
Abstract
Drug-induced liver injury (DILI) cannot be accurately predicted by animal models. In addition, currently available in vitro methods do not allow for the estimation of hepatotoxic doses or the determination of an acceptable daily intake (ADI). To overcome this limitation, an in vitro/in silico method was established that predicts the risk of human DILI in relation to oral doses and blood concentrations. This method can be used to estimate DILI risk if the maximal blood concentration (Cmax) of the test compound is known. Moreover, an ADI can be estimated even for compounds without information on blood concentrations. To systematically optimize the in vitro system, two novel test performance metrics were introduced, the toxicity separation index (TSI) which quantifies how well a test differentiates between hepatotoxic and non-hepatotoxic compounds, and the toxicity estimation index (TEI) which measures how well hepatotoxic blood concentrations in vivo can be estimated. In vitro test performance was optimized for a training set of 28 compounds, based on TSI and TEI, demonstrating that (1) concentrations where cytotoxicity first becomes evident in vitro (EC10) yielded better metrics than higher toxicity thresholds (EC50); (2) compound incubation for 48 h was better than 24 h, with no further improvement of TSI after 7 days incubation; (3) metrics were moderately improved by adding gene expression to the test battery; (4) evaluation of pharmacokinetic parameters demonstrated that total blood compound concentrations and the 95%-population-based percentile of Cmax were best suited to estimate human toxicity. With a support vector machine-based classifier, using EC10 and Cmax as variables, the cross-validated sensitivity, specificity and accuracy for hepatotoxicity prediction were 100, 88 and 93%, respectively. Concentrations in the culture medium allowed extrapolation to blood concentrations in vivo that are associated with a specific probability of hepatotoxicity and the corresponding oral doses were obtained by reverse modeling. Application of this in vitro/in silico method to the rat hepatotoxicant pulegone resulted in an ADI that was similar to values previously established based on animal experiments. In conclusion, the proposed method links oral doses and blood concentrations of test compounds to the probability of hepatotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accurate prediction of human drug-induced liver injury (DILI) based on animal experiments is difficult and is, therefore, the leading cause of drug withdrawal from the market (Godoy et al. 2013). In vitro methods with primary human hepatocytes (PHH) represent a well-established tool to identify concentrations of test compounds that induce toxicity or that cause gene expression alterations (Gebhardt et al. 2003; Vinken and Hengstler 2018; Braeuning et al. 2018). In addition, human hepatocytes engineered to allow in vitro expansion and cell lines are frequently used in this context (Tolosa et al. 2019; Wink et al. 2018; O’Brien et al. 2006). However, the predictive performance of four so far published in vitro studies with PHH in 2D and 3D spheroid culture is limited, resulting in sensitivities of 51, 66, 59 and 69% and accuracies of 71, 71, 67 and 82%, respectively (Xu et al. 2008; Khetani et al. 2013; Proctor et al. 2017; Vorrink et al. 2018). Several reasons may be responsible for the limited predictive performance in vitro. One is that some studies only used PHH from one donor per compound (Proctor et al. 2017; Vorrink et al. 2018), thus not taking interindividual variability into account. A second limitation is that data was analyzed using only the margin of safety (MoS) concept (e.g. O’Brien et al. 2006), where in vitro testing is performed at the maximum concentration of the compound in blood (Cmax) multiplied by a factor that usually ranges between 20 and 100. A positive or negative in vitro test result at this concentration leads to a prediction of hepatotoxicity in vivo that is then compared to the real in vivo situation. However, testing at concentrations that are more than one order of magnitude above in vivo relevant concentrations is potentially problematic, since the mechanisms of toxicity may differ between concentrations at Cmax and Cmax × 20. Moreover, it is difficult to correctly predict if a drug is safe at therapeutic doses when its therapeutic window is relatively narrow. If the ratio between toxic and therapeutic blood concentrations is 20 or less, for example for acetaminophen (APAP), testing at Cmax × 20 (or even Cmax × 100) will result in false positive in vitro test results, even if the compound would test negative at Cmax.
While previous in vitro studies focused on the question of whether a compound can be correctly classified as hepatotoxic or non-hepatotoxic, the present study additionally addresses the possibility of estimating blood concentrations and corresponding oral doses that are associated with a specific risk of hepatotoxicity. We propose the following three steps for human hepatotoxicity prediction: (1) determination of the lowest compound concentrations positive in an in vitro test relevant for in vivo hepatotoxicity, (2) extrapolation to in vivo blood concentration, and (3) reverse modeling to obtain the lowest oral hepatotoxic dose (Fig. 1a).

Strategy of the present study. a Concept of in vitro to in vivo extrapolation. b In vitro–in vivo relationship. The present study aims to predict the lowest compound concentrations in blood that induce hepatotoxicity based on the lowest concentrations in vitro that cause positive test results. c One-sided knowledge of human hepatotoxicity
Cytotoxicity is usually considered a fundamental readout for hepatotoxicity, when investigating the lowest positive concentration in in vitro testing (Step 1) (O’Brien et al. 2006; Godoy et al. 2013). However, alterations in gene expression or functional changes may already occur at concentrations lower than those causing cytotoxicity (Heise et al. 2012; Waldmann et al. 2014). For this reason, it may be advantageous that in vitro test batteries also include a well-chosen set of genes and/or functional readouts in addition to cytotoxicity (Leist et al. 2017). Step 2, which requires extrapolation to the lowest blood concentration that causes an increased risk of hepatotoxicity in vivo, assumes that the lowest positive concentration in vitro is indicative of the lowest blood concentration that causes an increased risk of hepatotoxicity (Louisse et al. 2017; Chen et al. 2018; Yoon et al. 2015). However, the relationship between the lowest positive in vitro and in vivo concentrations has not yet been systematically studied. Arguments in favor of this assumption are that cultivated PHH may import and export chemicals from the culture medium in a manner similar to hepatocytes in vivo, which import and export the same compounds from the blood. This also assumes that interactions of test compounds with cellular mechanisms occur similarly in vitro and in vivo once the test compound has entered the hepatocyte. However, it is highly likely that there are differences between hepatocytes within the intact liver and cultivated PHH (Godoy et al. 2009). Therefore, the exact relationship between the lowest positive concentrations in vitro and in vivo remains to be elucidated. Several mechanisms may complicate the establishment of this relationship, with pharmacokinetics playing a major role. In vitro concentrations of the compound in the culture medium may initially decrease quite rapidly due to adsorption, for example to the culture dish, usually followed by a relatively slower decrease due to its metabolism by hepatocytes (Kramer et al. 2015). In contrast, concentration changes in vivo are usually faster, with an initial increase due to absorption from the intestine, followed by a decrease due metabolism and renal as well as biliary excretion (Reif et al. 2017; Thiel et al. 2015). Moreover, blood has a higher protein concentration than culture media, which leads to differences between the free and protein-bound fraction of test compounds. Finally, cultivated PHH do not respond identically to hepatocytes in an intact liver, because the isolation and cultivation processes used to obtain PHH have been shown to induce changes in hepatocellular functions (Godoy et al. 2015, 2016). Extrapolation between blood concentrations and oral doses (step 3) can be performed by pharmacokinetic reverse modeling. However, it should be considered that this is still associated with a degree of uncertainty, although there has been much progress in physiologically based pharmacokinetic (PBPK) modeling in recent years (Jones et al. 2015; Wagner et al. 2015).
After determining the best strategy to predict hepatotoxicity, it is important to consider the type of data needed (Fig. 1b, c). Repeated oral dosing of mammalian species with test compounds leads to blood concentrations that can be readily described using pharmacokinetic parameters, such as Cmax, half-life, and steady state concentrations (Fig. 1b). Reliable information on pharmacokinetics in blood is available in humans for pharmaceutical drugs and some industrial chemicals. For in vitro to in vivo extrapolation it would be helpful to know the in vivo test compound concentration in the target cells (Fig. 1b). Such information is usually not available for hepatocytes and determining intracellular drug concentrations in vivo is challenging. However, for marketed drugs it is known if certain doses lead to hepatotoxicity or are non-hepatotoxic; literature and databases (e.g. https://livertox.nih.gov/) provide information on whether patients treated with specific drugs will suffer from mild or severe forms of hepatotoxicity. Using in vitro tests with PHH, the nominal, i.e., initially established, concentration in the culture medium applied to the cells, is known (Fig. 1b). Theoretically, it would be ideal to directly compare toxicity to hepatocytes in vitro and in vivo for known intracellular concentrations (dashed line in Fig. 1b). However, the lack of knowledge about intracellular concentrations in human livers in vivo, and the experimental effort for determining intracellular concentrations in vitro, makes this approach unfeasible. In contrast, it is possible to study the relationship between the lowest concentrations in vitro that cause positive test results (i.e., alteration of a measured parameter to a certain extent) and concentrations in blood that cause hepatotoxicity in vivo. If this relationship could be mathematically described, it would represent an important step for extrapolating in vitro data to the in vivo situation.
One of the challenges of this proposed strategy is that the lowest blood concentration of a test compound that causes an increased risk of hepatotoxicity in humans is often unknown. Pharmaceutical drugs are administered according to specific dosing regimens, and for these therapeutic doses, toxicity information is usually available from larger population cohorts. Therefore, three scenarios should be considered to address this challenge (Fig. 1c). In the first scenario, a specific dosing schedule that leads to known blood concentrations (e.g. Cmax) causes an increased risk of hepatotoxicity, and as a result higher doses and blood concentrations will also be hepatotoxic (Fig. 1c, upper panel). However, for most hepatotoxic drugs it is not known by how much a dose has to be reduced to decrease the risk of hepatotoxicity to zero, because patients only receive therapeutic doses. In other words, the lowest observed adverse effect level (LOAEL) that is routinely determined in animal experiments is not known for most hepatotoxic drugs in humans. In scenario two in which a specific dosing schedule and the resulting blood concentrations do not cause an increased risk of hepatotoxicity, it is usually unknown if higher doses would be hepatotoxic (Fig. 1c, middle panel). Therefore, it remains open whether 100-fold higher doses would be required to reach the hepatotoxic level or if a twofold dose escalation is sufficient. Scenario three represents the few exceptions for which the human LOAELs are precisely known, one example being acetaminophen (Fig. 1c, lower panel). Studies have shown that doses of up to 4 g per day do not cause an increased risk of hepatotoxicity (Mazaleuskaya et al. 2015; Civan et al. 2014), while higher doses of 7–10 g from accidental or suicidal intoxications do increase the risk of hepatotoxicity. However, for most drugs, the specific therapeutic window, hence the toxic threshold in humans remains, elusive.
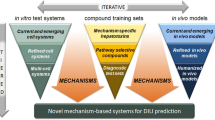
The goal of this study was to establish an in vitro/in silico method to estimate the risk of human hepatotoxicity associated with oral doses and blood concentrations of compounds for which this information is unknown. The steps required to reach this goal (Fig. 2) include the establishment of new performance metrics to optimize the in vitro test with cultivated hepatocytes concerning the input parameters cytotoxicity as well as gene expression, and to identify pharmacokinetic parameters that can be estimated best by the test method. Based on a logistic regression model the risk of hepatotoxicity associated with specific blood concentrations was calculated. Finally, reverse pharmacokinetic modeling was applied to simulate which oral doses lead to these blood concentrations. All experiments performed for primary human hepatocytes were replicated with HepG2 cells to study whether investment into the more expensive primary hepatocytes is justified. Although validation with higher numbers of compounds is still required, the data presented here shows that it is feasible to estimate the acceptable daily intake with regard to human hepatotoxicity of an unknown compound based on the lowest concentration that causes a positive result in the in vitro test.

Working pipeline. The goal of the present study is to estimate oral doses associated with a specific risk of human hepatotoxicity based on in vitro data and modeling. The workflow illustrates the individual milestones leading to this goal, and the figures with the corresponding key results
Materials and methods
Test compounds and concentrations
Detailed information about the test compounds, solvents, preparation of stock solutions and covered concentration ranges are given in Supplement 1. Background information on the individual test compounds, such as the suggested mechanisms of hepatotoxicity, type of liver injury, degree of DILI concern, and idiosyncratic mechanisms is also available in this supplement. The information whether individual drugs cause an increased risk of hepatotoxicity was obtained from the database https://livertox.nih.gov/ and from several other sources listed in Supplement 1 under “hepatotoxicity information” and is briefly summarized in Table 1. The rationale for the chosen concentration ranges was (1) to test high enough concentrations to induce cytotoxicity. First choice was to dissolve the test compounds in culture medium. If solubility in the culture medium was not sufficient to reach cytotoxic concentrations, DMSO was used as a solvent, whereby 0.1% DMSO served as a standard concentration. Only if 0.1% DMSO was not sufficient to induce cytotoxicity, higher concentrations up to 0.5% were applied. In all cases controls with identical DMSO concentrations were used; (2) to include the in vivo Cmax into the tested concentration range.
Primary human hepatocytes
Cryopreserved primary human hepatocytes (PHH) were purchased from BioIVT. Freshly isolated PHH were obtained from Hepacult, and the University of Leipzig. The isolation of freshly isolated PHH is described in Godoy et al. 2013. In total, PHH from 17 different donors were used. The donor characteristics are given in Supplement 2. Cytotoxicity experiments were performed exclusively with cryopreserved PHH. Gene expression experiments were performed with both freshly isolated and cryopreserved PHH.
Cell culture of PHH and HepG2 cells
In the current study, cryopreserved PHH, freshly isolated PHH, and the HepG2 cell line (ATCC number: HB-8065™) were used. The identity of the HepG2 cells was confirmed by short tandem repeat (STR) profiling once a year by DSZM (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures). Cryopreserved PHH were cultured according to a published standard operating procedure (SOP) (Gu et al. 2018 supplement 2). Culture of freshly isolated PHH followed a published standard protocol (Grinberg et al. 2014). The SOP for the cultivation of HepG2 cells is given in Supplement 3A.
Cytotoxicity analysis with PHH and HepG2 cells
The cytotoxicity assay with PHH was performed using the CellTitre-Blue (CTB) assay as described in Gu et al. (2018) according to the SOP in Supplement 3A. The same cytotoxicity assay was also used for HepG2 cells (Supplement 3A). William’s E medium for cultivation of PHH contained 11 mM glucose, while Dulbecco’s Modified Eagle’s Medium (DMEM) used for HepG2 cells contained 25 mM glucose. The glucose concentrations indicated in the results section were reached by adding glucose accordingly. All tested concentrations of the compounds and solvents are given in Supplements 1, 4 and 5.
Gene expression analysis in PHH and HepG2
Expression analysis of seven genes (CYP1B1, CYP3A7, SULT1C2, FBXO32, TUBB2B, G6PD and RGCC) was performed by real-time quantitative reverse transcription PCR (qRT-PCR). TaqMan probes for the seven genes, as well as two housekeeping genes (GAPDH and UBC), were obtained from Applied Biosystems. The SOPs for the treatment of cells, isolation of RNA, reverse transcription, primers, and qRT-PCR conditions are given in Supplement 3A. For the analysis of gene expression freshly isolated and cryopreserved hepatocytes were used (Supplement 2). Donor characteristics and the donor used for analysis of each compound and experiment are given in Supplement 2. Gene expression was calculated relative to the expression of the housekeeping genes GAPDH and UBC as described in Supplement 3A. Only samples with a stable expression of the housekeeping gene (deltahousekeeper ≤ 0.5) were further analysed.
Glutathione depletion assay in HepG2 for evaluating the oxidation stress
HepG2 cells (15,600 viable cells in 200 µl DMEM) were seeded in black, clear-bottomed 96-well plates (Sigma Aldrich), 24 h prior to exposure to test compounds as described in Supplements 1, 4 and 5. After 48 h of exposure, the cell culture medium was removed and the cells were incubated with 0.5 µg/ml Hoechst 33342 diluted in culture medium for 30 min at 37 °C, to stain the nuclei of the living cells. After the incubation period, fluorescence was measured at an excitation wavelength of 340 nm and emission wavelength of 450 nm using a spectrophotometer (TECAN SpectraFluor Plus). The media containing Hoechst 33342 were removed, the cells were washed once with warm PBS and then incubated with 100 µl of 40 µM dibromobimane (Sigma Aldrich) diluted in PBS for 30 min at 37 °C. To determine background fluorescence, wells without cells that were incubated with dibromobimane (40 µM) in PBS were included. Fluorescence was measured at an excitation wavelength of 393 nm and an emission wavelength of 477 nm. To determine the glutathione (GSH) levels, the background fluorescence for dibromobimane was subtracted from the values obtained in wells that contained cells, and these background-corrected fluorescence values were normalized to the live cell number reflected by the fluorescence signal of Hoechst 33342. The normalized values were then used to fit a dose–response curve, as described below. The concentrations used are given in Supplement 5. An SOP of the glutathione assay is given in Supplement 3A.
Statistical analysis
All statistical analyses were performed with the statistical programming language R-version 3.5.1 (https://www.R-project.org/).
Curve fitting and calculation of EC values
The raw data were processed as follows: CellTitre-Blue Reagent® was used to evaluate the cell viability. Background controls (fluorescence values from CellTiter-Blue Reagent® mixed with medium that was not in contact with cells) were subtracted from each data point. Replicates of control values were averaged for each biological replicate separately. Next, fluorescence values of all samples were divided by the corresponding averaged control values and multiplied by 100 to obtain percentages. Based on the assumption that the concentration–response relationship can be described by a sigmoidal curve, a four-parameter log-logistic model (4pLL) was fitted to the data using the R package drc version 3.0-1 (Ritz et al. 2015). For a concentration x and four parameters b, c, d, e, the response is given by:
where c and d are the values of the right and left asymptote, respectively, b is the slope and e is the concentration at which 50% of the overall effect is observed. For given concentrations and background-corrected, normalized response values, the parameters were estimated numerically according to the least square method with the Gauss–Newton algorithm. The above described curve fitting procedure can lead to a left asymptote that reaches values higher or lower than 100%. To ensure that the left asymptote attains a value of 100%, a refit-procedure was conducted. All response values were divided by the value of the left asymptote after the first fit and again multiplied by 100 to obtain percentages. The 4pLL model was again fitted to the data. ECk values were calculated as the concentration where the sigmoidal curve attains the value (100 − k)% (e.g. EC10 corresponds to the concentration where the sigmoidal curve reaches the value 90%). To assess the quality of the curve-fit, a goodness-of-fit statistic was calculated as 1 − [(the sum of squared differences between the data points and the fitted curve)/(the sum of squared differences between the data points and the mean response)], which gives values near 1 for a good fit and values near 0 for a poor fit; curves with goodness-of-fit of at least 0.55 were used to derive ECk values for further analysis. Confidence intervals of the EC values were calculated by the delta method which approximates the variance of the inverse function of f (Grinberg 2017).
The above described procedure for the determination of EC values may result in values lower or higher than the actually tested concentrations. The calculated EC values were only accepted if they were within the interval [concmin/5, concmax × 5], whereby the choice of a range of five serves to avoid EC-values too far from the lowest and highest tested concentrations. Values below concmin/5 were set to “< concmin/5” and values above concmax × 5 were set to “> concmax × 5”. Cases in which the respective EC value could not be calculated, because the function never reached the target value on the y-axis (e.g., 50% for the EC50), were set to “> concmax”.
In subsequent analyses, the median EC value of the three biological replicates was used for each compound. Alternatively, the minimum or the maximum of three EC values was used. When one or more of the three EC values were outside of the acceptable interval (< concmin/5 or > concmax × 5), the following rules were applied to ensure that minimum, median and maximum EC values were available: (i) if a donor had an EC value < concmin/5, it was replaced by concmin/5; (ii) if a donor had an EC value > concmax × 5, it was replaced by concmax × 5. The effective concentrations are given in Supplement 6, the fitted curves are shown in Supplement 7A-C and the goodness of fit is summarized in Supplement 8.
Calculation of toxicity separation and estimation indices
Two indices were established, the toxicity separation index (TSI), which quantifies how well a test method differentiates between hepatotoxic and non-hepatotoxic compounds, and the toxicity estimation index (TEI), which measures how well hepatotoxic blood concentrations in vivo can be estimated by an in vitro test system. Input data for the calculation of both TSI and TEI are in vitro (e.g. EC10, EC50) and in vivo (e.g. Cmax, AUC) concentrations. A detailed description how the TSI and TEI are calculated, with specific examples, is given in Supplement 3B.
Briefly, to calculate the TSI, the difference between the in vivo concentration for a given exposure scenario and the positively tested in vitro concentration is calculated on log10 scale for each compound. The differences are then sorted in ascending order and for each interval between two consecutive differences, a cutoff value, is chosen. For each cutoff value a prediction of the toxicity status (hepatotoxic or non-hepatotoxic) of each compound is performed and compared to the true toxicity status. Thereby, sensitivity and specificity can be calculated for each cutoff. 1-specificity is then plotted against sensitivity for each cutoff value and the TSI is calculated as the AUC, i.e., the area under the ROC curve. The R package pROC version 1.13 was used (Robin et al. 2011). A TSI of 1 is optimal since it coincides with the perfect separation of toxic and non-toxic compounds, while a TSI of 0.5 is obtained for a random class assignment to the compounds.
To calculate the TEI, non-toxic compounds are excluded. The TEI is calculated as:
where i = 1,…, n represent the compounds in question, x(i) and y(i) the in vitro value and the in vivo value of compound i, respectively, and \( 1_{{\left( {\text{condition}} \right)}} \)(i) the indicator function which takes the value 1 if the condition is fulfilled by the compound i, otherwise 0. A value of 1 represents the optimal value of the TEI. In case of missing in vitro alerts a penalty factor was used (Supplement 3B).
Hepatotoxicity prediction
A support vector machine (SVM) was used to classify compounds as hepatotoxic or non-hepatotoxic, employing cytotoxicity median EC10 values (48 h compound incubation) and Cmax (total concentration; 95% percentile) as input variables. The classification performance was assessed using leave-one-out cross validation with 30 iterations. With this approach, one compound is left out and a classifier is constructed using variables from the remaining compounds and their toxicity status (i.e., hepatotoxic or non-hepatotoxic) as input. Next, the classifier is used to predict whether the left-out compound was hepatotoxic or not. This procedure is then repeated until all compounds were left out once. The original data points can be considered to lie in a vector space where the number of dimensions corresponds to the number of input variables. The aim is to identify a hyperplane, which separates the observations of the two classes (hepatotoxic and non-hepatotoxic). Since only two input variables (in vivo Cmax and in vitro median EC10) were used in the analysis finally presented in Fig. 7d, the vector space was a two-dimensional coordinate system and the hyperplane a straight line. The line was chosen to maximize the size of the margin (i.e., the minimal distance from all data points to the line) and simultaneously constrain the sum of errors by a given constant C (i.e., misclassifications are allowed when the data cannot be perfectly separated by a straight line). The R package mlr version 2.13 (Bischl et al. 2016) was used as a framework for classifier training and evaluation, and the package kernlab version 0.9-27 (Karatzoglou et al. 2004) for the SVM classification. Default settings were used for the hyperparameter. In addition to the allocation of the compounds to one of the two groups, the probability of group membership was calculated using a logistic regression model fitted to the differences between the data points and the separating line (Platt 2000). The separating line between the two categories (hepatotoxic and non-hepatotoxic) corresponds to a probability of 50% to belong to the hepatotoxic compounds. Based on the fitted logistic regression model a distance from the separating line can be determined for any other probability. By this method lines parallel to the separating line with probabilities of 10, 5, 1, 0.5 and 0.1% were defined.
Simulation of pharmacokinetics
For each of the test compounds, a physiologically based pharmacokinetic (PBPK) model was constructed using the Simcyp Simulator (commercial software, Version 15; SimCyp, Sheffield, UK). The input parameters used to describe the compounds within the human PBPK models are given in Supplement 3C and Supplement 9). The performance of the PBPK model for each compound was assessed by comparing the simulated concentrations of the compounds in blood with available data. The simulations were performed in populations of virtual individuals who reflected a European population in terms of age and sex. In this exercise, if the drug or chemical was known to have a significant circulating metabolite (e.g., aspirin with its metabolite salicylic acid), both the parent and metabolite were simulated. If a compound was known to be significantly metabolised by an enzyme that is polymorphically expressed in the population, e.g., CYP2D6, simulations were conducted for both the extensive and poor metaboliser phenotypes. A potential limitation of the adopted approach in the fit for purpose models described here is that the role of hepatic uptake in the disposition of the compounds was not considered. Once the initial PBPK model was constructed and the performance checked against clinical data, models were refined as required to produce a model that better described the clinical data. Finally, simulations were conducted for each of the compounds at the specified doses in a population of 100 North European Caucasian subjects (age 20–50, 50% female). Multiple dose simulations were conducted for a long enough period to ensure that steady state concentrations were achieved. From the simulations, the Cmax, Cmax portal vein and average steady-state concentration cav, ss (AUC0−t/dosing interval) were calculated for both the first dose and at steady-state. The mean values in the population as well as the concentrations corresponding to the 5th and 95th percentiles of the population were calculated.
Results
Establishment of metrics for evaluation of in vitro test method performance
To evaluate the performance of an in vitro test method, two new metrics—the toxicity separation index (TSI) and toxicity estimation index (TEI)—were introduced, where TSI considers the separation of hepatotoxic from non-hepatotoxic compounds, and TEI estimates how well hepatotoxic blood concentrations in vivo can be estimated for hepatotoxic compounds (Fig. 3). Assessment of in vitro test methods using these two indices may be advantageous, because the general performance of different methodological alternatives can be compared, for example different cytotoxicity cutoffs or the inclusion of additional readouts for a given set of compounds. Once an optimized test method has been established, it can then be applied to independent compounds where the performance is assessed in standard terms, such as sensitivity and specificity.

a–d Concept of the toxicity separation (TSI) and toxicity estimation (TEI) indices (color figure online)
Both TSI and TEI are calculated based on the projection of a predefined battery of test compounds onto a two-dimensional coordinate system, where the x-axis indicates the lowest concentrations that cause a positive test result (‘in vitro alert’, such as decreased viability or increased expression of genes) of any test method, and the y-axis indicates the in vivo blood concentrations (e.g., Cmax) that result from a specific dosing schedule. In this in vitro-to-in vivo extrapolation plot (shortened: ‘extrapolation plot’), each test compound is represented by a symbol. Red and green color indicate whether the individual compounds cause an increased risk of hepatotoxicity (red) or are non-hepatotoxic (green) at the corresponding Cmax. For ease of understanding, the principles of TSI and TEI are illustrated with hypothetical scenarios (Fig. 3a–d). TSI measures how well a test method differentiates between hepatotoxic and non-hepatotoxic compounds. It covers a range from 0.5 to 1.0, where a TSI of 1.0 indicates a perfect separation, while 0.5 represents a random result. The hypothetical examples illustrate both good (Fig. 3a, b) and poor (Fig. 3c, d) separation of hepatotoxic and non-hepatotoxic compounds. The concept of separation in such a plot is based on the assumption that the difference between the in vitro alert concentration and its corresponding concentration in vivo is larger for non-hepatotoxic than for hepatotoxic compounds. The diagonal line in the extrapolation plot indicates a hypothetical situation where the in vitro alert concentration exactly corresponds to the in vivo hepatotoxic blood concentration for the hepatotoxic compounds (‘iso-concentration line’).
TEI measures how accurately an in vitro test method estimates hepatotoxic blood concentrations in vivo; in other words—it measures how far the red points are below the iso-concentration line, e.g., a TEI of 1 indicates a position where all hepatotoxic compounds (red points) are on the iso-concentration line or above. Therefore, shifting all points in Fig. 3a downwards leaves the TSI unchanged, but decreases the TEI, as shown in Fig. 3b. If all points lie on, or very close to, the iso-concentration line, as in Fig. 3c, the TEI is high, but the test method has no, or only little, discriminatory power (i.e., low TSI). Finally, shifting the points in Fig. 3c downwards results in both poor TSI and poor TEI (Fig. 3d). When designing a good in vitro method, priority is given to obtaining a high TSI, since the first objective is to discriminate whether a compound is hepatotoxic or not. A high TEI is also desirable but should not be achieved at the expense of a worse TSI, since this metric is only relevant once hepatotoxic compounds have been reliably identified.
Once an extrapolation plot has been established for a set of hepatotoxic and non-hepatotoxic compounds, it can then be used to assess additional substances without having information on their hepatotoxicity by adding them to the existing plot. The position on the x-axis is determined in vitro, while additional knowledge is required for the y-axis location.
In vitro data generation and PBPK modeling
This chapter gives an overview over the generated data, while the actual application as summarized in the working pipeline (Fig. 2) follows in the next chapters. For in vitro test system optimization with concrete TSI and TEI values, the lowest concentrations of 28 test compounds that caused a positive result in vitro were presented on the x-axis of the extrapolation plot, and the blood concentrations (Cmax) established by PBPK modeling were plotted on the y-axis. To generate the required data, PHH from three donors were used to perform concentration-dependent cytotoxicity analyses of the 28 compounds using the (CTB) assay according to a published standard operation procedure (Fig. 4a, left panel; Supplement 3A). After fitting a sigmoidal dose–response curve, EC values ranging from EC10 to EC80 with a stepwise increase of 10 were calculated. Figure 4b illustrates the example of the EC10 for one compound (clonidine, CLON) in PHH. The raw data generated from the 28 compounds tested in PHH from three donors at five concentrations plus solvent controls are available in Supplement 4. An overview of the EC10 values for all compounds is given in Table 1. In a subsequent step, expression of a previously published seven-gene panel (CYP1B1, CYP3A7, TUBB2B, SULT1C2, G6PD, RGCC and FBXO32) (Grinberg et al. 2014) was determined in a concentration-dependent manner in cultivated PHH from three donors for the 28 compounds. The results for one compound (valproic acid, VPA) are shown in Fig. 4c; data obtained for all compounds and donors are available in Supplement 4 and 6. Cytotoxicity and the expression of the seven genes was also determined in a concentration-dependent manner for HepG2 cells in three independent experiments, as done for PHH (Fig. 4a, right panel, d, e, Table 1, Supplement 5, 6). Moreover, GSH depletion was measured as an additional in vitro endpoint in HepG2 cells for evaluation in a pilot study (Fig. 4f) as described below. The processed data for PHH and HepG2, all fitted curves and the goodness of fit for these curves are given in Supplements 6, 7 and 8.

In vitro data generation and PBPK modeling. a Exposure schedules for cytotoxicity tests with cultivated primary human hepatocytes (PHH) and HepG2 cells. Schedules of all in vitro tests used in the present study and the corresponding standard operation procedures are in Supplement 3. b Concentration response curve of cytotoxicity in PHH for clonidine (CLON) as an example. The vertical lines indicate the EC10 value and its 95% confidence interval. Cytotoxicity data of all test compounds, including the raw data are in Supplement 7. c Expression data of the 7-gene signature, for the example of valproic acid (VPA) in PHH. The lowest positive test concentration is defined as the lowest concentration at which one of the seven genes exceeds expression of the corresponding solvent control by a factor of 2.5 (red line). The error ranges are standard errors of the mean (SEM) of three independent experiments. Expression data of all test compounds, including the raw data, are in Supplement 4, 5 and 6. d Example of cytotoxicity analysis (clonidine; CLON) in HepG2 cells. e Example of expression analysis in HepG2 cells for valproic acid (VPA). f Example of glutathione depletion analysis in HepG2 cells. g Illustration of pharmacokinetic parameters for the example carbamazepine. Cmax: maximal blood concentration (total concentration, i.e., free and protein bound); Cmax steady state: maximal blood concentration in the steady state; Cav steady state: average blood concentration in the steady state. h Correlation plots of Cmax and Cmax, in steady state, as well as Cmax in whole blood (of the general circulation) and Cmax in blood of the portal vein. Each symbol represents one test compound. The lines crossing the symbols indicate the ranges between 5th and 95th percentiles. The complete set of PBPK modeling data is given in Supplement 10 (color figure online)
Pharmacokinetic modeling was performed for oral dosing schedules used in clinical routine (Supplement 9). For all compounds, the blood Cmax, Cmax at the steady state (Cmax,ss) and the average concentration at steady state (Cav,ss) (Fig. 4g) were calculated for (a) total concentration (protein bound plus free compound) in blood from the general circulation, (b) free, non-protein bound concentrations in blood from the general circulation, and (c) total concentrations (protein bound plus free compound) in blood from the portal vein (Supplement 9). Besides pharmacokinetic modeling, a comprehensive literature search was performed for experimentally analyzed blood concentrations for the different test compounds (Supplement 6). All pharmacokinetic parameters correlated with one another (Supplement 10). One example of the correlation plots for total (protein-bound and unbound) concentrations in the general circulation of Cmax versus Cmax,ss is illustrated in Fig. 4h, where Cmax,ss was only slightly higher than Cmax for most compounds. In a second example, the correlation plot of Cmax in the general circulation versus the corresponding concentration in the portal vein shows that portal vein concentrations can be higher than concentrations in the general circulation, which is plausible for orally administered compounds with a high first pass effect (Fig. 4h). Correlation plots for pharmacokinetic parameters and physicochemical properties with blood concentrations of the study compounds are given in Supplement 10. As expected, the daily dose of the test compounds strongly correlates with the Cmax in blood (Supplement 10A); Cmax of the hepatotoxic compounds is higher compared to the non-hepatotoxic substances. Moreover, a weak inverse correlation between hydrophobicity and Cmax was observed (Supplement 10B), whereas Cmax showed a weak inverse correlation with the molecular weight of the tested compounds (Supplement 10C). Key parameters, including Cmax, EC10 (median of the three donors), and the lowest positively tested concentrations of the seven genes are summarized in Table 1, and the complete set of data is available in Supplements 1, 6, and 9.
An important aspect for test development is whether specific dosing regimens of drugs (or specific levels of exposure to environmental compounds) lead to an increased probability of hepatotoxicity. This information is given in Table 1 (sources and details in Supplement 1) for the dosing schedules summarized in Supplement 9. For most of the drugs in Table 1, reliable information was only available for one (or for a few similar) therapeutic dosing schedule. An exception is acetaminophen where not only non-hepatotoxic doses and therapeutic blood concentrations are available, but also comprehensive data from overdoses that lead to hepatotoxicity (Table 1 and, Supplement 9). Therefore, acetaminophen appears twice in Table 1, with a hepatotoxic and a non-hepatotoxic blood concentration. Besides pharmaceutical compounds, certain chemicals (ethanol, dimethyl sulfoxide, glucose monohydrate, methylparaben and triclosan) were also included (Table 1). Ethanol was considered, because large studies are available that provide information on doses, and associated blood concentrations, leading to liver damage when exposure continues over longer periods of time (Supplement 9). In contrast, the very low ethanol blood concentrations observed after transdermal exposure during hand disinfection can be considered non-hepatotoxic. Therefore, ethanol also appears in Table 1 with both a hepatotoxic and a non-hepatotoxic Cmax.
In vitro test optimization based on cytotoxicity
The above-introduced concept of TSI and TEI was applied to the 28 test compounds to determine which cytotoxicity parameter (EC-threshold, incubation period) is optimal (x-axis), while Cmax (total concentration; 95% population percentile; y-axis) was kept constant. When PHH from three donors were tested for cytotoxicity, the first question to be answered was from which donor the cytotoxicity data should be used, the median, minimum or maximum. A second important question was whether the often-used EC50 value is optimal or if other EC values (EC10up to EC80) are superior. To systematically address these questions, extrapolation plots were generated, considering all the different parameters for the x-axis, and the corresponding TSI and TEI were determined and plotted against each other (Fig. 5a). Higher TSI values were obtained when the median donor values were used, compared to the corresponding minima and maxima (Fig. 5a). Moreover, a consistent and relatively strong decrease in TEI was obtained when EC values were increased from EC10 to EC80. This was observed for the minimum, maximum, as well as the median values (Fig. 5a). Based on these results, the median EC10 value was chosen for further analysis of cytotoxicity.

Test system optimization for cytotoxicity. a Relationship between EC value (EC10, EC20, to EC80) and TSI as well as TEI. EC values are given for the hepatocytes from three donors, red color representing the most susceptible (minimum), blue the median, and green the most resistant (maximum) donor. The intensity of the dots represents the respective EC values with the darkest dots representing the respective EC10 values and the lightest dots representing the respective EC80 values. b Relationship between incubation period (1, 2 and 7 days) and TSI as well as TEI. c Extrapolation plot of the EC10 of the median donor. Each compound was tested with hepatocytes from three donors. To illustrate interindividual variability, the EC10 values for each individual donor are given by a diamond, and the three diamonds corresponding to one test compound are connected by a line. The vertical lines crossing each median EC10 value illustrate the ranges between 5th and 95th percentiles of Cmax (total maximal blood concentrations). The 95th percentile coincides with the median diamond, the horizontal dashes below are median values and 5th percentiles. Dashed diagonal line: iso-concentration line (identical concentrations in vivo and in vitro). The key for the abbreviated compound names is given in Supplement 1 (color figure online)
A third question was how long cultivated PHH should be exposed to the test compounds for cytotoxicity testing. In the present study, an incubation period of 48 h was used. Additionally, incubations of 24 h and 7 days (with repeated culture medium changes with fresh test compound) were performed as previously reported (Gu et al. 2018). A higher TSI was obtained for the 48 h compared to the 24 h and 7 day incubation periods (Fig. 5b). This observation was independent of the EC cutoff, as exemplified for EC10, EC20 and EC50 in Fig. 5b. Therefore, the median EC10 with 48 h of compound exposure were used to generate an extrapolation plot (Fig. 5c). Each compound was identified by an abbreviation defined in Table 1, where red and green symbols indicate hepatotoxic and non-hepatotoxic compounds, respectively. In general, hepatotoxic compounds were located above the non-hepatotoxic compounds. This resulted in an almost optimal TSI of 0.996 but a lower TEI of 0.844, since most of the hepatotoxic compounds clustered below the iso-concentration line (Fig. 5c).
Integration of gene expression into the in vitro test
Next, we evaluated if adding gene expression as an additional readout to the optimized version of the test method obtained above could further improve its performance. For this purpose, seven genes (Fig. 4c, e) were selected from a previously published study analyzing genome-wide expression data in cultivated human hepatocytes of 143 compounds (Grinberg et al. 2014). The selection criteria were (1) gene expression increased by many compounds; (2) gene expression increased in human liver disease (steatosis, fibrosis and cirrhosis) to support in vivo relevance; and (3) gene expression not altered by the hepatocyte isolation and cultivation procedure. Although the goal was to include gene expression into the test battery together with cytotoxicity, the seven genes were initially analyzed alone and in combination (without cytotoxicity). Extrapolation plots were generated, which included each of the seven genes individually, with the lowest as well as the median and the maximum compound concentrations that induced gene expression plotted on the x-axis, and the Cmax on the y-axis (Fig. 6a–g). To identify an appropriate cutoff, an analysis of TSI and TEI was performed where all seven genes were considered. Gene expression at a specific test compound concentration was defined as positive if the expression of the most sensitive gene increased 1.5-fold; this cutoff value was increased to 5.0-fold in steps of 0.1 (Fig. 6i). A maximal TSI was obtained for cutoffs ranging between 2.1 and 2.6; therefore, a cutoff of 2.5 was further used to define a positive test result. A comparison of the median and the minimum alert concentration among the three donors (with a 2.5-fold cutoff) demonstrated that using the median leads to a higher TSI (Fig. 6j). The maximum gene expression alert concentrations (the most resistant donor) are not shown, because more compounds did not reach the cutoff of 2.5-fold, which would lead to the disadvantage of a high number of compounds without in vitro alert. Together, these results justify the use of a 2.5-fold gene expression cutoff for the median donor for further analyses.

Expression of seven genes in relation to human hepatotoxicity. a–g Results for each of the seven genes, CYP3A7, CYP1B1, G6PD, SULT1C2, FBXO32, RGCC, and TUBB2B. The x-axis gives the lowest positive test compound concentrations, based on an at least 2.5-fold median increase over solvent controls. The y-axis represents the Cmax (total maximal blood concentration, 95% CI). Red and green symbols represent hepatotoxic and non-hepatotoxic compounds, respectively. Compounds that did not increase expression of the corresponding genes by at least 2.5-fold are listed under “no alert” in the right column of each plot. h Combination of the seven genes. The x-axis gives the lowest alert (at least 2.5-fold increase of the median) achieved by the most responsive of the seven genes. i Variation of the cutoff for definition of positive test results for the lowest alert resulting from expression analysis of the seven genes. The cutoff (median) was varied between 1.5 and 5.0 in intervals of 0.1. j TSI and TEI for each of the seven genes. Circles represent the analysis of the median, triangles of the most susceptible donor (minimum). Dashed lines in a–h are iso-concentration lines (color figure online)
The data of the individual genes (Fig. 6a–g) showed that only some of the compounds generated a positive test result in the gene expression assay. Despite this limitation, a relatively good separation of hepatotoxic and non-hepatotoxic properties was possible for the substances that did reach an alert concentration. Therefore, a combined analysis of all seven genes was performed, and a concentration defined as positive when at least a 2.5-fold increase was obtained for the median alert concentration per compound, for at least one gene (Fig. 6h). Even under these conditions, three hepatotoxic (ETOHhigh, LAB, LEV) and seven non-hepatotoxic (ETOHlow, FAM, GLC, HYZ, MEL, PPL, and TSN) compounds did not generate an alert. The results show that gene expression may support the differentiation of hepatotoxic and non-hepatotoxic compounds, but only if an alert concentration is observed; however, with only seven genes not all test compounds can be assessed.
A subsequent goal was to study if TSI or TEI can be improved by combining cytotoxicity (median EC10, 48 h incubation) with gene expression. For this purpose, all possible combinations (n = 128) of gene expression—for zero up to seven genes—with cytotoxicity were analyzed (Supplement 11). In each combination, the readout (alert of median gene expression or EC10) that resulted in the lowest positively tested concentration was considered. None of the combinations improved TSI beyond 0.996, which was already achieved by the median EC10 alone. However, the TEI of EC10 (0.844) was improved by additionally considering gene expression (Fig. 7a). CYP1B1 and CYP3A7 were of particular relevance in the combined scenario. When three genes were considered—CYP1B1 and CYP3A7 and a third gene (G6PD, SULT1C2, or TUBB2B)—a maximal TEI of 0.887 was obtained. Adding further genes, up to all seven, did not further improve the TEI (Fig. 7a). In conclusion, a combination of cytotoxicity (median EC10, 48 h incubation) and the expression of three genes as specified above resulted in optimal TSI and TEI for the analyzed set of compounds, as illustrated in Fig. 7b.

Gene expression, pharmacokinetic parameters and classifier construction. a Improvement of the TEI by combining cytotoxicity with gene expression. The TEI of cytotoxicity (EC10) alone is indicated by the red dot. Combination of cytotoxicity (EC10, median) with three genes, CYP3A7, CYP1B1 and a third gene (G6PD, SULT1C2 or TUBB2B) leads to the highest TEI. b Extrapolation plot combining cytotoxicity (EC10) and expression of CYP3A7, CYP1B1 as well as G6PD, one of the optimal gene combinations of gene expression with cytotoxicity. The EC10 of the three donors is indicated by diamonds. Gene expression is indicated by triangles (lack of a triangle means that no alert for gene expression was obtained). The dashed line represents the iso-concentration line. c Analysis of TSI and TEI based on cytotoxicity (EC10) for the following pharmacokinetic parameters: Cmax, total (protein bound and free) concentration after a single dose (WB Cmax total); Cmax, total concentration, steady state after repeated doses (WB Cmax ss total); average concentration, total, after repeated doses (WB av ss total); Cmax, free (non-protein bound) concentration after a single dose (WB Cmax free); Cmax, free concentration, steady state, after repeated doses (WB Cmax ss free); average, free concentration, steady state, after repeated doses (WBav ss free); portal vein Cmax, total concentration after a single dose (PV Cmax total); portal vein steady state Cmax after repeated doses, total concentration (PV Cmax ss total); average concentration in the portal vein, steady state after repeated doses total concentration (PVav ss total). All parameters are given for the mean, 5% as well as 95% percentile of a population, indicated by the circles, squares and triangles, respectively. The Cmax and Cmax ss is based on blood the cav,ss and portal vein is based on plasma. d Support vector machine (svm) classifier and performance metrics based on cytotoxicity (EC10). Training compounds were assessed by leave-one-out cross-validation, while the test compounds were assessed by the classifier built on the training compounds. The vertical dotted line indicates 0.5 probability of toxicity; compounds with lower or higher probabilities of toxicity are classified as non-hepatotoxic or hepatotoxic, respectively. Performance measures refer to the training compounds. Abbreviations of the training and test compounds are defined in Supplement 1 (color figure online)
Choice of pharmacokinetic parameter to represent in vivo blood concentration
To determine the best factor to represent in vivo blood concentration, the pharmacokinetic parameter on the y-axis was varied, while keeping the cytotoxicity parameter (median EC10, 48 h incubation) constant on the x-axis. Key questions were to identify which of the following parameters are superior (Fig. 7c): total or free concentrations; concentrations in blood of the general circulation or in the portal vein; the use of pharmacokinetic parameters (e.g. Cmax) of the 95% percentile, mean or 5% percentile of a population; maximal blood concentrations (Cmax), maximal blood concentrations in the steady state (Cmax ss) or average blood concentrations in the steady state (Cav, ss). The modeled pharmacokinetic parameters are available in Supplement 9. Plotting TSI against TEI for all pharmacokinetic parameters, led to the following findings for the analyzed set of compounds (Fig. 7c): (1) the use of total blood concentrations (orange symbols) resulted in higher TSI and TEI than free drug concentrations (blue symbols); (2) blood concentrations of the test compounds in the general circulation (e.g., the orange and red symbols) resulted in higher TSI, but lower TEI than concentrations in the portal vein after oral uptake (green symbols); (3) PBPK modeling allows simulation of interindividual differences, differentiating for example, the mean blood concentrations, as well as concentrations in the highest 5% (95th percentile) and the lowest 5% (5th percentile) of a human population. The use of the 95th percentiles led to higher TSI and TEI than the corresponding 5th percentiles and the mean; and (4) the use of Cmax led to a slightly higher TSI than Cmax ss and clearly higher TSI and TEI than Cav, ss. In summary, the use of total (free and protein bound) Cmax of the 95th percentile leads to the best TSI based on in vitro data (cytotoxicity, EC10, median), while portal vein concentrations lead to higher TEI at the expense of a reduced TSI (Supplement 11). Therefore, the total Cmax of the 95th percentile was used for the next step in the pipeline (Fig. 2), the establishment of the classifier for the prediction of hepatotoxicity status.
Prediction of hepatotoxicity and non-hepatotoxicity by SVM classification
Once the optimized parameters had been established based on TSI and TEI, the test system was used to evaluate whether compounds were hepatotoxic or non-hepatotoxic. When projected onto the known compounds in the extrapolation plot, the location of a compound with unknown hepatotoxicity status allows for a visual assessment (qualitative) of its potential toxicity. However, an objective categorization of compounds as hepatotoxic or non-hepatotoxic requires the use of a classification algorithm. Here, a SVM classifier was used to classify compounds as either toxic and non-toxic by finding a separation line that maximizes the minimal distance to any of the compounds while constraining the errors by a constant. Based on the 28 training compounds (Fig. 7d) the classification performance was assessed using leave-one-out cross-validation.
The in vitro EC10 (median, 48 h incubation) and Cmax (total, general circulation, 95% CI) were used as input parameters. This resulted in 28 out of 30 correct predictions (Fig. 7d), and thus a sensitivity, specificity and accuracy of 100, 87.3, and 93.3%, respectively. The incorrect predictions were for APAP, which at a therapeutic dose (14 mg/kg) was falsely predicted as hepatotoxic. The second false positive was glucose. A rich meal may increase blood glucose levels from approximately 90 to 219 mg/dl (5.0–12.2 mM), which despite a prediction of ‘hepatotoxic’, is not expected to have adverse effects on the liver. The accuracy was not improved when expression of the seven genes was additionally included as an input parameter (Supplement 11), which agrees with the observation that TSI did not improve when gene expression was considered in addition to cytotoxicity.
An overall classifier was obtained by fitting an SVM classifier on all 30 compounds using the same input variables (EC10 median, 48 h incubation and Cmax, total, general circulation, 95% percentile) as for the leave-one-out classifiers. This classifier was applied to eight independent test substances known to be either hepatotoxic (leflunomide, nevirapine, tolcapone and troglitazone) or non-hepatotoxic (ethyl-, propyl-, butyl- and isobutylparaben) at specific blood concentrations (Fig. 7d). The classifier properties reflect the proportion of hepatotoxic compounds (here: 14 of total 30). Therefore, the present classifier was calibrated for test data with a similar number of hepatotoxic and non-hepatotoxic compounds. If this proportion will differ in future studies, adjustment of the predicted probabilities derived from the SVM will be required. Tested concentrations, toxicity information, pharmacokinetics, raw data and EC10 values are given in Supplement 12. The eight independent test compounds were added to the optimized extrapolation plot (Fig. 8a). All non-hepatotoxic compounds were located in a region at least three orders of magnitude below the iso-concentration line in the extrapolation plot. Blood concentrations of ethyl-, propyl-, butyl- and isobutylparaben are known from biomonitoring studies, and such exposure levels are not expected to cause an increased risk of hepatotoxicity (Azzouz et al. 2016; Frederiksen et al. 2011; Shekar et al. 2016; Sandanger et al. 2011; Mulla et al. 2015; Nellis et al. 2013). In contrast, the four hepatotoxic compounds were located in the hepatotoxic area delineated by the original set of compounds (Fig. 8a). Using the SVM classifier trained on the 28 original compounds, the independent eight compounds were all correctly classified as either hepatotoxic or non-hepatotoxic (Fig. 7d).

Predictions and comparison with published data. a Analysis of eight further hepatotoxic and non-hepatotoxic compounds not included in the first set of test substances in PHH. The red and green symbols correspond to the training compounds, similar to Fig. 5c but without the illustration of error ranges. The additional non-hepatotoxic substances (ethylparaben, butylparaben, isobutylparaben, propylparaben) are represented by black triangles, the additional hepatotoxic compounds (leflunomide, nevirapine, tolcapone, troglitazone) by black dots. b Ab initio risk evaluation of pulegone. The red and green symbols show the training compounds similar to a. The black diagonal line (0.5) indicates optimal separation of hepatotoxic (red) and non-hepatotoxic (green) compounds. If a substance lies on this line its probability to either belong to the hepatotoxic or non-hepatotoxic compounds is 0.5. Correspondingly, the 0.1, 0.05, 0.01 and 0.005 probabilities are indicated by dashed diagonal lines. Data of pulegone are given in blue color. 1.27 mM is the EC10 (median; 48 h incubation) of the cytotoxicity of pulegone. The dashed blue lines indicate that pulegone intersects the 0.01 and 0.05 probability lines at blood concentrations (Cmax) of 0.299 and 1.57 µM, respectively. The blue line represents the iso-concentration line (Iso-line). c Derivation of an acceptable daily intake (ADI) for the 0.05 probability line and comparison to established ADIs derived from hepatotoxicity in rats. d Linear equations, and Cmax in blood for pulegone for the individual probability lines. Moreover, single and repeated oral doses are given that would result in the corresponding Cmax in blood (color figure online)
The purpose of the analysis with eight independent compounds was to check whether the separation line between hepatotoxic and non-hepatotoxic compounds established by the SVM classifier is plausible. A real validation of the predictive performance in terms of sensitivity, specificity, etc., would require the testing of more compounds with different mechanisms of action and varying degrees of hepatotoxicity, not chosen to be only on the opposite extremes of the spectrum with regard to hepatotoxicity as for the present set of compounds. Nevertheless, the successful separation of the hepatotoxic and non-hepatotoxic compounds studied here allowed us to proceed with the next step in the working pipeline (Fig. 2), the extrapolation from in vitro alert concentrations to in vivo blood concentrations.
Estimation of the risk of hepatotoxicity at specific blood concentrations of test compounds
An important question is whether the probability of hepatotoxicity caused by specific in vivo blood concentrations can be extrapolated based on in vitro alert concentrations. The systematic degree of separation of hepatotoxic and non-hepatotoxic compounds observed across the entire in vitro concentration range (Fig. 5c) suggests that such an extrapolation may be possible. As described above, a SVM classifier was used to identify the line that best separates the hepatotoxic and the non-hepatotoxic compounds. A compound located exactly on this line has a 50% probability of belonging to the hepatotoxic category (Fig. 8b). As a consequence, the intersection of the in vitro concentration (EC10, median, 48 h incubation) with this line can be used to estimate an in vivo concentration (Cmax) with a probability of 50% that it belongs to the hepatotoxic category (red symbols). It should be considered that the 50% probability of hepatotoxicity scenario does not mean that 50% of the individuals will suffer from hepatotoxicity; belonging to the hepatotoxic category means a risk far below 50% for individual patients. The risk for each hepatotoxic compound has been defined in Supplement 1 (‘hepatotoxicity information’). For example, oral doses of ketoconazole (one of the hepatotoxic compounds) caused hepatotoxicity in 0.007–0.05% and liver enzyme elevations in 4–20% of all treated patients.
Application of the extrapolation procedure to the four hepatotoxic test compounds (leflunomide, nevirapine, tolcapone and troglitazone) led to calculated in vivo blood concentrations that are related with a 50% probability of hepatotoxicity of 0.050, 2.55, 1.46, and 0.61 µM, respectively. Using a similar procedure, blood concentrations with a lower probability of hepatotoxicity, e.g., based on a 5% or 1% probability of hepatotoxicity, can also be calculated (Fig. 8b).
Estimation of an acceptable daily intake based on in vitro data
For all compounds studied so far, human hepatotoxicity and associated blood concentrations were known. However, often this knowledge is not available (ab initio toxicity evaluation). Pulegone was chosen as an example to establish an acceptable daily intake concerning hepatotoxicity. Pulegone is a naturally occurring organic compound used in flavoring agents and in the fragrance industry. High doses caused hepatotoxicity in rats (Khojasteh et al. 2012; Chen et al. 2011). Therefore, knowing which concentrations in blood increase the risk of human hepatotoxicity is of interest. Cytotoxicity testing in PHH from three donors resulted in a median EC10 of 1.27 mM (Fig. 8b, c). Application of the above-mentioned extrapolation procedure identified 30.3 µM as the blood concentration (Cmax) corresponding to a 50% probability of belonging to the hepatotoxic category, and 1.57 µM as the concentration corresponding to a 5% probability (Fig. 8c, d). The latter concentration associated with a 5% probability of hepatotoxicity may serve as a basis for derivation of an acceptable daily intake dose (ADI). Reverse PBPK modeling for repeated oral doses indicated that a blood concentration (Cmax) of 1.57 µM corresponds to 268 µg pulegone/kg body weight/day (Fig. 8c), which may be considered an in vitro derived ADI. This extrapolation can also be performed for stricter probability levels, for example, a 1% probability will result in an in vitro derived ADI of 51 µg/kg/day (Fig. 8d). ADIs based on 28 days oral toxicity studies in rats by the established method ranged between 100 and 750 µg/kg/day (HMPC 2016; CEFS/SCF/CS 2002). Tested concentrations, raw data and EC10 values, as well as reverse modelling of the pulegone case study are given in Supplement 12.
Comparison with publicly available data and analysis by margin of safety
Recently, the cytotoxicity of 110 compounds (69 hepatotoxic and 41 non-hepatotoxic) was tested in spheroid cultures of human hepatocytes (Proctor et al. 2017). Besides information on hepatotoxicity, the EC50 of cytotoxicity and the Cmax of the test compounds were also published (Supplementary material 2 in Proctor et al. 2017). For comparison, we prepared an extrapolation plot using the data provided by Proctor et al. (2017) in their supplementary information (Fig. 9a), resulting in a TSI of 0.773 and a TEI of 0.788. Although the compounds were not perfectly separated, the trend is that the non-hepatotoxic compounds are further below the iso-concentration line compared to the hepatotoxic compounds.

a Extrapolation plot of recently published cytotoxicity data generated with spheroids of primary human hepatocytes (data from Proctor et al. 2017). Red and green symbols indicate compounds with and without DILI concern, respectively. In the data set of Proctor et al., exact EC50 values (e.g., 100 µM) were given for some compounds, indicated by circles in this figure; for other compounds EC50 values could not be reached (data given e.g., as > 100 µM) indicated by triangles in the present extrapolation plot. b Presentation of the data of Proctor et al. (2017) by the ‘margin of safety’ (MoS) method. The y-axis presents the ratio of in vitro cytotoxicity (EC50) in PHH and the Cmax in blood. Compounds with DILI concern were grouped into severe, high and low (red symbols). For controls (no DILI concern), compounds with and without elevated liver enzymes (EE) were differentiated (green symbols). The numbers on top of the panel, e.g., (17/23), represent the number of compounds below a MoS of 20 and the total number of compounds. c Data of the present study presented by the MoS method, using the EC10 as a measure of cytotoxicity. Data for the maximum, median and minimum EC10 (min, med, max) of three donors are shown. d Corresponding data to d for EC50 values to allow a direct comparison to the data of Proctor et al. (2017) in d who also used EC50 values. 1EE indicates a transient elevation of liver enzymes without histological tissue damage (Proctor et al. 2017) (color figure online)
Previous studies have mainly used the margin of safety (MoS) method to evaluate hepatotoxicity in vitro. To allow a comparison, our data was also analyzed by MoS, where the ratio of the EC50 or EC10 to the Cmax in blood were plotted on the y-axis; whereas, hepatotoxic compounds (DILI concern) and controls (no DILI concern) are shown on the x-axis (Fig. 9b–d). This method was applied to plot the published data of Proctor et al. (2017), together with the data used in the present study. Proctor et al. subdivided “DILI concern” compounds into “severe”, “high”, and “low” (Fig. 9b). While the most severe DILI concern compounds showed a MoS lower than 20, a relatively high fraction of the high and low DILI concern compounds were above 20. Data from the present study (Fig. 9c, d) showed that using the median EC10 of three donors allows for a better differentiation between hepatotoxic and non-hepatotoxic compounds than the EC10 minimum or maximum (Fig. 9c). Moreover, EC10 values (Fig. 9c) differentiated better than EC50 values (Fig. 9d); the EC50 resulted in more hepatotoxic compounds with a MoS > 20 compared to the corresponding EC10 data.
HepG2 instead of PHH for in vitro testing
The human hepatocyte-based test method established in this study successfully distinguished hepatotoxic and non-hepatotoxic compounds. However, PHH are expensive, compared to HepG2 cells. It is unknown whether using the same approach with HepG2 cells would generate comparable results to those obtained with PHH. Therefore, the same experiments as described for PHH were performed using HepG2 cells, including cytotoxicity and expression of the seven genes. Extrapolation plots using the median EC10 (Fig. 10a) and the combination of median EC10 with CYP3A7, CYP1B1 and G6PD (Fig. 10b) showed that lower metrics for test performance were obtained for HepG2 than for PHH. The TSI for PHH was 0.996 compared to 0.911 for HepG2. The TEI was also lower for HepG2 (0.844 and 0.810 for PHH and HepG2, respectively). The difference between HepG2 cells and PHH is illustrated in Fig. 10c where ratios of the median EC10 values of HepG2 and PHH are plotted for each compound (corresponding analysis for EC50: Supplement 6). The highest ratios were obtained for NAC, CBZ, BPR and ETOH, where PHH were at least tenfold more susceptible than HepG2 (Fig. 10c). In contrast, HepG2 cells were more susceptible to other compounds, including TSN and VPA. The results show that 6 out of 28 tested compounds resulted in more than tenfold differences between PHH and HepG2.

Hepatotoxicity prediction based on HepG2 cells and comparison to PHH. a Extrapolation plot of HepG2 cells using cytotoxicity (EC10, median, 48 h incubation) as in vitro parameter (x-axis) and the Cmax (total concentration, 95% CI) on the y-axis. b Extrapolation plot with combined analysis of cytotoxicity (EC10, median) and gene expression (CYP3A7, CYP1B1, G6PD). c Ratio plot comparing cytotoxicity in HepG2 and PHH. Ratios were calculated by dividing EC10 (median) values measured with HepG2 cells by those obtained with of PHH. d Influence of the additional consideration of glutathione (GSH) depletion on TSI and TEI. Cytotoxicity (EC10, median), as well as a combination of cytotoxicity and the expression of CYP3A7, CYP2B1 and G6PD were compared with and without the additional consideration of GSH depletion using the median EC10. The dashed line in a and b represents the iso-concentration line
Despite the inferior performance metrics of the HepG2-based in vitro test method, HepG2 cells may still be used for exploratory studies. In an attempt to improve the performance of the HepG2-based test, a glutathione (GSH) assay was performed with all test compounds. A combined analysis of cytotoxicity (median EC10) and GSH depletion (median EC10), where combination means the use of the more sensitive of the two readouts for each compound, improved TSI and TEI compared to either GSH depletion or cytotoxicity alone (Fig. 10d). In addition, addition of measuring GSH content to the combined cytotoxicity- and gene expression-based test battery (CYP3A7, CYP1B1 and G6PD) improved TSI and TEI (all analyses based on median values) (Fig. 10d). Therefore, it should be evaluated in the future, whether the GSH assay also improves hepatotoxicity analysis in PHH.
Discussion
We present a method for the prediction of hepatotoxicity of test compounds and for estimation of the probability of hepatotoxicity at specific blood concentrations based on in vitro test results with cryopreserved or freshly isolated human hepatocytes. The test performance was optimized for a training set of compounds based on the here established metrics, TSI and TEI. An optimized set of parameters was then used for the analysis of independent compounds to estimate blood concentrations associated with an increased probability of hepatotoxicity. Based on reverse modeling, corresponding oral doses and an acceptable daily intake can thereafter be derived. Advantages of the method established here are that (1) metrics-based test system optimization allows systematic integration of further test parameters and iterative cycles of optimization and validation to improve the test performance; (2) based on in vitro data it is possible to derive oral doses that either cause an increased risk of toxicity or that can be considered as acceptable.
One application of the test method is to evaluate test compounds for which intended blood concentrations are already known. Even early in drug development when pharmacokinetic studies are not yet available, an approximation of the required blood concentration is often available, e.g., the bactericidal concentration in vitro when novel antibiotics are developed. The test method introduced here could then estimate whether this concentration leads to an increased risk of hepatotoxicity or can be considered acceptable. Moreover, biomonitoring studies in environmental toxicology identify concentrations of xenobiotics in blood; the test method established here will inform about the associated risk of hepatotoxicity. A second application comprises novel compounds for which only little information is available, where the method allows estimation of acceptable oral doses. A limitation of this approach is that reverse modeling for novel compounds is only possible with some degree of uncertainty.
An almost optimal differentiation between the analyzed hepatotoxic (n = 18) and non-hepatotoxic (n = 20) compounds (including training and test sets) was achieved based on the EC10 of the cytotoxicity test after a 48 h incubation period both using the extrapolation plots and the classifier. Therefore, it is not surprising that a 7-gene signature, which on its own also differentiated the hepatotoxic from non-hepatotoxic compounds, did not further improve the already close to optimal TSI, and only slightly increased the TEI, when combined with cytotoxicity. It is plausible that cytotoxicity plays a major role for the prediction of hepatotoxic blood concentrations. However, the present data should not be interpreted to mean that gene expression cannot add independent information when predicting hepatotoxic blood concentrations. Therefore, to show that gene expression can contribute to the predictive performance, it will be important to identify compounds where hepatotoxic blood concentrations are strongly overestimated based on cytotoxicity alone. Such compounds, which induce liver toxicity based on mechanisms other than cytotoxicity, will then have to be investigated using methods that identify alternative mechanisms, for example attraction of immune cells by cytokine release or compromised bile salt transport.
A SVM classifier with cytotoxicity (EC10, median, 48 h incubation) and Cmax (total concentration, general circulation, 95% percentile) as input variables correctly predicted whether a compound is hepatotoxic or not for 28 out of 30 compound-exposure scenarios in a leave-one-out cross validation procedure, and correctly predicted further eight independent test compounds as either hepatotoxic or non-hepatotoxic. A false positive result was obtained for a non-hepatotoxic blood concentration of the analgesic acetaminophen (APAP). APAP is not hepatotoxic at therapeutic doses of 14 mg/kg (corresponding to a Cmax of 0.109 mM), but causes a high risk of hepatotoxicity at accidental or suicidal doses of 140 mg/kg or higher (https://livertox.nih.gov/Acetaminophen.htm). This corresponds to a modeled Cmax of 1.21 mM. Cytotoxicity analysis of APAP identified 2.2 mM as the EC10 and an alert for G6PD at 5 mM. However, the small range between non-toxic and cytotoxic doses leads to a situation where the non-cytotoxic APAP blood concentration still clusters into the hepatotoxic range of the extrapolation plot, which results in a false positive classification. The second false positive compound was glucose. After the consumption of a rich standard meal, blood glucose may increase from approximately 90–219 mg/dl (95% CI) (Freckmann et al. 2007), which we considered a non-hepatotoxic exposure when designing the study.
Establishment of the method described in the present study depends on existing knowledge of the blood–concentration–time profile resulting from a specific exposure scenario. Available information is most comprehensive for pharmaceutical drugs, for which both precise pharmacokinetics and hepatotoxicity data for large numbers of patients are available. Therefore, most of the compounds analyzed in the current work were pharmaceutical drugs. However, to study a wide range of diverse substances other chemicals were also included. Use of body lotions with the preservative methylparaben (MePa) may lead to blood concentrations of 0.009 µM MePa (Guo et al. 2014), which is not hepatotoxic in vivo and also orders of magnitude below hepatotoxic concentrations according to the in vitro method with cultivated hepatocytes. Dimethyl sulfoxide (DMSO) has been used in gels (Roth and Shainhouse 2004) which may lead to blood concentrations of 108 µM that are clearly below cytotoxic levels. Alcohol abuse with a daily oral consumption of more than 16 g ethanol leads to steatosis in approximately 90% of individuals (Crabb 1999; Massey and Arteel 2012). For this exposure scenario, a blood peak concentration (total concentration, whole blood, 95% percentile) of 10.1 mM was modeled. In contrast, transdermal exposure caused by disinfection aerosol spray leads to an exposure of up to 9.72 g ethanol (Pendlington et al. 2001), for which a blood peak concentration (whole blood, total, 95% percentile) of 5.76 µM was modeled, which is considered non-toxic (Kramer et al. 2007). The extrapolation plot and the SVM classifier could separate hepatotoxic and non-hepatotoxic ethanol blood concentrations.
The novel metrics, TSI and TEI enable quantitative assessment of in vitro test performance and enable a comparison of how performance is influenced by individual parameters. A basic question addressed with this approach was which cutoff should be used to define a cytotoxic effect. In toxicology, the EC50 defined as the concentration that reduces viability by 50% is often applied. However, systematically increasing the EC cutoff revealed a strong decrease in the TEI in the range from EC10 to EC80, with a concomitant decrease in the TSI. Therefore, it appears to be important to identify concentrations where cytotoxicity first becomes evident. Another practical question addressed was whether the median, maximum or minimum EC10 value perform better when hepatocytes from several donors are analyzed. Should, for example, the minimum EC10 yield higher TSI and TEI values, then the test performance would be improved by identifying particularly susceptible hepatocyte batches. Moreover, cryopreserved hepatocyte batches are available where cells of several donors have been pooled (Hengstler et al. 2000; Hewitt et al. 2007). However, this pooled approach was avoided in the present study to determine whether it was relevant to use particularly susceptible hepatocytes. The present results show that the median values of cytotoxicity from three donors led to a higher TSI than the corresponding minima or maxima values. Conversely, the minima (e.g., EC10 of the most susceptible donor) yielded a higher TEI. Since, however, a higher TSI is preferred over a higher TEI, the median EC10 currently represents the best available parameter to describe cytotoxicity in the data set analyzed here. The median value works best for the overall strategy, since it does not seem necessary to identify particularly susceptible donors to gain an optimal TSI. On the other hand, pooled hepatocyte batches (Hengstler et al. 2000) should still be considered with caution, because the data obtained will represent a mean which—in contrast to the median—is outlier-sensitive.
Another question addressed in this study was how long cultivated hepatocytes should be exposed to test compounds, and whether repeated exposures are superior to a single addition of test compound (Gu et al. 2018; Godoy et al. 2013). Our results demonstrated that a two-day exposure period of the test compound (with only one addition of test compound at the morning after thawing and plating) lead to a higher TSI and TEI compared to 24 h or 7-day exposure with repeated culture medium changes containing fresh test compound. The better results obtained after 2 days compared to a single day of exposure is plausible since some test compounds require more than 24 h to develop their full toxic potential (Gu et al. 2018). However, it was surprising that the longer incubation protocol (7 days) and repeated exposures did not further improve the test performance. An explanation may be that hepatocytes exhibit gene expression alterations that increase with the length of the cultivation period, leading to a decrease in the hepatocyte differentiation status (Godoy et al. 2015; Zellmer et al. 2010; Rowe et al. 2013). Moreover, hepatotoxic effects caused by repeated doses in vivo often require the involvement of cell types other than hepatocytes, such as stellate cells (Hammad et al. 2014, 2017), endothelial cells (Hoehme et al. 2010; Ghallab et al. 2016, 2018), and cholangiocytes (Jansen et al. 2017; Vartak et al. 2016; Ghallab et al. 2018), which are not represented adequately in currently available in vitro methods. If, however, hepatocytes are the primary target of a hepatotoxic compound, it seems that this type of toxicity can be adequately identified by a single compound exposure for 48 h, at least for the compound set tested in the present study. Since this approach is easier and enables a higher throughput than long-term cultures with hepatocytes in more complex systems (e.g. Frey et al. 2014; Godoy et al. 2013), it was used in the present study. Furthermore, it was tested whether an inexpensive and easy-to-handle permanent human hepatocyte cell line may be used for the in vitro test method instead of the more challenging PHH. Regarding TSI and TEI, the HepG2 cell line used in the present study performed worse compared to PHH and is, therefore, not considered a PHH replacement.
In previous studies, the blood or plasma peak concentration (Cmax) has routinely been used to extrapolate between in vitro data and the in vivo situation concerning DILI (e.g. Proctor et al. 2017), as well as toxicity of other organs, e.g., nephrotoxicity (Sjogren et al. 2018; Sjogren and Hornberg 2018). However, a comprehensive analysis to determine which pharmacokinetic parameter is optimal for this purpose has not yet been performed. The present study demonstrated that total (i.e., protein bound and free) concentrations improve in vitro–in vivo extrapolation compared to free (non-protein bound) concentrations, based on the TSI/TEI concept. It should be considered that the cell culture medium for hepatocytes used in the present study is serum-free without albumin (Schug et al. 2013; Godoy et al. 2013); whereas, human blood contains 35–53 g albumin/l. A further result from the analysis of pharmacokinetic parameters was that the 95th percentile of Cmax of a population led to a higher TEI than the mean or the 5th percentile. An explanation may be that patients with the highest drug concentrations in blood are at the highest risk of hepatotoxicity, and a better agreement between in vitro and in vivo data may be obtained if the extrapolation is based on 95th percentile concentrations. It is also plausible that Cmax in the portal vein resulted in a higher TEI than the compound concentration in the general circulation. After oral administration, concentrations in the portal vein are usually higher due to the first pass effect of the liver (Mielke et al. 2011). However, a higher TEI for portal vein Cmax was obtained at the expense of a worse TSI. An explanation may be that liver tissue is exposed longer to the blood from the general circulation than to the higher but transient concentrations in the portal vein during compound absorption from the intestine. Altogether, the 95th percentile of the total, maximal blood concentration in the general circulation allowed for the best separation (TSI) of hepatotoxic and non-hepatotoxic compounds of all analyzed pharmacokinetic parameters for the here studied set of compounds.
Once established, it was necessary to determine whether the method resulted in correct predictions of independent compounds. We tested additional hepatotoxic and non-hepatotoxic compounds (n = 8) that all clustered to the expected regions of the extrapolation plot and were correctly predicted by the classifier. In the future, a greater number of compounds will have to be tested; it will also be necessary to study whether in vitro prediction by the method established here depends on the mechanisms of action of test compounds. Nevertheless, the favorable results encouraged us to use the blood concentrations estimated to correspond to a 50% or 5% probability of hepatotoxicity for the compound to perform a case study for de novo risk evaluation of pulegone, an organic compound present in teas, spices and perfumes (Zárybnický et al. 2018; Gordon and Khojasteh 2015). Since pulegone has been reported as hepatotoxic in rats (Khojasteh et al. 2012; Chen et al. 2011), there is some interest in human risk evaluation of hepatotoxicity. Pulegone is a relatively non-toxic compound with a Cmax of 1.27 mM (EC10, median, 48 h incubation). Using the technique established here, this EC10 value corresponds to a Cmax of 1.57 µM that is associated with a 5% probability to belong to the hepatotoxic category, which after reverse modeling for repeated oral doses corresponds to 268 μg/kg/day. Acceptable daily intake (ADI) derived by the established method based on toxicity studies in rats ranged between 100 and 750 µg/kg/day (CEFS/SCF/CS 2002; HMPC 2016). Therefore, the ADI derived by the novel in vitro method would be in the range of ADIs established in animal studies. However, a limitation is that it has not yet been systematically studied which probability (of compound to belong to the hepatotoxic category) is most appropriate for the establishment of an ADI in vitro. The use of an estimated blood concentration corresponding to a 1% probability would, for example, result in an ADI of 51 µg/kg/day. Therefore, a next important step will be to compare a larger number of in vitro and conventionally established ADIs and identify the most appropriate probability for extrapolation.
Four prior studies used PHH to predict human hepatotoxicity (Proctor et al. 2017; Xu et al. 2008; Khetani et al. 2013; Vorrink et al. 2018). Proctor et al. (2017) provided in vivo Cmax and EC50 values of the cytotoxicity tests for individual compounds. This allowed us to calculate a TSI of 0.773 and a TEI of 0.788, which are lower compared to the corresponding values (TSI 0.996, TEI 0.887) of the present study. This difference is not necessarily explained only by the different types of in vitro methods, but may also be due to the tested concentration ranges. One difference to our study is that we escalated the concentrations of test compounds until either cytotoxicity was obtained or the limits of solubility were reached. Using this strategy, an EC10 could be derived for 27 of the 28 tested compounds, and the only negative compound still showed a positive result when gene expression was used. Therefore, it was possible to identify positive in vitro test concentrations for all compounds, including the negative controls. In contrast, 24 of 69 hepatotoxic compounds and 31 of 41 non-hepatotoxic compounds studied by Proctor et al. (2017) were not tested up to cytotoxic levels (and were reported as EC50 > ”highest tested concentration”); therefore, the difference in results between both studies may also be due to the choice of test compound concentrations.
A further limitation of previous studies is that hepatocytes of only one human donor were tested per compound (Proctor et al. 2017; Vorrink et al. 2018); whereas, the present study analyzed three human donors. This difference is likely to have an impact on the accuracy of the data. In contrast to the study by Proctor et al., no raw data, EC50 or EC10 data of the individual compounds were provided by Vorrink et al. (2018), which made it impossible to directly compare their finding to ours. Such comparisons would have been of interest, because there were some atypical dose response relationships, where toxicity was observed at lower concentrations, but decreased or disappeared at higher concentrations (Vorrink et al. 2018). For instance, iproniazid was reported to be cytotoxic at 1 × Cmax but not at 20 × Cmax; similarly, propranolol was reported to be cytotoxic at 5 × Cmax but not at 20 × Cmax. This is different to our study where we did not observe a loss in cytotoxicity at higher concentrations for any of the tested compounds. Moreover, the authors reported a specificity of 100% (Vorrink et al. 2018), which from the data presented is difficult to interpret. One of the DILI negative compounds (propranolol) was reported as positive at 5 × Cmax, but the test result was nevertheless interpreted as negative. Another potential concern is that the highest concentration (20 × Cmax) was not tested for some DILI negative compounds, for example, aminoglutethimide, practolol and ternozolomide, possibly because of solubility problems. However, this was not explicitly stated. These “DILI negative” compounds were predicted to be negative based only on the two lowest tested concentrations (Cmax and 5 × Cmax); however, some “severe DILI” compounds also tested negative at the two lowest concentrations, only exhibiting cytotoxicity at the highest (20 × Cmax) concentration, for example alpidem, clomipramine, dihydralazine, disulfiram, etc. Therefore, it is difficult to differentiate these compounds from the aforementioned negatively predicted compounds, where data at 20 × Cmax are not available. Another critical aspect is that APAP was considered a true positive. However, this analysis was based on a Cmax for APAP of 136 µM, which corresponds to a therapeutic dose of approximately 14 mg/kg—a safe dose which does not cause DILI (https://livertox.nih.gov/Acetaminophen.htm).
In conclusion, the workflow established in the present study allows for systematic test optimization by quantitatively evaluating the influence of individual test parameters on the performance of the in vitro method by the novel performance metrics TSI and TEI. It allows classification of compounds as hepatotoxic or non-hepatotoxic and provides an extrapolation procedure to estimate the probability of hepatotoxicity for specific blood concentrations and the associated oral doses of test compounds for ab initio evaluation of chemicals.
References
Azzouz A, Rascón AJ, Ballestros E (2016) Simultaneous determination of parabens, alkylphenols, phenylphenols, bisphenol A and triclosan in human urine, blood and breast milk by continuos solid-phase extraction and gas chromatography–mass spectrometry. J Pharm Biomed Anal 119:16–26
Bischl B, Lang M, Kotthoff L, Schiffner J, Richter J, Studerus E, Casalicchio G, Jone ZM (2016) mlr: Machine Learning in R. J Mach Learn Res 17(170):1–5
Braeuning A, Oberemm A, Heise T, Gundert-Remy U, Hengstler JG, Lampen A (2018) In vitro proteomic analysis of methapyrilene toxicity in rat hepatocytes reveals effects on intermediary metabolism. Arch Toxicol. https://doi.org/10.1007/s00204-018-2360-3 (Epub ahead of print)
CEFS/SCF/CS (2002) Opinion of the Scientific Committee on Food on pulegone and menthofuran. https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out133_en.pdf
Chen XW, Serag ES, Sneed KB, Zhou SF (2011) Herbal bioactivation, molecular targets and the toxicity relevance. Chem Biol Interact 192(3):161–176. https://doi.org/10.1016/j.cbi.2011.03.016
Chen L, Ning J, Louisse J, Wesseling S, Rietjens IMCM (2018) https://www.ncbi.nlm.nih.gov/pubmed/29634986Use of physiologically based kinetic modelling-facilitated reverse dosimetry to convert in vitro cytotoxicity data to predicted in vivo liver toxicity of lasiocarpine and riddelliine in rat. Food Chem Toxicol 116(Pt B):216–226. https://doi.org/10.1016/j.fct.2018.04.012 (Epub 2018 Apr 7)
Civan JM, Navarro V, Herrine SK, Riggio JM, Adams P, Rossi S (2014) Patterns of acetaminophen use exceeding 4 grams daily in a hospitalized population at a tertiary care center. Gastroenterol Hepatol (N Y) 10(1):27–34
Crabb DW (1999) Pathogenesis of alcoholic liver disease: newer mechanisms of injury. Keio J Med 48:184–188. https://doi.org/10.2302/kjm.48.184
Freckmann G, Hagenlocher S, Baumstark A, Jendrike N, Gillen RC, Rössner K, Haug C (2007) Continuous glucose profiles in healthy subjects under everyday life conditions and after different meals. J Diabetes Sci Technol 1(5):695–703
Frederiksen H, Jørgensen N, Andersson AM (2011) Parabens in urine, serum and seminal blood from healthy Danish men determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS). J Expos Sci Environ Epidemiol 21(3):262–271
Frey O, Misun PM, Fluri DA, Hengstler JG, Hierlemann A (2014) Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat Commun 5:4250. https://doi.org/10.1038/ncomms5250
Gebhardt R, Hengstler JG, Müller D, Glöckner R, Buenning P, Laube B, Schmelzer E, Ullrich M, Utesch D, Hewitt N, Ringel M, Hilz BR, Bader A, Langsch A, Koose T, Burger HJ, Maas J, Oesch F (2003) New hepatocyte in vitro systems for drug metabolism: metabolic capacity and recommendations for application in basic research and drug development, standard operation procedures. Drug Metab Rev 35(2–3):145–213
Ghallab A, Cellière G, Henkel SG, Driesch D, Hoehme S, Hofmann U, Zellmer S, Godoy P, Sachinidis A, Blaszkewicz M, Reif R, Marchan R, Kuepfer L, Häussinger D, Drasdo D, Gebhardt R, Hengstler JG (2016) Model-guided identification of a therapeutic strategy to reduce hyperammonemia in liver diseases. J Hepatol 64(4):860–871
Ghallab A, Hofmann U, Sezgin S, Vartak N, Hassan R, Zaza A, Godoy P, Schneider KM, Guenther G, Ahmed YA, Abbas AA, Keitel V, Kuepfer L, Dooley S, Lammert F, Trautwein C, Spiteller M, Drasdo D, Hofmann AF, Jansen PLM, Hengstler JG, Reif R (2018) Bile Microinfarcts in cholestasis are initiated by rupture of the apical hepatocyte membrane and cause shunting of bile to sinusoidal blood. Hepatology. https://doi.org/10.1002/hep.30213
Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Müller A, Tuschl G, Mueller SO, Dooley S (2009) Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology 49(6):2031–2043
Godoy P, Hewitt NJ, Albrecht U et al (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87(8):1315–1530. https://doi.org/10.1007/s00204-013-1078-5
Godoy P, Schmidt-Heck W, Natarajan K, Lucendo-Villarin B, Szkolnicka D, Asplund A, Björquist P, Widera A, Stöber R, Campos G, Hammad S, Sachinidis A, Chaudhari U, Damm G, Weiss TS, Nüssler A, Synnergren J, Edlund K, Küppers-Munther B, Hay DC, Hengstler JG (2015) Gene networks and transcription factor motifs defining the differentiation of stem cells into hepatocyte-like cells. J Hepatol. 63(4):934–942. https://doi.org/10.1016/j.jhep.2015.05.013 (Epub 2015 May 25. Erratum in: J Hepatol. 2016 Feb;64(2):525–526)
Godoy P, Widera A, Schmidt-Heck W, Campos G, Meyer C, Cadenas C, Reif R, Stöber R, Hammad S, Pütter L, Gianmoena K, Marchan R, Ghallab A, Edlund K, Nüssler A, Thasler WE, Damm G, Seehofer D, Weiss TS, Dirsch O, Dahmen U, Gebhardt R, Chaudhari U, Meganathan K, Sachinidis A, Kelm J, Hofmann U, Zahedi RP, Guthke R, Blüthgen N, Dooley S, Hengstler JG (2016) Gene network activity in cultivated primary hepatocytes is highly similar to diseased mammalian liver tissue. Arch Toxicol 90(10):2513–2529
Gordon P, Khojasteh SC (2015) A decades-long investigation of acute metabolism-based hepatotoxicity by herbal constituents: a case study of pennyroyal oil. Drug Metab Rev 47(1):12–20. https://doi.org/10.3109/03602532.2014.990032 (Epub 2014 Dec 16. Review)
Grinberg M (2017) Statistical analysis of concentration-dependent high-dimensional gene expression data, Dissertation, Department of Statistics of the University of Dortmund, Dortmund
Grinberg M, Stöber RM, Edlund K, Rempel E, Godoy P, Reif R, Widera A, Madjar K, Schmidt-Heck W, Marchan R, Sachinidis A, Spitkovsky D, Hescheler J, Carmo H, Arbo MD, van de Water B, Wink S, Vinken M, Rogiers V, Escher S, Hardy B, Mitic D, Myatt G, Waldmann T, Mardinoglu A, Damm G, Seehofer D, Nüssler A, Weiss TS, Oberemm A, Lampen A, Schaap MM, Luijten M, van Steeg H, Thasler WE, Kleinjans JC, Stierum RH, Leist M, Rahnenführer J, Hengstler JG (2014) Toxicogenomics directory of chemically exposed human hepatocytes. Arch Toxicol 88(12):2261–2287. https://doi.org/10.1007/s00204-014-1400-x
Gu X, Albrecht W, Edlund K, Kappenberg F, Rahnenführer J, Leist M, Moritz W, Godoy P, Cadenas C, Marchan R, Brecklinghaus T, Pardo LT, Castell JV, Gardner I, Han B, Hengstler JG, Stoeber R (2018) Relevance of the incubation period in cytotoxicity testing with primary human hepatocytes. Arch Toxicol 92(12):3505–3515. https://doi.org/10.1007/s00204-018-2302-0
Guo Y, Wang L, Kannan K (2014) Phthalates and parabens in personal care products from China: concentrations and human exposure. Arch Environ Contam Toxicol 66:113–119. https://doi.org/10.1007/s00244-013-9937-x
Hammad S, Hoehme S, Friebel A, von Recklinghausen I, Othman A, Begher-Tibbe B, Reif R, Godoy P, Johann T, Vartak A, Golka K, Bucur PO, Vibert E, Marchan R, Christ B, Dooley S, Meyer C, Ilkavets I, Dahmen U, Dirsch O, Böttger J, Gebhardt R, Drasdo D, Hengstler JG (2014) Protocols for staining of bile canalicular and sinusoidal networks of human, mouse and pig livers, three-dimensional reconstruction and quantification of tissue microarchitecture by image processing and analysis. Arch Toxicol 88(5):1161–1183. https://doi.org/10.1007/s00204-014-1243-5
Hammad S, Braeuning A, Meyer C, Mohamed FEZA, Hengstler JG, Dooley S (2017) A frequent misinterpretation in current research on liver fibrosis: the vessel in the center of CCl4-induced pseudolobules is a portal vein. Arch Toxicol 91(11):3689–3692
Heise T, Schug M, Storm D, Ellinger-Ziegelbauer H, Ahr HJ, Hellwig B, Rahnenfuhrer J, Ghallab A, Guenther G, Sisnaiske J, Reif R, Godoy P, Mielke H, Gundert-Remy U, Lampen A, Oberemm A, Hengstler JG (2012) In vitro–in vivo correlation of gene expression alterations induced by liver carcinogens. Curr Med Chem 19(11):1721–1730
Hengstler JG, Utesch D, Steinberg P, Platt KL, Diener B, Ringel M, Swales N, Fischer T, Biefang K, Gerl M, Böttger T, Oesch F (2000) Cryopreserved primary hepatocytes as a constantly available in vitro model for the evaluation of human and animal drug metabolism and enzyme induction. Drug Metab Rev 32(1):81–118
Hewitt NJ, Lechón MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG (2007) Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev 39(1):159–234
HMPC/EMA138386/2005 Rev. 1 Committee on Herbal Medicinal Products (HMPC) (2016) Public statement on the use of herbal medicinal products1 containing pulegone and menthofuran Final, 2016. https://www.ema.europa.eu/en/documents/scientific-guideline/public-statement-use-herbal-medicinal-products1-containing-pulegone-menthofuran-revision-1_en.pdf
Hoehme S, Brulport M, Bauer A, Bedawy E, Schormann W, Hermes M, Puppe V, Gebhardt R, Zellmer S, Schwarz M, Bockamp E, Timmel T, Hengstler JG, Drasdo D (2010) Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc Natl Acad Sci USA 107(23):10371–10376
Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, Hengstler JG (2017) The ascending pathophysiology of cholestatic liver disease. Hepatology 65(2):722–738. https://doi.org/10.1002/hep.28965
Jones HM, Chen Y, Gibson C et al (2015) Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther 97(3):247–262
Karatzoglou A, Smola A, Hornik K, Zeileis A (2004) kernlab—an S4 Package for Kernel Methods in R. J Stat Softw. https://doi.org/10.18637/jss.v011.i09
Khetani SR, Kanchagar C, Ukairo O, Krzyzewski S, Moore A, Shi J, Aoyama S, Aleo M, Will Y (2013) Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci 132(1):107–117. https://doi.org/10.1093/toxsci/kfs326
Khojasteh SC, Hartley DP, Ford KA, Uppal H, Oishi S, Nelson SD (2012) Characterization of rat liver proteins adducted by reactive metabolites of menthofuran. Chem Res Toxicol 25(11):2301–2309. https://doi.org/10.1021/tx300144d
Kramer A, Below H, Bieber N, Kampf G, Toma CD, Huebner NO, Assadian O (2007) Quantity of ethanol absorption after excessive hand disinfection using three commercially available hand rubs is minimal and below toxic levels for humans. BMC Infect Dis 7:117
Kramer NI, Di Consiglio E, Blaauboer BJ, Testai E (2015) Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicol In vitro 30(1 Pt A):217–224. https://doi.org/10.1016/j.tiv.2015.09.005
Leist M, Ghallab A, Graepel R, Marchan R, Hassan R, Bennekou SH, Limonciel A, Vinken M, Schildknecht S, Waldmann T, Danen E, van Ravenzwaay B, Kamp H, Gardner I, Godoy P, Bois FY, Braeuning A, Reif R, Oesch F, Drasdo D, Höhme S, Schwarz M, Hartung T, Braunbeck T, Beltman J, Vrieling H, Sanz F, Forsby A, Gadaleta D, Fisher C, Kelm J, Fluri D, Ecker G, Zdrazil B, Terron A, Jennings P, van der Burg B, Dooley S, Meijer AH, Willighagen E, Martens M, Evelo C, Mombelli E, Taboureau O, Mantovani A, Hardy B, Koch B, Escher S, van Thriel C, Cadenas C, Kroese D, van de Water B, Hengstler JG (2017) Adverse outcome pathways: opportunities, limitations and open questions. Arch Toxicol 91(11):3477–3505. https://doi.org/10.1007/s00204-017-2045-3
Louisse J, Beekmann K, Rietjens IM (2017) https://www.ncbi.nlm.nih.gov/pubmed/27768849Use of physiologically based kinetic modeling-based reverse dosimetry to predict in vivo toxicity from in vitro data. Chem Res Toxicol 30(1):114–125. https://doi.org/10.1021/acs.chemrestox.6b00302
Massey VL, Arteel GE (2012) Acute alcohol-induced liver injury. Front Physiol 3:1–8. https://doi.org/10.3389/fphys.2012.00193
Mazaleuskaya LL, Sangkuhl K, Thorn CF, FitzGerald GA, Altman RB, Klein TE (2015) PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genom 25(8):416–426
Mielke H, Anger LT, Schug M, Hengstler JG, Stahlmann R, Gundert-Remy U (2011) https://www.ncbi.nlm.nih.gov/pubmed/21046363A physiologically based toxicokinetic modelling approach to predict relevant concentrations for in vitro testing. Arch Toxicol 85(6):555–563. https://doi.org/10.1007/s00204-010-0612-y
Mulla H, Yakkundi S, McElnay J, Lutsar I et al (2015) An observational study of blood concentrations and kinetics of methyl- and propyl-parabenes in neonates. Pharm Res 32:1084–1093
Nellis G, Lutsar I, Varendi H, Turner MA et al (2013) Blood concentrations of methyl and propylparabens in neonates treated with gentamicin as the most commonly used antibiotic. In: 31th Annual meeting of ESPID (European Society of Pediatric Infectious Diseases). Milan, Italy; 28.05–01.06.2013
O’Brien PJ, Irwin W, Diaz D, Howard-Cofield E, Krejsa CM, Slaughter MR, Gao B, Kaludercic N, Angeline A, Bernardi P, Brain P, Hougham C (2006) High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol 80(9):580–604
Pendlington RU, Whittle E, Robinson JA, Howes D (2001) Fate of ethanol topically applied to skin. Food Chem Toxicol 39:169–174. https://doi.org/10.1016/S0278-6915(00)00120-4
Platt JC (2000) Probabilistic outputs for support vector machines and comparison to regularized likelihood methods. Adv Large Margin Classif. https://www.bibsonomy.org/bibtex/2b13a556c2a6c1a3a2a30fe889ea9b738/zeno
Proctor WR, Foster AJ, Vogt J, Summers C, Middleton B, Pilling MA, Shienson D, Kijanska M, Ströbel S, Kelm JM, Morgan P, Messner S, Williams D (2017) Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch Toxicol 91(8):2849–2863. https://doi.org/10.1007/s00204-017-2002-1 (Epub 2017 Jun 13)
Reif R, Ghallab A, Beattie L, Günther G, Kuepfer L, Kaye PM, Hengstler JG (2017) In vivo imaging of systemic transport and elimination of xenobiotics and endogenous molecules in mice. Arch Toxicol 91(3):1335–1352. https://doi.org/10.1007/s00204-016-1906-5
Ritz C, Baty F, Streibig JC, Gerhard D (2015) Dose-response analysis using R. PLoS One 10(12):e0146021. https://doi.org/10.1371/journal.pone.0146021
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform 12:77. http://www.biomedcentral.com/1471-2105/12/77
Roth SH, Shainhouse JZ (2004) Efficacy and safety of a topical diclofenac solution (pennsaid) in the treatment of primary osteoarthritis of the knee: a randomized, double-blind, vehicle-controlled clinical trial. Arch Intern Med 164(18):2017–2023
Rowe C, Gerrard DT, Jenkins R, Berry A, Durkin K, Sundstrom L, Goldring CE, Park BK, Kitteringham NR, Hanley KP, Hanley NA (2013) Proteome-wide analyses of human hepatocytes during differentiation and dedifferentiation. Hepatology 58(2):799–809. https://doi.org/10.1002/hep.26414
Sandanger TM, Huber S, Moe MK, Braathen T, Leknes H, Lund E (2011) Blood concentrations of parabens in postmenopausal women and self-reported use of personal care products: the NOWAC postgenome study. J Expo Sci Environ Epidemiol 21:595–600
Schug M, Stöber R, Heise T, Mielke H, Gundert-Remy U, Godoy P, Reif R, Blaszkewicz M, Ellinger-Ziegelbauer H, Ahr HJ, Selinski S, Günther G, Marchan R, Blaszkewicz M, Sachinidis A, Nüssler A, Oberemm A, Hengstler JG (2013) https://www.ncbi.nlm.nih.gov/pubmed/23274394Pharmacokinetics explain in vivo/in vitro discrepancies of carcinogen-induced gene expression alterations in rat liver and cultivated hepatocytes. Arch Toxicol 87(2):337–345. https://doi.org/10.1007/s00204-012-0999-8
Shekar S, Sood S, Showkat S, Lite C, Chandrasekhar A, Vairamani M, Barathi S, Santosh W (2016) Detection of phenolic endocrine disupting chemicals (EDCs) from maternal blood and amniotic fluid in Indian population. Gen Comp Endocrinol. https://doi.org/10.1016/j.ygcen.2016.05.025
Sjogren AK, Hornberg JJ (2018) Compound selection and annotation to validate the predictivity of in vitro toxicity assays for use in drug discovery, in response to Commentary by Dr. Zink (Zink, D. Arch Toxicol (2018)). Arch Toxicol 93(1):225–226
Sjogren AK et al (2018) A novel multi-parametric high content screening assay in ciPTEC-OAT1 to predict drug-induced nephrotoxicity during drug discovery. Arch Toxicol 92:3175–3190
Thiel C, Schneckener S, Krauss M, Ghallab A, Hofmann U, Kanacher T, Zellmer S, Gebhardt R, Hengstler JG, Kuepfer L (2015) A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross-species extrapolation. J Pharm Sci 104(1):191–206. https://doi.org/10.1002/jps.24214
Tolosa L, Jiménez N, Pelechá M, Castell JV, Gómez-Lechón MJ, Donato MT (2019) Long-term and mechanistic evaluation of drug-induced liver injury in Upcyte human hepatocytes. Arch Toxicol 93(2):519–532. https://doi.org/10.1007/s00204-018-2349-y (Epub 2018 Nov 13)
Vartak N, Damle-Vartak A, Richter B, Dirsch O, Dahmen U, Hammad S, Hengstler JG (2016) Cholestasis-induced adaptive remodeling of interlobular bile ducts. Hepatology 63(3):951–964. https://doi.org/10.1002/hep.28373
Vinken M, Hengstler JG (2018) Characterization of hepatocyte-based in vitro systems for reliable toxicity testing. Arch Toxicol 92(10):2981–2986
Vorrink SU, Zhou Y, Ingelman-Sundberg M, Lauschke VM (2018) Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol Sci 163(2):655–665
Wagner C, Zhao P, Pan Y (2015) Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacomet Syst Pharmacol 4(4):226–230
Waldmann T, Rempel E, Balmer NV, König A, Kolde R, Gaspar JA, Henry M, Hescheler J, Sachinidis A, Rahnenführer J, Hengstler JG, Leist M (2014) Design principles of concentration-dependent transcriptome deviations in drug-exposed differentiating stem cells. Chem Res Toxicol 27(3):408–420. https://doi.org/10.1021/tx400402j
Wink S, Hiemstra SW, Huppelschoten S, Klip JE, van de Water B (2018) Dynamic imaging of adaptive stress response pathway activation for prediction of drug induced liver injury. Arch Toxicol 92(5):1797–1814. https://doi.org/10.1007/s00204-018-2178-z (Epub 2018 Mar 3)
Xu JJ, Henstock PV, Dunn MC, Smith AR, Chabot JR, de Graaf D (2008) Cellular imaging predictions of clinical drug-induced liver injury. Toxicol Sci 105(1):97–105. https://doi.org/10.1093/toxsci/kfn109
Yoon M, Blaauboer BJ, Clewell HJ (2015) Quantitative in vitro to in vivo extrapolation (QIVIVE): an essential element for in vitro-based risk assessment. Toxicology 332:1–3. https://doi.org/10.1016/j.tox.2015.02.002 (Epub 2015 Feb 11)
Zárybnický T, Boušová I, Ambrož M, Skálová L (2018) Hepatotoxicity of monoterpenes and sesquiterpenes. Arch Toxicol 92(1):1–13. https://doi.org/10.1007/s00204-017-2062-2 (Epub 2017 Sep 13. Review)
Zellmer S, Schmidt-Heck W, Godoy P, Weng H, Meyer C, Lehmann T, Sparna T, Schormann W, Hammad S, Kreutz C, Timmer J, von Weizsäcker F, Thürmann PA, Merfort I, Guthke R, Dooley S, Hengstler JG, Gebhardt R (2010) Transcription factors ETF, E2F, and SP-1 are involved in cytokine-independent proliferation of murine hepatocytes. Hepatology 52(6):2127–2136. https://doi.org/10.1002/hep.23930
Acknowledgements
This work has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 681002 (EU-ToxRisk), from TransQST (no. 116030), LivSysTransfer (BMBF, 031L0119) with contributions from StemCellNet (BMBF, 01EK1604A), LiSyM (BMBF, 031Loo45031L0045) and InnoSysTox (BMBF/EU, 031L0021A) and the European Regional Development Fund (CP16/00097). We thank the Permanent Senate Commission on Food Safety (SKLM), Egon Willighagen, Sylvia Escher, Christian Desaintes and Hennicke Kamp for valuable discussion and support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Marianna Grinberg, Wolfgang Moritz, Reinhard Kreiling, Susann Fayyaz, Heidrun Ellinger-Ziegelbauer, Thomas Steger-Hartmann, and Lars Kuepfer are employees of the companies listed under the affiliations. The parabens analysed in this study were obtained from Clariant Produkte (Deutschland) GmbH. Iain Gardner and Mian Zhang are employees of Certara UK Limited, a company that develops the pharmacokinetic software used in this study. No financial support was obtained from any of the involved companies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Albrecht, W., Kappenberg, F., Brecklinghaus, T. et al. Prediction of human drug-induced liver injury (DILI) in relation to oral doses and blood concentrations. Arch Toxicol 93, 1609–1637 (2019). https://doi.org/10.1007/s00204-019-02492-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02492-9