD cloning (Clontech). In short, the 840 bp atria-specific sequence was amplified by PCR using 5′-CGGGCTGCAGGAATTCCACCACAGTGGCCCCATCC-3′ and 5′-ACCATGGATCCTGCACAATCCTGCTGCTGCCCCAC-3′. The TdTomato-pA gene was amplified by PCR using 5′-TGCAGGATCCATGGTGAGCA-3′ and 5′ TAAACAAGTTCCGAGGCTGGGTACCGGGCCCAATG-3′, whereas the PGK-Neo cassette was amplified using 5′-CTCGGAACTTGTTTATTGCAGC-3′ and 5′-GCTTGATATCGAATTAAAGGGAACAAAAGCTGGAG-3′. Primer regions not in bold were added to facilitate HD fusion cloning. The three fragments were cloned into EcoRI-digested pBluescript II KS( +) using the HD fusion cloning kit (Clontech). The plasmid was sequenced to ensure fidelity.
Myl2 gene targeting in mESCs
The targeting vector and TALEN-encoding plasmids were introduced into E14 mESCs cultured in feeder-free conditions using the Xfect transfection reagent (Clontech). Correctly targeted mESC clones were identified using 5′ junction PCR genotyping primers 5′-TTGAGAAGAGACGCAGGGTG-3′ and 5′-GCTGAACTTGTGGCCGTTTAC-3′, which amplify a PCR product encompassing the entire 5′ homology arm and part of the eGFP-bGH-PGK-Puro cassette. Additionally, 3′ junction PCR primers 5′-TCCATCAGAAGCTGGTCGAG-3′ and 5′-CCTCTATGATGGCCGCTGTC-3′ were used to identify positive clones. Clones positive from initial genotyping experiments were expanded and re-genotyped after six passages. A region flanking the TALEN targeting site was amplified from genomic DNA of expanded mutant cell lines using primers 5′-TTTCGCCTCGCATGTTGTCTGGG-3′ and 5′-GGATCGCTACCCAGCGCCTT-3′ to identify heterozygous mutations. This band was sequenced, and a clone with a heterozygous knock-in (Myl2eGFP/w) was used for further experiments.
Random integration of atrial-specific transgene
Myl2eGFP/w mESCs were transfected with the linearized SMyHC3-TdTomato-PGK-Neo construct and clones with stable integration were manually isolated after 10 days of treatment with G418 (250 µg/mL). Stable integration in individual clones was confirmed by PCR genotyping using primers 5′-GGAGATAAGGAGGCCAGAAATAGC-3′ and 5′-CTCACGTAGAACCCCCTTTGC-3′, which amplify a sequence within the SMyHC3-TdTomato cassette. Positive clones were tested for differentiation potential, and expression of the eGFP and TdTomato reporter genes was assessed by flow cytometry and microscopy after differentiation to cardiomyocytes.
Generation of reporter mESC-derived embryos
Reporter mESCs were used to generate reporter embryos using eight-cell morula injection essentially as described (Poueymirou et al. 2007). Embryos were dissected and imaged on a fluorescent stereomicroscope (Zeiss Axio Zoom.V16).
Cardiomyocyte differentiation in hanging drops with small molecules
For differentiation of reporter mESCs, cells were plated to V-bottomed 96-well plates at 500 cells/well in 20 µL embryoid body differentiation medium (DMEM, 20% fetal bovine serum, non-essential amino acids, glutamax, 2-mercaptoethanol). Plates were inverted until day 2, when 200 µL medium was added. On day 5, EBs were then manually transferred to a 12-well plate coated with 0.1% gelatin at 6 EBs/well. Small-molecule compounds were diluted in DMSO and added to the cell culture medium at the indicated time points. All compounds were purchased from Sigma-Aldrich. D12 spontaneous differentiation cultures were dissociated with TrypLE for 20 min on a shaker. TrypLE was inactivated with serum and cells were washed and resuspended in 1× PBS before analysis on a BD LSRFortessa flow cytometer. Wild-type EBs and Myl2-eGFP EBs were used as negative controls for gating. Analysis was performed using Flowjo (Flowjo LLC). For fluorescent microscopy of differentiated clones, cells were fixed in 4% PFA, permeabilized with 0.1% Triton-X and stained with anti-RFP antibody (Rockland) and Alexa Fluor secondary (Thermo) to increase the fluorescent signal. Reporter mouse pluripotent stem cells were characterized by immunocytochemistry with an anti-Oct4 antibody (sc-8628). Characterization of sarcomeric protein expression in reporter cells was performed by immunocytochemistry for Mhc/MF20 (Developmental Studies Hybridoma Bank), and α-actinin (Sigma-Aldrich). Cells were imaged by epifluorescence (Zeiss Axioimager) or inverted fluorescence (Leica DMi8) microscopy.
Directed cardiomyocyte differentiation and high-throughput detection
E14 wild-type, Myl2eGFP/w, and Myl2eGFP/w, SMyHC3-TdTomato dual reporter mESCs were cultured in feeder-free conditions on 0.1% gelatin in LIF-containing medium. Directed differentiation into cardiomyocytes was performed as described with some modifications (Kattman et al. 2011). mESCs were allowed to form EBs in suspension culture and grown at 75,000 cells/mL in media composed of the following: IMDM, Ham’s F12, BSA, Glutamax, B27 supplement, N2 supplement, monothioglycerol, and ascorbic acid. After 2 days of suspension culture, EBs were dissociated with TrypLE and then re-aggregated for 40 h in media containing activin A (R&D Systems) and BMP4 (R&D systems). On day 4, EBs were again dissociated and replated at 125,000 cells in 250 µL of StemPro, a serum-free medium (SP34, ThermoFisher Scientific #10639011) supplemented with VEGF (R&D systems), FGF10 (R&D systems), and FGFb (R&D systems) in ultra-low attachment 24-well plates. Day 5 EBs were dissociated with the neonatal mouse dissociation kit (Milenyi) and plated to 384-well clear-bottomed black plates. Compounds were added to adherent cells at the indicated time points, and the differentiation efficiency was measured using a Pherastar fluorescent plate reader (BMG). Data consist of at least four biological replicates and four technical replicates within each experiment. For characterization of D6 cells by immunocytochemistry, cells were fixed in 4% PFA, permeabilized with 0.5% Triton-X and stained with anti-Isl antibody (Developmental Studies Hybridoma Bank) and Alexa Fluor secondary (Thermo).
Results
Gene expression analysis of the newly formed embryonic heart
We selected a genetic approach for the efficient detection of anterior–posterior marker expression during in vitro differentiation. To assess specificity of markers to the anterior and posterior heart, we conducted a qPCR analysis of native, embryonic tissues to determine expression patterns of published chamber-specific markers. We also analyzed markers of stromal cell differentiation, progenitor cells, and signaling pathways to identify relevant differences in the newly formed chambers and inform assay development. We selected E10.5 hearts, as at this developmental time point the two atria and two ventricles have clearly formed. By choosing an early developmental stage, we sought to identify markers which would be suitable for in vitro differentiation, which produces immature cardiomyocytes. Known ventricle-specific genes such as Hey2, Irx4, Kcne1, and Myl2 were also upregulated in the ventricular compartment in our study (Fig. 1a). Importantly, Myl2 was the most upregulated ventricular gene (50-fold higher than atria), suggesting its suitability for reporter cell generation (Fig. 1a). Published atrial markers Nr2f2, Myl7, Gja5, and Pitx2 were detected at increased levels in the atria (Fig. 1b), but also showed expression in the ventricular compartment, confirming the lack of robust, native atrial markers during early embryogenesis in the mammalian system (Xavier-Neto et al. 1999). Sarcolipin was specifically expressed in the atrial compartment and absent in the ventricles (Fig. 1b), however, the presence of this marker in skeletal muscle makes it less suitable for differentiation analysis of mESCs (Babu et al. 2007). Importantly, the pan-cardiac marker Tnnt2 showed no difference between atrial and ventricular compartments, suggesting equal composition of cardiomyocytes in the atria and ventricles (Fig. 1c).

qRT-PCR profiling of the newly formed four-chambered mouse heart (E10.5). Published markers of a ventricular: Irx4, Hey2, Kcne1, Myl2 b atria: Nr2f2, Myl7, Gja5, Sln, Pitx2 c pan-cardiac: Tbx5, Tnnt2, Gata4, Nppb, Nppa d progenitor: Kit, Isl1, Kdr, Pdgfra, Nkx2.5 e Axin2, Alcam, Cyp26a1, Cyp26c1, Aldh1a2. Data are presented as mean + SEM (n = 3)
In addition to known cardiomyocyte markers, we identified differences between the nascent compartments in stromal cell type composition. Interestingly, fibroblast markers Cdh2 and Thy1 were upregulated in the ventricular compartment, whereas smooth muscle marker Myh11 was upregulated in the atrial compartment (Supplementary Fig. S1). Also, endothelial marker Pecam1 was upregulated within the ventricular compartment. These suggest the early ventricle has a more fibrotic/endothelial character, whereas the atria have more cells of a smooth muscle identity.
No difference in cardiac progenitor marker Nkx2-5 was observed between the embryonic atria and ventricles (Fig. 1d). However, progenitor markers Kit and Kdr were upregulated in the ventricular compartment (Fig. 1d), indicating the presence of endothelial progenitors in the developing ventricle (van Berlo et al. 2014). Additionally, the Isl1 transcription factor, a marker of the second heart field and later sinoatrial nodal cells, was upregulated within the atria (Fig. 1d).
Signaling pathway markers also displayed differences between atria and ventricles (Fig. 1e). To distinguish between canonical and non-canonical Wnt signaling, Axin2 and Alcam, respectively, were used as target genes (Cizelsky et al. 2014; Yan et al. 2001). While canonical Wnt signaling (Axin2) showed increased expression in atria, non-canonical Wnt signaling (Alcam) was upregulated in the ventricles (Fig. 1e). Additionally, retinoic acid signaling genes Cyp26a1 and Aldh1a2 were upregulated in the atria compared to the ventricles (Fig. 1e). These confirm the presence of developmental signaling pathways across the cardiac chambers of the newly formed embryonic heart.
Generation of dual reporter mESC line with genome editing
TALEN-mediated genome editing was used to generate a mESC line from which anterior and posterior lineages could be efficiently analyzed after in vitro differentiation. To mark cells of the anterior (ventricular) lineage, Myl2 was selected for genomic modification, as Myl2 has been shown to be specific to ventricular segments of the heart during embryonic and adult stages (O'Brien et al. 1993).
TALEN-encoding plasmids were constructed to encode nucleases which induce a double-strand break in the second exon of the mouse Myl2 locus. Additionally, a donor plasmid for in-frame integration of an eGFP-PGK-Puro cassette was generated (Fig. 2a). After transfection with TALEN-encoding and donor plasmids, clones with an integration event were purified via puromycin selection, and correctly targeted mESC clones were identified by junction PCR (Supplementary Fig. S2a). Selected clones with undifferentiated morphology were used for further experiments. Upon spontaneous differentiation in hanging drops, Myl2-eGFP expression was observed at D9–D10 of differentiation in areas with spontaneous beating. A single clone was selected for further targeting, hereafter referred to as Myl2eGFP/w.

Generation of reporter mESC line. a Targeting strategy for generation of TALEN-mediated knock-in at the ventricle-specific Myl2 locus and random integration of atrial-specific SMyHC3-TdT into mESCs. b Fluorescent expression of ventricular and atrial reporters upon spontaneous differentiation to cardiomyocytes, ×10 magnification. c Representative flow cytometry analysis of wild-type, Myl2eGFP/w, and dual reporter mESCs upon directed differentiation to cardiomyocytes and retinoid treatment
To our knowledge, no gene has been described which has expression only in mouse atrial cardiomyocytes during both embryonic and adult stages. Our qRT-PCR analysis of embryonic hearts also failed to identify endogenous markers of atrial cardiomyocytes not expressed in other mammalian cell types. We thus selected the atrial-specific enhancer/proximal promoter of the quail slow myosin heavy chain (SMyHC3), previously shown to mark atrial cells during all developmental stages and adulthood (Xavier-Neto et al. 1999). A construct was assembled in which the SMyHC3 promoter drives the fluorescent protein TdTomato (SMyHC3-TdT-PGK-Neo) (Fig. 2a). After transfection and antibiotic selection with G418, stably transfected clones were identified by PCR (Supplementary Fig. S2b). A total of ten clones were differentiated in hanging drops, and one mESC clone was selected for further experiments, based on cardiomyocyte differentiation capacity and expression of both eGFP and TdTomato detectable by fluorescent microscopy and flow cytometry (Fig. 2b, c). For simplicity, these markers are hereafter referred to as venGFP (ventricular, anterior) and atrRFP (atrial, posterior). Immunocytochemistry of dual reporter mESCs indicates expression of the Oct-4 protein, a pluripotency marker, as well as characteristic pluripotency morphology, further confirming that dual reporter mESCs represent cells in the pluripotent state (Supplementary Fig. S3). Importantly, differentiated venGFP and atrRFP cardiomyocytes also expressed sarcomeric proteins myosin heavy chain (Mhc/MF20) and α-actinin, indicating the overlap of reporter expression with the contractile phenotype in vitro (Supplementary Fig. S4a–b). Live cell imaging also revealed the presence of venGFP and atrRFP expression in spontaneously beating, differentiated cardiomyocytes (Supplementary Fig. S5). These suggest the utility of reporter expression as an endpoint for assays which have previously used the detection of spontaneously beating cells.
Embryonic expression of venGFP and atrRFP in reporter mESC-derived embryos
To confirm the correct expression of fluorescent reporters in the developing embryo, chimeric embryos were generated via laser-assisted morula injection of dual reporter mESCs. Embryos were dissected during early stages of cardiac development from stages spanning the formation of the cardiac crescent to the fully looped heart. Reporter embryos are apparent in Fig. 3a–l. We observed robust venGFP expression at the cardiac crescent stage (E7.5) (Fig. 3a), pointing to reporter expression prior to the formation of the linear heart tube. Myl2 is known to be expressed when the lateral plate mesoderm forms into the linear tube and marks the primitive ventricle (future left ventricle), but we have observed venGFP expression from the midline towards the distal ends of the cardiac crescent but not in the distal portion that will give rise to other fragments of the heart (Fig. 3a, b). atrRFP expression was not observed in this stage. In E8.25 embryos, we observed venGFP expression in the first beating chamber of the primitive ventricle (also reported by previous studies) and atrRFP expression in the forming venous pole region and the myocardial sinus horns of the embryonic heart (Fig. 3c, d). In E8.5 embryos, we observed venGFP at the primitive left ventricle, and atrRFP in the primitive, common atrium at the junction of the primitive ventricle and inflow region (Fig. 3e, f). In the rightward looping heart (E8.75), venGFP expression was observed in the future left ventricle (Fig. 3g, h). Interestingly, the future atrioventricular canal was marked by double positive cells, and atrRFP signal alone was visible in the sinus venosus region (Fig. 3g, h). In the E9.5 embryo, robust atrial and ventricular expression were observed (Fig. 3i–l). Collectively, we observed venGFP in the forming left ventricle, atrioventricular canal, and right ventricle/outflow tract. Moreover, we recorded atrRFP expression on the forming left and the right sides of the common atria at the junction of the dorsal aorta and atrioventricular canal. Our results mirror published reports of the expression of venGFP(Myl2-eGFP) and atrRFP (SMyHC3), confirming our fluorescent reporter expression is specific to atrial and ventricular regions during the earliest stages of cardiogenesis (O’Brien et al. 1993; Xavier-Neto et al. 1999). However, in contrast to previous studies, the use of a fluorescent reporter shows detail at the cellular level, in addition to a dynamic range of expression. Importantly, the onset and duration of fluorescent reporter expression in reporter cell-derived hearts coincides with spontaneous beating in differentiated cardiomyocytes of the embryonic heart. Furthermore, the ability of reporter mESCs to contribute to all embryonic structures and specifically express fluorescent reporters only in the heart is strong in vivo evidence for both pluripotency of reporter mESCs and specificity of reporter expression.

Anatomical localization of fluorescent reporter expression in chimeric embryos. a, b E7.5 embryo, venGFP (Myl2, green) expression in the cardiac crescent, bright field image showing cardiac crescent (cc) which will form the primitive ventricle (pv) of the linear heart tube, which is depicted by black lines. c, d E8.25 embryo, venGFP expression in the primitive ventricle, atrRFP (red) in the inflow region of the linear heart tube, bright field image showing sinus horns (sh), inflow region (inf) of the linear heart tube which together makes the venous pole of the heart and primitive ventricle. The horizontal line depicts the axis of the embryo prior to looping. e, f E8.5 embryo, venGFP expression in the primitive ventricle/future left ventricle, atrRFP expression in the inflow region of the developing heart and developing primitive atrium (pa) observed by a small ballooning at the junction of the horizontal line which is angled by the starting of looping. g, h E8.75 embryo, the horizontal line shows advancement of looping observed by the elliptic right side of the primitive ventricle and the left side of the primitive left ventricle which connects to the primitive atrium. i, j E9.5 embryo, right side, advancement of the looping outflow tract (oft)/ right ventricle (rv) showing venGFP expression and the left part of the common atria (ra) showing atrRFP expression. k, l E9.5 embryo, right side, venGFP expression in left ventricle (lv), atrRFP expression in the left part of the common atria (la) and in the connection of the left ventricle, left atria. Red–green regions (orange) in the atrioventricular canal (AVC) regions are visible (color figure online)
Gene expression dynamics during spontaneous differentiation of reporter mESCs
To gauge the suitability of in vitro differentiation of reporter mESCs to cardiac chamber development, we conducted gene expression analyses of differentiating cells at three time points: D6 (before the onset of spontaneously beating cells), D9 (first appearance of small numbers of spontaneously beating cells), and D12 (large numbers of beating clusters of cells) (Fig. 4a). Gene expression of ventricular, atrial, and pan-cardiac cardiomyocyte markers increased progressively from D6–D12, in line with spontaneous beating observations (Fig. 4b–e). Similar expression dynamics were observed for smooth muscle (Myh11) and fibroblast (Thy1, Vim, Acta2) markers, suggesting that differentiation of these cell types occurs simultaneously with cardiomyocyte differentiation (Supplementary Fig. S6). Multipotent, cardiac progenitor markers (Kit, Isl1, Kdr, Pdgfra, Nkx2-5) were also observed, with highest expression levels at day 9 (Fig. 4d). Early mesodermal marker (Brachyury) was more strongly expressed at D6 of differentiation, suggesting that D6–D12 of differentiation covers both early and late mesodermal progenitor stages (Fig. 4d). Wnt targets (Axin2, Alcam) and retinoic acid components (Aldh1a2, Cyp26a1, Cyp26c1) were also expressed during the D6–D12 window (Fig. 4f). Interestingly, canonical Wnt (Axin2) and non-canonical Wnt (Alcam) displayed inverse temporal expression, in line with previous observations that non-canonical Wnt replaces canonical Wnt during cardiomyocyte differentiation (Mazzotta et al. 2016). Retinoid degrading enzymes Cyp26a1 and Cyp26c1 also showed inverse temporal expression, as Cyp26c1 replaced Cyp26a1 during D6–D12 differentiation (Fig. 4f). To our knowledge, this has not been described previously, and has potential implications for teratogenic effects of retinoid treatment. Endogenous retinoic acid signaling (Aldh1a2) was nearly absent at D6 of differentiation, and increased at D9 and D12 time points. As retinoic acid signaling is thought to be a principal component of anterior–posterior patterning of the heart (Xavier-Neto et al. 1999), this suggests that in the absence of exogenous retinoid treatment, in vitro retinoic acid patterning of multipotent cardiac progenitor cells occurs during D6–D12 spontaneous differentiation of reporter mESCs. Collectively, these results show that embryonic cardiac chamber differentiation genes are expressed during D6–D12 spontaneous differentiation of mESCs, suggesting the suitability of the in vitro model for compound testing.

qRT-PCR profiling of spontaneous differentiation in reporter mESCs. a Differentiation schematic for sampling at D6, D9, and D12 of differentiation for detection of markers of b atria: Nr2f2, Myl7, Gja5, Sln, Pitx2, TdTomato c ventricles: Irx4, Hey2, Kcne1, Myl2 d progenitor cells: Kit, Isl1, Kdr, Pdgfra, Nkx2.5, Brachyury e pan-cardiac: Tbx5, Tnnt2, Gata4, Nppb, Nppa f signaling pathways: Axin2, Alcam, Cyp26a1, Cyp26c1, Aldh1a2. Data are presented as mean ± SEM for each time point (n = 3)
Stage-specific effects of retinoids on expression of anterior–posterior reporters during spontaneous differentiation of reporter mESCs
To assess the functionality of a dual reporter differentiation assay, we tested commercially available retinoids with known teratogenic properties for effects on fluorescent reporter expression (Table 2). Initial experiments showed that D2–D10 addition of high concentrations of all-trans retinoic acid (10 µM) completely eliminates the cardiac lineage and expression of both venGFP and atrRFP. As our aim was to detect more subtle defects in patterning, we selected shorter time windows and smaller doses for our experiments based on the appearance of spontaneous beating, venGFP/atrRFP expression, and temporal gene expression analysis. Time points for compound addition were selected shortly before (D6–D9) and after the appearance of beating cardiomyocytes (D9–D12). As the linear heart tube of E8 embryos beat robustly, we estimate these earliest myocytes roughly approximate E8–E10 hearts.
Results of differentiation experiments are shown in Fig. 5. Regardless of compound treatment window, cultures were measured on D12 of differentiation for %venGFP and %atrRFP cells, mean fluorescent intensity (MFI) of venGFP and atrRFP, and atrRFP/venGFP ratio. Both anterior and posterior cell populations were strikingly sensitive to retinoid treatment during early time windows. A significant decrease in the %venGFP cells was observed for D6–D9 treatment with ATRA (1 µM and 10 µM) and 9CRA (0.1 µM, 1 µM, 10 µM), but not by BX (Fig. 5a). A decrease in venGFP-MFI was observed during D6–D9 treatment with ATRA (1 µM, 10 µM), 9CRA (1 µM, 10 µM), and BX (1 µM, 10 µM) (Fig. 5b). Similar to D6–D9 treatment, a decrease in %venGFP + cells was induced by D9–D12 treatment with ATRA (1 µM, 10 µM) and 9CRA (1 µM, 10 µM), but not by BX (Fig. 5a). The venGFP-MFI showed an increase in venGFP levels with high levels of 9CRA (10 µM) and BX (10 µM) during D9–D12 treatment. These results clearly show the stage-specific effects of retinoids on fluorescent reporter expression.

Effects of retinoids on reporter expression during spontaneous differentiation of reporter mESCs. D12 measurements of a %venGFP + cells b venGFP-mean fluorescent intensity c %atrRFP + cells d atrRFP-mean fluorescent intensity e %atrRFP/%venGFP f atrRFP-MFI/venGFP-MFI. Fluorescence activity was normalized to DMSO and is presented as mean + SEM (n = 5). **P < 0.01, *P < 0.05 (Wilcoxon test vs DMSO control)
Expression of the atrRFP reporter was also affected by retinoids (Fig. 5c, d). We observed a statistically significant decrease in %atrRFP cells upon D6–D9 treatment with ATRA (1 µM, 10 µM) and 9CRA (1 µM), but no statistically significant effects from BX treatment. atrRFP-MFI decreased upon D6–D9 treatment with ATRA (0.1 µM, 1 µM, 10 µM) and 9CRA (1 µM, 10 µM), but was unaffected by BX treatment. A decrease in %atrRFP cells was observed in D9–D12 treatment with ATRA (1 µM), while a decrease in atrRFP-MFI during D9–D12 treatment was observed for ATRA (0.1 µM, 1 µM, 10 µM) and 9CRA (10 µM). Non-statistically significant increases in %atrRFP+ cells were observed for early (D6–D9) 9CRA (0.1 µM) and BX treatment (0.1 µM, 1 µM), as well as late (D9–D12) 9CRA (0.1 µM) and BX treatment (0.1 µM).
The effects of atrialization of the total cardiac progenitor pool by retinoids were explored via quantification of atrial/ventricular ratio and atrRFP/venGFP-MFI ratio (Fig. 5e, f). Increases in atrial/ventricular ratio were observed for nearly all retinoids at early time points D6–D9, though only ATRA (0.1 µM) was statistically significant. This suggests that the atrializing potential of ATRA exists prior to the onset of spontaneous beating, as observed previously (Lee et al. 2017). atrRFP-MFI/venGFP-MFI levels were increased at early time points D6–D9 by ATRA (10 µM) and BX (10 µM), but decreased at later time points by ATRA (0.1 µM, 1 µM, 10 µM) and 9CRA (10 µM). Collectively, these results indicate increased severity of cardiovascular developmental toxicity from dual RAR/RXR agonists (ATRA, 9CRA) than RXR agonists (BX). These results also confirm the complex time and spatial characteristics of retinoic acid signaling during cardiogenesis, and indicate this can be partially recapitulated in vitro with reporter cell lines using a spontaneous differentiation model. Interestingly, these results mirror the time-dependent effects of retinoid acid signaling in the zebrafish model (Keegan et al. 2005).
Chemical modulation of retinoic acid, Wnt, and TGFβ pathways in high-throughput ventricular differentiation
To assess the feasibility of a high-throughput differentiation assay (384-well format), we developed a protocol for generation of a large number of cardiac progenitors in chemically defined conditions. We modified a widely used protocol requiring serial dissociation of EBs and treatment with BMP4/activin A to generate large enough number of multipotent cardiac progenitors for efficient toxicological screening (Kattman et al. 2011). The most significant modification was plating mesodermal progenitors (D4 differentiation) to ultra-low attachment plates instead of to adherent conditions, allowing the generation of a large number of D5 multipotent cardiac progenitors (~ 20 million). These were plated to 384-well plates for total fluorescence analysis on D10 of differentiation. Total fluorescence of venGFP could be measured, but not atrRFP, likely reflecting the difference in strength of these two promoters, the preferential induction of ventricular cardiomyocytes via the protocol, and the reduced sensitivity of the plate reader format compared to flow cytometry. Indeed, flow cytometric analysis from cells differentiated via this protocol generated a strong preference for cells of the ventricular lineage (Supplementary Fig. S7). This is also in line with a recent study which found the necessity of titrating BMP4/activin A-induced mesodermal specification to generate cells capable of differentiation to the atrial lineage (Lee et al. 2017).
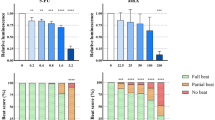
Spontaneous beating occurs at D7 in this assay and was used as a reference point for compound addition points. Importantly, venGFP cells beat robustly in cell sheets in the 384-well format, and the abundance of beating correlates qualitatively with the amount of venGFP cells. In conditions with highly efficient differentiation, the cells beat synchronously in a unified cell sheet. Compounds modulating retinoic acid (ATRA), TGF-β (SB431542), and Wnt (XAV939) signaling pathways were tested in this assay system during two windows, one in which compounds are added before the onset of beating (D6–D8), and one in which compounds are added after the onset of beating (D7–D9) (Fig. 6a). D6 cells also express the multipotent cardiac progenitor marker Isl1, confirming their undifferentiated, progenitor state (Fig. 6b). Early addition (D6–D8) of ATRA caused a decrease in venGFP fluorescence, whereas late addition resulted in an increase in venGFP fluorescence (D7–D9) (Fig. 6c, d). This suggests biphasic effects of retinoic acid signaling on late progenitor specification to ventricular cardiomyocytes. Inhibition of both Tgf-β (SB431542) and canonical Wnt (XAV939) increased venGFP fluorescence when added prior to spontaneous beating, but showed no statistically significant effects when added after the onset of beating (Fig. 6c, d). This also suggests divergent roles for these pathways in multipotent cardiac progenitors and differentiated cardiomyocytes. Overall, these results indicate the potential for detection of stage-specific differentiation modulation of chamber-specific cardiomyocytes in a high-throughput system. Additionally, they further confirm that the in vitro commitment of cells to anterior–posterior lineages largely occurs before the onset of spontaneous beating.

384-well plate screening in reporter mESCs in serum-free conditions. a Schematic of serum-free differentiation system for high-throughput screening. b Immunocytochemistry of cardiac progenitor marker Isl1 on D6 of the differentiation protocol, ×40 magnification. c Representative wells of compound treatment showing effects of compounds on Myl2 promoter activation upon D6-D8 treatment, ×5 magnification. d D10 total fluorescence measurements of venGFP upon treatment either before (D6–D8) or after (D7–D9) the onset of beating. Fluorescence activity was normalized to DMSO and is presented as mean + SEM (n = 4). **P < 0.01, *P < 0.05 (Wilcoxon test vs DMSO control)
Discussion
To facilitate the detection of teratogens which are deleterious to fetal health, we developed an in vitro model based on the anterior–posterior patterning of the early heart. Using qRT-PCR analysis of early four-chambered hearts, we have identified suitable markers for genome modification and gained insight into the spatially distinct cell composition of the embryonic heart. TALEN-mediated genome editing was used to generate a dual reporter mESC line for more efficient detection of differentiated lineages, and expression of reporter alleles was observed in chimeric embryos and during both spontaneous and directed differentiation of mESCs at the onset of spontaneous beating. To our knowledge, this is the first reporter cell line in the mammalian system including markers for both anterior and posterior cardiac lineages simultaneously. Furthermore, the reporter system described here presents an alternative readout to the counting of beating clusters currently used as part of the EST, and allows for adaptation to high-throughput screening for developmental toxicity.
Using our reporter system, we delineated the windows of action of a panel of retinoids, demonstrating their effects on the induction of expression of anterior–posterior markers in vitro. Retinoic acid is a potent teratogen, and in humans principally causes malformation of the heart, nervous system, and craniofacial tissues (Lammer et al. 1985). In zebrafish and mouse models, retinoic acid signaling has been implicated in both limiting the number of cardiac progenitors in the cardiac crescent and patterning progenitors into anterior–posterior compartments (Keegan et al. 2005; Ryckebusch et al. 2008; Xavier-Neto et al. 1999). Our in vitro results also reflect biphasic retinoid acid signaling, as we observed both inhibition and promotion of anterior–posterior lineages which were dependent upon the time of treatment (D6–D9 vs D9–12). These results suggest lineage commitment of cells to anterior–posterior fates precedes the onset of beating, and this coincides with replacement of retinoic acid-degrading enzyme Cyp26a1 with Cyp26c1. To our knowledge, this has not been described previously, and has potential implications for teratogenic effects of retinoid treatment. We also observed that low concentrations of retinoic acid administered before the onset of beating preferentially inhibited the ventricular lineage but did not affect the atrial lineage, mirroring the atrialization seen in zebrafish models (Keegan et al. 2005).
For large-scale toxicological screening, detection of reporter alleles with a high-throughput plate reader would be desirable. In the present study, we adapted a directed differentiation protocol to a high-throughput, 384-well plate format. We utilized a directed differentiation protocol in chemically defined medium to generate cardiac progenitor populations suitable for fluorescent protein detection. This screening system was capable of detecting expression from the venGFP (Myl2), but not atrRFP (SMyHC3) promoters, and this can be partially explained by differences in the strength of these promoters in the mouse system. Re-engineering of reporter alleles to express the more easily detectable luciferase instead of fluorescent proteins might resolve this issue. Additionally, the directed differentiation system we employed mostly produces cardiomyocytes of the ventricular lineage (Lee et al. 2017). Further optimization of our directed differentiation protocol to Cyp26a1+ and Cyp26a1− mesoderm would likely be necessary to achieve detection of the SMyHC3 promoter in a high-throughput format (Lee et al. 2017). However, the screening system we employed was successful in detecting the effects of Wnt, TGF-β, and retinoid signaling pathway alteration on differentiation to the ventricular lineage.
Though developmental toxicity was the aim of this study, aberrant expression of atrial/ventricular identity genes might also predict physiological defects arising from developmental exposure to teratogens. Ventricular upregulation of the SMyHC3 transgene during embryogenesis was shown to precede adult-onset pathological hypertrophy upon deletion of the transcription factor Irx4 in the murine model (Bruneau et al. 2001). Perturbed Wnt signaling during development can also lead to chamber-specific arrhythmias in adulthood due to underlying defects in chamber-specific gene expression (Li et al. 2018). These effects are overlooked by the existing EST, and cannot be feasibly routinely tested in vivo, as pups from pregnant mothers in embryonic toxicity studies would necessarily be brought to term and subjected to ECG and telemetry recordings. Our assays, which use chamber-specific gene expression as a readout of differentiation, could potentially uncover subtle developmental toxicity which results in adult disease due to altered chamber-specific gene expression.
Teratogenic activities have not been determined in humans/human cell models for more than 90% of drugs, and rodent studies may not always be predictive (van Gelder et al. 2010). Though the present study was conducted in mESCs, in line with previous iterations of the EST, a clear goal would be to develop a reliable system in human embryonic stem cells (hESCs)/human-induced pluripotent stem cells (hIPSCs). One limitation is that there is no feasible method of validating transgene expression in human embryos, in contrast to transgenic mouse methods in which chimeric embryos show a clear overlap of anatomical expression, as in the current study. Other barriers to establishment of hPSC methods include the ethical issues surrounding hESCs, and the oncogenic mutations known to exist in most hIPSC lines (Merkle et al. 2017). mESC models based on transgenic lines thus still hold advantages in terms of cost, validation, and the existence of well-characterized reporter alleles (Kugler et al. 2016a, b).
In the present study, only a small set of compounds in a small number of time windows were tested. Future efforts must focus on testing a larger set of well-characterized reference compounds for their effects on anterior–posterior patterning in shorter time windows. As some non-familial CHD cases could be due to non-medical exposure to environmental pollutants and industrial byproducts, these could also be tested for safety windows in low-cost, high-throughput differentiation assays (Ou et al. 2017). Additionally, the same time windows presented here could be tested in other reporter lines which have been developed for toxicological assessment, such as a recently described Wnt/Β-catenin reporter (Kugler et al. 2016a, b).
As the heart is the first organ to form, and its development includes most key developmental signaling pathways, interruption of cardiac differentiation in vitro might not only reflect interference with in vivo cardiogenesis, but could also be predictive of a general disruption of in vivo organogenesis. Indeed, defects in the patterning of the heart might be predictive of defects in patterning of limbs and craniofacial tissue, as significant overlap exists in those developmental programs (Diogo et al. 2015). Thus, by developing adequately validated, stepwise cardiac differentiation assays, the need for conducting differentiation assays to other lineages (endodermal, ectodermal) could be eliminated. We also envision such stage-specific analyses of different reporter alleles in the context of cardiac differentiation of mESCs will allow the study of previously misunderstood mechanisms of developmental toxicity. Mechanistic information gained from high-throughput assays could also aid in the identification of off-target effects early in the drug development process and an overall reduction in the congenital disease burden.
References
Babu GJ, Bhupathy P, Carnes CA, Billman GE, Periasamy M (2007) Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J Mol Cell Cardiol 43(2):215–222
Beekhuijzen M (2017) The era of 3Rs implementation in developmental and reproductive toxicity (DART) testing: current overview and future perspectives. Reprod Toxicol 72:86–96
Bérard A, Ramos E, Rey E, Blais L, St-André M, Oraichi D (2007) First trimester exposure to paroxetine and risk of cardiac malformations in infants: the importance of dosage. Birth Defects Res B Dev Reprod Toxicol 80(1):18–27
Bruneau BG, Bao ZZ, Fatkin D, Xavier-Neto J, Georgakopoulos D, Maguire CT, Berul CI, Kass DA, Kuroski-de Bold ML, de Bold AJ, Conner DA, Rosenthal N, Cepko CL, Seidman CE, Seidman JG (2001) Cardiomyopathy in Irx4-Deficient Mice Is Preceded by Abnormal Ventricular Gene Expression. Molecular and Cellular Biology 21(5):1730–1736
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39(12):e82
Cizelsky W, Tata A, Kühl M, Kühl SJ (2014) The Wnt/JNK signaling target gene alcam is required for embryonic kidney development. Development 141(10):2064–2074
Cooper WO, Hernandez-Diaz S, Arbogast PG, Dudley JA, Dyer S, Gideon PS, Hall K, Ray WA (2006) Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med 354(23):2443–2451
DeSesso JM (2017) Future of developmental toxicity testing. Curr Opin Toxicol 3:1–5
Diav-Citrin O, Shechtman S, Bar-Oz B, Cantrell D, Arnon J, Ornoy A (2008) Pregnancy outcome after in utero exposure to valproate: evidence of dose relationship in teratogenic effect. CNS Drugs 22(4):325–334
Diogo R, Kelly RG, Christiaen L, Levine M, Ziermann JM, Molnar JL, Noden DM, Tzahor E (2015) A new heart for a new head in vertebrate cardiopharyngeal evolution. Nature 520(7548):466–473
Donofrio MT, Moon-Grady AJ, Hornberger LK, Copel JA, Sklansky MS, Abuhamad A, Cuneo BF, Huhta JC, Jonas RA, Krishnan A, Lacey S, Lee W, Michelfelder EC Sr, Rempel GR, Silverman NH, Spray TL, Strasburger JF, Tworetzky W, Rychik J, American Heart Association Adults With Congenital Heart Disease Joint Committee of the Council on Cardiovascular Disease in the Young, and Council on Clinical Cardiology, Council on Cardiovascular Surgery, and Anesthesia, Council on Cardiovascular, and Stroke Nursing (2014) Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation 129(21):2183–2242
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ (2012) TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acid Res 40:W117–W122
Gottardis MM, Bischoff ED, Shirley MA, Wagoner MA, Lamph WW, Heyman RA (1996) Chemoprevention of mammary carcinoma by LGD1069 (targretin): an RXR-selective ligand. Cancer Res 56(24):5566–5570
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461(7264):614–620
Inman GJ, Nicolás FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 62(1):65–74
Jain R, Epstein JA (2018) Competent for commitment: you've got to have heart! Genes Dev 32(1):4–13
Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G (2011) Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8(2):228–240
Keegan BR, Feldman JL, Begemann G, Ingham PW, Yelon D (2005) Retinoic acid signaling restricts the cardiac progenitor pool. Science 307(5707):247–249
Kugler J, Kemler R, Luch A, Oelgeschläger M (2016a) Identification and characterization of teratogenic chemicals using embryonic stem cells isolated from a Wnt/β-catenin-reporter transgenic mouse line. Toxicol Sci 152(2):382–392
Kugler J, Luch A, Oelgeschläger M (2016b) Transgenic mouse models transferred into the test tube: new perspectives for developmental toxicity testing in vitro? Trends Pharmacol Sci 37(10):822–830
Kwon C, Arnold J, Hsiao EC, Taketo MM, Conklin BR, Srivastava D (2007) Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. PNAS 104(26):10894–10899
Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix AW Jr, Lott IT, Richard JM, Sun SC (1985) Retinoic acid embryopathy. N Eng J Med 313(14):837–841
Lee JH, Protze SI, Laksman Z, Backx PH, Keller GM (2017) Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 21(2):179–194
Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A et al (1992) 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR alpha. Nature 355(6358):359–361
Li G, Khandekar A, Yin T, Hicks SC, Guo Q, Takahashi K, Lipovsky CE, Brumback BD, Rao PK, Weinheimer CJ, Rentschler SL (2018) Differential Wnt-mediated programming and arrhythmogenesis in right versus left ventricles. J Mol Cell Cardiol 123:92–107
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP (2012) Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. PNAS 109(27):E1848–E1857
Mazzotta S, Neves C, Bonner RJ, Bernardo AS, Docherty K, Hoppler S (2016) Distinctive roles of canonical and noncanonical Wnt signaling in human embryonic cardiomyocyte development. Stem Cell Rep 7(4):764–776
Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, Saphier G, Handsaker RE, Genovese G, Bar S, Benvenisty N, McCarroll SA, Eggan K (2017) Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545(7653):229–233
Moons P, Sluysmans T, De Wolf D, Massin M, Suys B, Benatar A, Gewillig M (2009) Congenital heart disease in 111 225 births in Belgium: birth prevalence, treatment and survival in the 21st century. Acta Paediatr 98(3):472–477
O'Brien TX, Lee KJ, Chien KR (1993) Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. PNAS 90(11):5157–5161
Ou Y, Bloom MS, Nie Z, Han F, Mai J, Chen J, Lin S, Liu X, Zhuang J (2017) Associations between toxic and essential trace elements in maternal blood and fetal congenital heart defects. Environ Int 106:127–134
Poueymirou WT, Auerbach W, Frendewey D, Hickey JF, Escaravage JM, Esau L, Doré AT, Stevens S, Adams NC, Dominguez MG, Gale NW, Yancopoulos GD, DeChiara TM, Valenzuela DM (2007) F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat Biotechnol 25(1):91–99
Russell WMS, Burch RL (1959) The principles of humane experimental technique. Methuen, London
Ryckebusch L, Wang Z, Bertrand N, Lin SC, Chi X, Schwartz R, Zaffran S, Niederreither K (2008) Retinoic acid deficiency alters second heart field formation. PNAS 105(8):2913–2918
Seiler AE, Spielmann H (2011) The validated embryonic stem cell test to predict embryotoxicity in vitro. Nat Protoc 6(7):961–978
Stern RS, Rosa F, Baum C (1984) Isotretinoin and pregnancy. J Am Acad Dermatol 10(5 Pt 1):851–854
van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD (2014) c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509(7500):337–341
van Gelder MM, van Rooij IA, Miller RK, Zielhuis GA, de Jong-van den Berg LT, Roeleveld N (2010) Teratogenic mechanisms of medical drugs. Hum Reprod Update 16(4):378–394
Van Dorp DA, Arens JF (1946) Biological activity of vitamin A acid. Nature 158:60
Xavier-Neto J, Neville CM, Shapiro MD, Houghton L, Wang GF, Nikovits W Jr, Stockdale FE, Rosenthal N (1999) A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 126(12):2677–2687
Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, Sakamoto D, Caothien RH, Fuller JH, Reinhard C, Garcia PD, Randazzo FM, Escobedo J, Fantl WJ, Williams LT (2001) Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta-catenin signaling is activated in human colon tumors. Proc Natl Acad Sci USA 98(26):14973–14978
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. We thank Dr. Jose Xavier-Neto for generously providing the SMyHC3 plasmid. We also thank Juhani Tepsell and Sanna Sihvo for technical assistance. We are grateful to Kirmo Wartiovaara for valuable advice on preparing transgenic lines. We recognize the Biomedicum functional genomics unit (University of Helsinki) for use of qRT-PCR equipment. This research was funded by R.S.L, grants from Business Finland (Tekes, 3iRegeneration, project 40395/13), Biocenter Finland, ILS Doctoral Program; H.J.R. grants from the Academy of Finland (Project 266661), Business Finland (Tekes, 3iRegeneration, project 40395/13), Sigrid Juselius Foundation and Finnish Foundation for Cardiovascular Research; B.L.K, grants from Biocentrum Helsinki, Biocenter Finland, and Business Finland (Tekes, 3iRegeneration, project 40395/13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
204_2019_2632_MOESM5_ESM.mp4
Supplementary file5 (MP4 9544 kb) Video of spontaneously beating reporter mESC-derived cardiomyocytes. Note the presence of venGFP and atrRFP expression in spontaneously beating cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leigh, R.S., Ruskoaho, H.J. & Kaynak, B.L. A novel dual reporter embryonic stem cell line for toxicological assessment of teratogen-induced perturbation of anterior–posterior patterning of the heart. Arch Toxicol 94, 631–645 (2020). https://doi.org/10.1007/s00204-019-02632-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02632-1