
Abstract
The goal of the present study was to obtain an in vivo relevant prioritization method for the endocrine potencies of different polycarbonate monomers, by combining in vitro bioassay data with physiologically based kinetic (PBK) modelling. PBK models were developed for a selection of monomers, including bisphenol A (BPA), two bisphenol F (BPF) isomers and four different bisphenol A diglycidyl ethers (BADGEs), using in vitro input data. With these models, the plasma concentrations of the compounds were simulated, providing means to estimate the dose levels at which the in vitro endocrine effect concentrations are reached. The results revealed that, whereas the in vitro relative potencies of different BADGEs (predominantly anti-androgenic effects) can be up to fourfold higher than BPA, the estimated in vivo potencies based on the oral equivalent doses are one to two orders of magnitude lower than BPA because of fast detoxification of the BADGEs. In contrast, the relative potencies of 2,2-BPF and 4,4-BPF increase when accounting for the in vivo availability. 4,4-BPF is estimated to be fivefold more potent than BPA in humans in vivo in inducing estrogenic effects and both 2,2-BPF and 4,4-BPF are estimated to be, respectively, 7 and 11-fold more potent in inducing anti-androgenic effects. These relative potencies were considered to be first-tier estimates, particularly given that the potential influence of intestinal metabolism on the in vivo availability was not accounted for. Overall, it can be concluded that both 2,2-BPF and 4,4-BPF are priority compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bisphenol A (BPA) is a well-known monomer that is used in the production of polycarbonate plastics as well as epoxy resins that are applied as protective linings in, for example, cans or fruit juice packages. The use as monomer leads to ubiquitous exposure to BPA, with the average daily intake in Europe ranging from 0.4 to 1.4 µg kg bw−1 per day (EFSA 2015). Since this exposure is about fourfold lower than the tolerable daily intake (TDI) of 4 µg kg bw−1 per day, it is not considered a human health risk (EFSA 2015). However, the overall public acceptance of BPA is declining because of its estrogenic and anti-androgenic properties. To this end, alternatives to BPA are being sought. Substitution of BPA can either be done using other polymers (e.g. polypropylene or polyether instead of polycarbonate) or by the replacement with chemicals that have similar chemical functionality (so-called ‘drop-in’ replacement) (Bakker et al. 2016). Particularly, the latter may not be a desirable approach, as analogues that are structurally and functionally close to BPA can be expected to express comparable biological activities (Rosenmai et al. 2014).
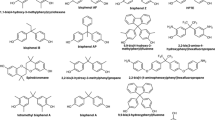
In a recent study, van Leeuwen et al. (2019) determined the estrogenic, anti-estrogenic, androgenic, and anti-androgenic potencies of 30 bisphenols (BPs) and bisphenol diglycidyl ethers (BDGEs) using yeast-based bioassays. In addition, the presence of these analogues in different canned foods and beverages was shown. BPA was most frequently detected, occurring in seven out of the ten analysed food and beverage samples, ranging from 0.03 to 68 µg kg−1 or L−1 product. Other BPs that were found included 2,2-BPF and 4,4-BPF, present in a soft drink sample at concentrations of 0.007 and 0.64 µg L−1, respectively. Finally, several bisphenol A diglycidyl ethers (BADGEs) were found, including BADGE, BADGE·H2O, BADGE·2H2O and BADGE·HCl, occurring in a range of 0.08–3.3 µg kg−1 or L−1 in different products such as soft drink and tomato soup. The structures of these different BPs and BADGEs are given in Fig. 1. Among the detected analogues, 4,4-BPF was observed to be equally potent as BPA in inducing estrogenic and anti-androgenic effects in vitro. Other analogues (2,2-BPF, BADGE, BADGE·H2O, and BADGE·HCl) mainly exhibited anti-androgenic activity with potencies that are comparable to BPA or up to fourfold higher (in case of 2,2-BPF and BADGE·HCl). BADGE-2H2O did not induce any (anti)estrogenic or (anti)androgenic effects.

Molecular structure of the BPs and BADGEs previously observed in different products by van Leeuwen et al. (2019) and included in the present study
The presence of 2,2-BPF, 4,4-BPF and different BADGEs in food products and the observed in vitro endocrine activities provide arguments for prioritization of these compounds for further testing as the in vitro biological activities, as such, are not indicative for potential adverse effects in vivo (OECD 2018). The ultimate biological activity will also depend on the systemic availability of the chemical. Given that follow-up in vivo animal experiments pose animal welfare issues and raise questions with respect to relevance to humans, there is a clear need for alternative approaches to assess in vivo relevant endocrine potencies. A significant way forward is the use of physiologically based kinetic (PBK) modelling-based reverse dosimetry for the quantitative in vitro-to-in vivo extrapolation (QIVIVE) of in vitro EC50/IC50 values to equivalent oral doses (Yoon et al. 2012; Dancik et al. 2015; Wetmore et al. 2015; Louisse et al. 2017; Strikwold et al. 2017; Fabian et al. 2019).
The goal of the current study was to obtain an in vivo relevant prioritization method of the BPs and BADGEs by extrapolating the in vitro potency data to equivalent oral doses with PBK-based reverse dosimetry. As starting point the in vitro endocrine potencies of the compounds as previously determined with yeast bioassays were used. These bioassays are based on a clone of a Saccharomyces cerevisiae expressing either the human estrogen receptor α (hERα) or the human androgen receptor (hAR) and when specifically activated drive the expression of a yeast-enhanced green fluorescent protein (yEGFP) (Bovee et al. 2004, 2007). For the development of the PBK models, in vitro human hepatic metabolic clearance of the bisphenol analogues was determined with HepaRG cells in suspension, while absorption of the analogues was measured with Caco-2 cells in a transwell system. Integration of these data with a generic PBK model allowed to simulate the peak concentrations of the chemicals in plasma at different oral doses. In this way, for each chemical, the dose that results in a peak plasma concentration that is equal to the EC50/IC50 value could be estimated. The outcomes were used to define the expected in vivo human relative potencies of the different BPs and BADGEs compared to BPA.
Materials and methods
Chemical compounds
BPA (CAS 80-05-7), 4,4-BPF (CAS 620-92-8), BADGE (CAS 1675-54-3), BADGE·H2O (CAS 76002-91-0), BADGE·2H2O (CAS 5581-32-8), BADGE·HCl (CAS 13836-48-1), and BADGE·HCl·H2O (CAS 227947-06-0) were purchased from Sigma-Aldrich (Steinheim, Germany). 2,2-BPF (CAS 2467-02-9) was purchased from TCI America (Portland, USA) and BPA glucuronide (CAS 267244-08-6) was purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Dimethyl sulfoxide (DMSO) was purchased from Merck (Darmstadt, Germany) and used for preparing stock solutions of the compounds. BPA glucuronide was dissolved in UPLC-MS grade methanol, purchased from Actu-All (Oss, the Netherlands).
Caco-2: in vitro intestinal transport study
Caco-2 cells (passage 21–27) were cultured in DMEM (Gibco, Life technologies; New York, USA) containing 4.5 g L−1d-glucose, l-glutamine, and 25 mM HEPES, supplemented with 10% (v/v) FBS (Gibco, Life technologies, New York, USA), 1% (v/v) minimal essential medium non-essential amino acids (Gibco, Life technologies; New York, USA), and 10,000 U mL−1 penicillin and 10 mg mL−1 streptomycin (Sigma-Aldrich, Steinheim, Germany). The treatment and exposure of the Caco-2 cells are based on the procedure as described in Strikwold et al. (2017). Cells were seeded at a concentration of approximately 22.4 × 104 cells mL−1 onto 12-well transwell plates containing a polycarbonate membrane (12 mm inserts, 0.4 µm pore size, Corning Incorporated, New York, USA). No significant contribution of BPs or BADGEs from the polycarbonate membrane was observed given that the mass balance during the experiment did not exceed 100% (see Table 2 of the “Results”). The seeded cells were maintained for 21–22 days in a 5% CO2-humidified atmosphere at 37 °C during which the medium in the apical and basolateral compartments (0.5 and 1.5 mL, respectively) was changed every 2 or 3 days and always 1 day before exposure.
Prior to the start of the transport experiment, the cell culture medium was removed, and the cells were equilibrated in HBSS (Gibco) for 30–45 min. The BPs and BADGEs, described under “Chemical compounds”, were diluted to a final concentration of 100 µM in HBSS medium (without phenol red) supplemented with 10 mM HEPES, pH 6.5. The final DMSO level in this apical transport medium was 0.2%. The basolateral transport medium consisted of pre-warmed (37 °C) HBSS supplemented with 30 mg mL−1 bovine serum albumin (Sigma-Aldrich, Steinheim, Germany) to reduce non-specific binding at pH 7.4. The 1.5 mL basolateral transport medium was first added to the basolateral compartment, followed by 0.5 mL apical transport medium to the apical compartment. The transwells were incubated under the same circumstances as for culturing the cells. For the time lapse experiment, 75 µL of the basolateral compartment was taken after 15, 30, 60, 90, and 120 min of incubation, and added to 150 µL of ice-cold methanol. This 75 µL was replaced by the same amount of pre-warmed basolateral transport medium to keep the compartment volume constant.
Given that each BP and BADGE analogue tested in the Caco-2 transport experiment showed linear absorption over 120 min (data not shown), the final permeability coefficients were determined at an incubation time of 30 min based on three independent replicates. For one replicate, the mass balance was determined by also taking a sample of the apical compartment and the membrane. For the latter, the membranes from the inserts were cut, added to 250 µL of ice-cold 65% methanol in water and sonicated for 15 min with a Branson 5510 water bath sonicator (Emerson, USA).
HepaRG: in vitro liver metabolism study
HepaRG cryopreserved cells (HPRGC10) and Williams’ medium E (WME) were purchased from ThermoFisher Scientific (Waltham, USA). HepaRG cells were thawed from a liquid nitrogen stock according to the protocol of the manufacturer and re-suspended in WME (serum- and phenol-free) to a final concentration of 0.5 × 106 cells mL−1. The test compounds were diluted from the stock solution to a concentration of 10 mM in DMSO. Subsequently, this solution was diluted to 2 mM in methanol–water (1:1) and further diluted to 2 µM in WME.
The treatment and exposure procedure of the HepaRG cells was based on the protocol as described in Zanelli et al. (2011). Each incubation was prepared by mixing an equal amount of cell suspension and compound dilution in a V shaped 96-well plate (Greiner Bio-one) to obtain the final conditions of 0.05% of total organic solvent (DMSO and methanol), 1 µM compound and 0.25 × 106 cells mL−1. The incubations were carried out in an incubator at 37 °C, 5% CO2, 19% O2 on top of a shaker running at 50 rpm. The reactions were stopped by transferring 100 µL of the incubation volume to an equal volume of ice-cold methanol after 0, 10, 20, 40, and 80 min of incubation in a new 96-well plate. Control incubations were performed in the presence of boiled HepaRG cells (obtained by heating in a microwave oven). After the incubations, the plates were centrifuged for 5 min, 300 rcf, at 4 °C and the supernatant was transferred to a new 96 well-plate with inserts, suitable for LC–MS/MS injection (Waters, Milford, USA). Four replicates were performed on 2 independent days.
Sample analysis
Samples from the metabolic clearance experiments with HepaRG cells and intestinal absorption studies with Caco-2 transwell system were analysed as described by Leeuwen et al. (2019). In short, samples were analysed on a Waters Acquity I-Class UPLC (Milford, USA) system that was equipped with a Waters Acquity UPLC BEH C18 (100 × 2.1 mm, 1.7 µm) column. The column heater was kept at 50 °C. The vial compartment of the autosampler was kept at 10 °C and a 10 µL injection volume was used. Mobile phase A was water and B was acetonitrile/water (90:10 v/v %), both containing 6.7 mM ammonium hydroxide (pH = 11) at a flow rate of 0.6 mL min−1. The gradient started at 0% B, was kept at 0% for 1 min and was then increased linearly to 40% B in 1 min and then linearly increased to 100% B in 5 min This mobile phase composition was kept for 1 min and subsequently returned to 0% B in 0.2 min. An equilibration time of 1.8 min was allowed before the next injection.
Mass spectrometric detection was performed with a Waters Xevo TQS tandem mass spectrometer (Waters, Wilmslow, UK) equipped with an electrospray ionization interface (ESI). Two separated injections were done with the MS operating separately in ESI− (BPs) and in ESI+ (BADGEs). A capillary voltage of 2.2 kV (ESI−) or 3.0 kV (ESI+), a desolvation gas temperature of 600 °C at a N2 flow of 800 L h−1, a source temperature of 150 °C and a cone gas (N2) flow of 150 L h−1 were used. Argon was used as collision-induced dissociation (CID) gas. The cone voltage and collision energy were optimized by direct infusion experiments under alkaline conditions.
Two product ions were selected for each BP and BADGE, to allow quantification as well as identification of the specific BP and BADGE compounds. Parent ions of the expected metabolites were monitored as well. These include the glucuronide and sulphate conjugates of BPA and BPF isomers and the hydrolysis products of the different BADGEs. Given that BADGE converts to BADGE·H2O and BADGE·2H2O, which are also included as model compounds in the present study, the conversion of BADGE to these metabolites could be quantified based on the included standard curves for BADGE·H2O and BADGE·2H2O. The same is true for BADGE·H2O, which converts directly to BADGE·2H2O. For comparison, a calibration curve of BADGE·HCl·H2O was included as well. No standard curves of the glucuronide and sulphate conjugates of BPA and BPF isomers were included and the formation of these metabolites could, therefore, not be quantified. Further details on the quantification and identification of the BPs and BADGEs can be found elsewhere (van Leeuwen et al. 2019).
Estimation of the oral equivalent doses from the EC50/IC50 values using QIVIVE
The in vitro EC50/IC50 values for estrogenic and anti-androgenic activity of the selected BPs and BADGEs were extrapolated to equivalent in vivo human oral doses by PBK model-based reverse dosimetry to derive the in vivo relevant potency ranking.
A generic PBK model defined by Jones and Rowland-Yeo (2013) was used to estimate the free peak plasma concentrations of the different BPs and BADGEs in human plasma at different oral doses. The model equations were coded and numerically integrated in Berkeley Madonna 8.3.18 (Macey and Oster, UC Berkeley, CA, USA), using the Rosenbrock’s algorithm for stiff systems. The model code of the generic model, containing the input data for BPA, is provided as supporting information 1. The generic PBK model simulates plasma and tissue concentrations in humans based on: (1) the intrinsic hepatic metabolic clearance, (2) tissue partition coefficients and binding data (fraction unbound in plasma and tissue, blood plasma ratio, fraction unbound in hepatocytes), (3) renal clearance (L h−1) and (4) the absorption rate ka (h−1) and fraction absorbed (unitless). These chemical-specific input data were obtained in the following way:
-
(Ad 1)
The in vitro intrinsic hepatic metabolic clearance of the different BP and BADGE compounds was determined by measuring substrate depletion of the chemicals in incubations with HepaRG cells as described above. The in vitro intrinsic clearances data (expressed as µL min−1 per 106 hepatocytes) were converted in the model to in vivo intrinsic clearances (CLint, L hr−1 kg bw−1) based on a hepatocyte per gram liver yield (HPGL) of 117.5 × 106 hepatocytes per gram liver (Simcyp 2016) and a liver weight of 1.47 kg (2.1% of a BW of 70 kg, Jones and Rowland-Yeo, 2013).
-
(Ad 2)
Tissue partition coefficients and binding data were estimated based on the physicochemical properties of the different BP and BADGE compounds using available calculators. First, the log P, log D and pKa values for the BPs and BADGEs were estimated with Chemicalize (www.chemicalize.com, ChemAxon). Based on these data, the fraction unbound in plasma (fup) and the blood/plasma (B/P) ratio of these compounds were estimated with Simcyp toolbox (Simcyp 2016) and PK-SIM (Version 6.3.2, Bayer Technology Services), respectively. From the fraction unbound in plasma, the fraction unbound in tissue (fut) was calculated according to the method of Poulin and Theil (2002) to simulate the free concentration in the liver that is available for metabolic clearance. The partition coefficients were calculated according to Rodgers and Rowland (2006) with PK-SIM (Version 6.3.2, Bayer Technology Services). The fraction unbound in the hepatocyte incubation (fuhep) was calculated applying a method described by Kilford et al. (2008) and using log D as input parameter. The final tissue partition coefficients and binding data are provided in Table 1.
Table 1 Physicochemical properties and calculated unbound plasma fractions, blood/plasma ratios and partition coefficients -
(Ad 3)
Renal clearance was assumed to be equal to the glomelular filtration rate (6.7 L h−1) times the unbound venous plasma concentration (Yoon et al. 2012).
-
(Ad 4)
The in vivo absorption rate constants (ka) of the compounds were derived from the in vitro apparent permeability (Papp_Caco-2) values that were scaled to in vivo log Papp values according to the following formula: log Papp_in vivo (cm s−1) = 0.6836 × log Papp_Caco-2− 0.5579 (Sun et al. 2002). The Papp in vivo values are related to ka as follows: ka = Papp_in vivo × intestinal surface area/intestinal volume = Papp_in vivo × (2 × pi × r × l)/(pi × r2) = Papp_in vivo × 2/r, where r and l are the radius and length of the intestinal section, respectively. The radius of the human intestine is set at 1 cm (Peters 2008, 2012). Based on the above equation, the length of the intestine is not a required input. BPA and the two isomers of BPF were assumed to be fully absorbed (Fa = 1), whereas the fractions absorbed of the different BADGEs were assumed to be equal to the observed recovery rates at 30 min to account for the metabolic conversion of these compounds as observed in the Caco-2 experiment.
Results
In vitro Caco-2 absorption
Table 2 provides the Papp values obtained for the different BPs and BADGEs in the Caco-2 transwell absorption experiments. In case of BPA, 2,2-BPF and 4,4-BPFa rapid absorption was observed with little differences in Papp values between these compounds. The observed Papp_Caco-2 values ranged from 28 to 72 × 10−6 cm s−1. No metabolic conversion of BPs by Caco-2 cells occurred, with the recoveries ranging between 82 and 98% (i.e. levels of the parent compound that were found back in the apical compartment, filter, cells, and basolateral compartment, see Table 2).
In case of BADGE, BADGE·H2O and BADGE·HCl, the Papp_Caco-2 values ranged from 1.4 to 8.2 × 10−6 cm s−1, which seems lower than that for the BPs. However, apart from transport, extensive metabolism of these BADGEs by Caco-2 cells was observed, resulting in low recoveries (Table 2). The metabolites could be identified and quantified by LC–MS based on the included standard curves (see “Materials and methods”). BADGE first converts to BADGE·H2O and then to BADGE·2H2O, whereas BADGE·H2O and BADGE·HCl directly convert to BADGE·2H2O and BADGE·HCl·H2O, respectively (similar to that observed in incubations with HepaRG cells). By accounting for these metabolites, the recoveries increase (Table 2). Also, BADGE·2H2O was assumed to be metabolically converted given its low recovery of 19% (Table 2); however, the metabolite(s) that were formed could not be detected with the targeted LC–MS/MS method. The low recovery of BADGE·2H2O was not due to matrix suppression as the standard curve is prepared and analysed in the same matrix (HBSS) as the samples from the Caco-2 experiment. Correcting Papp_Caco-2 of the different BADGEs for the recoveries (assuming the starting concentrations of the parent compound are not 100 µM, but proportionally lower because of metabolism) allows to obtain an indication of the permeability of the parent compounds. These results show that the apparent permeability of the BADGEs, when there would be no metabolism, is comparable with the BPs, with Papp_Caco-2 values, ranging from 49 to 78 × 10−6 cm s−1. These latter values were used in the PBK model for the rate of absorption (ka) of the parent chemicals, whereas the recovery rates were used in the model as a measure of the fraction that is absorbed (Fa).
In vitro metabolic clearance with HepaRG cells
Figure 2 shows the observed substrate depletion of BPA, 2,2-BPF, and 4,4-BPF. The conversion of these three BPs primarily resulted in the formation of glucuronide and sulphate conjugates (supporting information 2). These conjugates were not further quantified. The determined half-life of the three BPs was similar (Table 3), though 4,4-BPF was observed to be slightly slower metabolized, resulting in a somewhat longer half-life. No conversion of the BPs was observed in the control incubations with boiled HepaRG cells (data not shown).

Substrate depletion of BPA (a), 2,2-BPF (b), and 4,4-BPF (c) in incubations with HepaRG cells
The conversion of the different BADGEs into their corresponding dihydrodiols occurred almost instantaneously (Fig. 3). With the exception of BADGE·2H2O, 90% conversion is already achieved at incubation time zero, where the reaction is stopped directly after mixing the compounds with HepaRG. No conversion of BADGE, BADGE·H2O and BADGE·HCl was observed in the control incubations with boiled HepaRG cells (data not shown). The exact half-life could not be determined but can be expected to be less than 1 min (Table 3). Further metabolic conversion of BADGE·2H2O did not take place.

Substrate depletion and metabolite formation of BADGE (a), BADGE·H2O (b), BADGE·2H2O (c), and BADGE·HCl and BADGE·HCl·H2O (d) in incubations with HepaRG cells
Estimation of the oral equivalent doses using QIVIVE
Using the generic PBK model defined by Jones and Rowland-Yeo (2013) to integrate the in vitro metabolic clearance data and Caco-2 absorption data of the present study, the expected plasma concentrations in humans of each of the different analogues were simulated. In case of BPA, this simulation of the plasma concentration could be evaluated against available human literature data, i.e. from Thayer et al. 2015 and Teeguarden et al. 2015. Figure 4 shows that the peak plasma concentration (Cmax) of BPA predicted by the PBK model is in line with the observed in vivo human peak concentrations as reported by Thayer et al. (2015). The model overestimates the in vivo peak concentrations as reported by Teeguarden et al. (2015) by 3- to 6-fold. However, previous findings from Yang et al. (2015) suggest that the absorption efficiency in the latter study was limited to 25%. Such a reduced absorption efficiency from food was not accounted for in the model of BPA to obtain worst-case estimates of the plasma concentrations.

PBK-model simulated total BPA plasma concentrations compared with the literature reported data from a Thayer et al. (2015) obtained at an oral BPA dose of 0.1 mg kg bw−1 and b Teeguarden et al. (2015) obtained at an oral dose of 0.03 mg kg bw−1. The line presents the simulated plasma concentration and the data points the observed levels in the different individuals of the two studies
In the previous study by van Leeuwen et al. (2019), BPA and 4,4-BPF showed estrogenic activity with both an EC50 value of 20 µM. All compounds, except BADGE·2H2O, showed anti-androgenic activity with IC50 values of, respectively, 30, 10, 20, 20, 50 and 8 µM for BPA, 2,2-BPF, 4,4-BPF, BADGE, BADGE,H2O and BADGE·HCl.
Based on the developed PBK models for the different analogues, the peak free plasma concentrations (Cmax, free) of each compound were first estimated for an arbitrary selected reference dose of 1 mg kg bw−1. From these data, the doses that are needed to achieve the in vitro EC50/IC50 values in plasma could be calculated by linear extrapolation. Given that the in vitro potency data were derived with yeast-based bioassays that do not include foetal bovine serum, being a source for protein binding (Bovee et al. 2004, 2007), the EC50/IC50 were assumed to best be compared with the unbound (free) effective concentrations. Table 4 provides the overview of the estimated free peak plasma concentrations and the absolute in vivo equivalent doses and estimated in vivo relative potencies. The estimated in vivo potencies of the compounds relative to BPA are depicted in Fig. 5.

The predictions of the relative potencies reveal that the fast conversion of BADGE, BADGE·H2O and BADGE·HCl will reduce the endocrine activity of these compounds in vivo. Though the exact half-life for liver metabolism could not be determined, an estimated in vitro half-life of 1 min for hepatic metabolic conversion resulted in estimated peak blood concentrations of BADGE, BADGE·H2O and BADGE·HCl (at a dose of 1 mg kg bw−1) that are one to two orders of magnitude lower than for the different BPs. As a result of the fast-metabolic conversion of these BADGEs to metabolites that do not have anti-estrogenic or anti-androgenic activity (i.e. BADGE·2H2O) or lower potencies than the parent (i.e. BADGE·HCl·H2O), the relative in vivo potencies are expected to be low (Table 4, and Fig. 5).
In contrast to the different BADGEs, the in vivo relative potencies of 2,2-BPF and 4,4-BPF increase when accounting for the in vivo availability of the compounds. Though 4,4-BPF is equally potent as BPA in inducing estrogenic effects in vitro, the in vivo potency is estimated to be fivefold higher than BPA. In addition, whereas in vitro 2,2-BPF was threefold more potent and 4,4-BPF 1.5-fold more potent than BPA in the yeast anti-androgen bioassay, these compounds become 7- and 11-fold more potent in vivo than BPA after conversion of the in vitro potencies to oral equivalent doses using the developed PBK models. Given that metabolic clearance and Caco-2 absorption of 2,2-BPF and 4,4-BPF were similar to BPA (Tables 2 and 3), this increase in relative potency can only be explained by the differences in lipophilicity of these compounds compared to BPA and resulting partition coefficients that were used in the PBK model (Table 1). The lower partition coefficients of both 2,2-BPF and 4,4-BPF compared to BPA result in a higher plasma concentration compared to BPA.
Discussion
The goal of the current study was to obtain an in vivo relevant prioritization method for BPs and BADGEs by converting their in vitro endocrine effect concentrations, i.e. EC50 values for estrogenicity and IC50 values for anti-androgenicity, into human equivalent doses using in vitro-based PBK models. Some of the compounds (i.e. 2,2-BPF, BADGE, and BADGE·HCl) have also been reported to induce anti-estrogenic effects (van Leeuwen et al. 2019). However, as these anti-estrogenic effects occur at higher concentrations than the anti-androgenic effects by these compounds, they were considered to be less relevant and were not accounted for in the current study.
For the development of the PBK models, the metabolic conversion of the compounds was determined with HepaRG cells in suspension. In contrast to other liver cell lines, this hepatoma cell line has a metabolic capacity that resembles primary hepatocytes, although this still needs to be fully elucidated (Guillouzo et al. 2007; Zanelli et al. 2011). The observed metabolic clearance of BPA in the present study of 135 µL min−1 per 106 hepatocytes is slightly higher than literature reported hepatic clearances obtained with primary human hepatocytes, ranging from 22 µL min−1 per 106 hepatocytes to 93 µL min−1 per 106 hepatocytes (Kuester and Sipes 2007; Kurebayashi et al. 2010). After scaling and integration of the in vitro metabolic clearance data into the generic PBK model, predictions could be made of the plasma concentrations of BPA. Comparison of the outcome with in vivo measured plasma concentrations supports the adequacy of the PBK approach and the usefulness of the HepaRG cell line in measuring the clearance of BPs.
The rapid conversion of BADGE, BADGE·H2O and BADGE·HCl into their corresponding diols (BADGE·2H2O and BADGE·HCl·H2O) also confirms the proficiency of these human liver cells in epoxide hydrolases, which catalyse this hydrolysis reaction (Bentley et al. 1989). Interestingly, this conversion of the epoxides was also observed in the absorption experiments with the human intestinal Caco-2 cells. Indeed, it has been reported that Caco-2 cells are proficient in epoxide hydrolases (Borlak and Zwadlo 2003). Although conversion of both BADGE and BADGE·H2O into BADGE·2H2O was observed in HepaRG cells, the further metabolic conversion of BADGE·2H2O did not take place in these cells. This may indicate an insufficiency of the HepaRG cells but may also suggest that metabolism of BADGE·2H2O does not occur in the liver. The poor recovery of BADGE·2H2O in the Caco-2 cell experiment of 19% suggests that an intestinal metabolic conversion of this compound might occur. However, given the targeted screening by LC–MS/MS analysis, no metabolite(s) of BADGE·2H2O could be identified in the absorption experiments with Caco-2 cells. Because BADGE·2H2O does not express any endocrine activity (Fic et al. 2014; van Leeuwen et al. 2019), the metabolic conversion of this compound was not further explored.
The in vitro effective concentrations (EC50/IC50) of BPA translate into equivalent oral doses in humans of 5000 (estrogenic activity) and 7500 mg kg bw−1 (anti-androgenic activity). These dose levels are many orders of magnitude higher than the estimated daily intake (EDI) of BPA of 0.4–1.4 µg kg bw−1 per day (EFSA 2015) and this suggests that the endocrine active activity of BPA is not likely to occur in vivo at the EDI. Nonetheless, questions can be raised with respect to the predictive value of the absolute estimated oral equivalent doses. Starting point in the extrapolation are the EC50/IC50 values obtained with yeast-based bioassays. Recently, Zhang et al. (2018) and Fabian et al. (2019) revealed that results obtained with a yeast estrogen screen (YES) provide a good prediction of the in vivo potency of BPA, after conversion of the in vitro effective concentration of this compound to equivalent oral doses in rats using PBK-based reverse dosimetry. Zhang et al. (2018) even reported a better predictive value using data from the yeast bioassay than using the MCF-7/BOS proliferation or the U2OS ER-CALUX bioassays. Though these results suggest that relevant absolute potency estimates can be made with in vitro assays like the yeast-based bioassay used in the present study, the goal of the present work was mainly to determine the in vivo relative potencies of different bisphenol analogues compared to BPA.
The results revealed that despite the comparable in vitro endocrine activities (mainly anti-androgenic) of BPA, 2,2-BPF, 4,4-BPF, BADGE, BADGE·H2O, and BADGE·HCl, the estimated in vivo potency ranking is quite different. Particularly as a result of the fast-metabolic conversion of the different BADGEs, the in vivo anti-androgenic potencies for these types of compounds are estimated to be one to two orders of magnitude lower than BPA. Both BADGE and BADGE·H2O convert to BADG·2H2O, which does not display (anti)estrogenic or (anti)androgenic activity (van Leeuwen et al. 2019). BADGE·HCl converts to BADGE·HCl·H2O. This metabolite does not display any estrogenic or androgenic activity and is fivefold less potent then its precursor BADGE·HCl in inducing anti-estrogenic and anti-androgenic effects (van Leeuwen et al. 2019). The fast-metabolic conversion of the BADGEs is, therefore, considered to lead to detoxification of the compounds.
In contrast to the different BADGEs, the relative potencies of 2,2-BPF and 4,4-BPF compared to BPA are higher when accounting for the in vivo availability of the compounds. For example, 4,4-BPF is equally potent as BPA in inducing estrogenic effects in vitro but is estimated to be 5-fold more potent in vivo. In case of the anti-androgenic effects, 2,2-BPF and 4,4-BPF are 3 and 1.5-fold more potent in vitro than BPA but become 7 and 11-fold more potent than BPA in vivo. This higher in vivo relative potencies of the BPF isomers were primarily due to lower partitioning into tissue and lower plasma binding of these compounds. Both the hepatic clearance and apparent permeability in the Caco-2 absorption were observed to be similar to BPA and did not significantly contribute to the difference between in vitro and in vivo relative potencies of the BPF isomers. It should be noted that intestinal metabolic clearance of 2,2-BPF, 4,4-BPF, and BPA was not accounted for in the reverse dosimetry approach. Compared with BPA, 4,4-BPF has been reported to being more prone to intestinal conversion based on in vitro experiments with human intestine and liver microsomes and recombinant UGT enzymes (Gramec Skledar et al. 2015). Given that Caco-2 cells do not express sufficient glucuronosyltransferase and sulfotransferase enzymes to catalyse the metabolism of the BPs (Meinl et al. 2008), these differences in intestinal metabolism could not be observed in the present study. The estimated in vivo relative potencies are, therefore, likely to provide a worst-case estimate with respect to the relative potencies of the two BPF isomers compared to BPA.
Comparison of the results of the present study with available in vivo data is not straight forward. Apart from BPA, there are few in vivo data available on the estrogenic or anti-androgenic potencies of the BPF isomers and BADGEs. To this end, the relative potencies of BPA analogues have mainly been based on in vitro data (Rochester and Bolden 2015; van Leeuwen et al. 2019). Moreover, data obtained with rodent studies cannot be directly compared with the results of the present study which presents estimates of human relative potencies. Nonetheless, some support for the estimated lower potency of the BADGEs comes from the EFSA evaluation on BADGE and its hydrolysis products (i.e. BADGE·H2O and BADGE·2H2O), reporting no reproductive effects in rats, or teratogenic or adverse effects on embryonal and foetal development in studies with rats and rabbits (EFSA 2004).
Overall, the results of the study show the importance of accounting for the toxicokinetics of chemicals when extrapolating relative potencies based on in vitro data to in vivo. By including information on metabolism, absorption and partitioning in the body, it could be shown that both 2,2-BPF and 4,4-BPF are priority compounds with potential higher in vivo endocrine (particularly anti-androgenic) activity than BPA.
References
Bakker J, Hakkert BC, Hessel EVS, Luit RJ, Piersma AH, Sijm DTHM, Rietveld AG, van Broekhuizen FA, van Loveren H, Verhoeven JK (2016) Bisphenol A Part 2. Recommendations for risk management. Report number 2015-0192. Bilthoven, The Netherlands: National Institute for Public Health and the Environment (RIVM). https://www.rivm.nl/bibliotheek/rapporten/2015-0192.pdf. Accessed May 2019
Bentley P, Bieri F, Kuster H, Muakkassah-Kelly S, Sagelsdorff P, Stäubli W, Waechter F (1989) Hydrolysis of bisphenol A diglycidylether by epoxide hydrolases in cytosolic and microsomal fractions of mouse liver and skin: inhibition by bis epoxycyclopentylether and the effects upon the covalent binding to mouse skin DNA. Carcinogenesis 10:321–327
Borlak J, Zwadlo C (2003) Expression of drug-metabolizing enzymes, nuclear transcription factors and ABC transporters in Caco-2 cells. Xenobiotica 33:927–943. https://doi.org/10.1080/00498250310001614286
Bovee TFH, Helsdingen RJR, Koks PD, Kuiper HA, Hoogenboom RLAP, Keijer J (2004) Development of a rapid yeast estrogen bioassay, based on the expression of green fluorescent protein. Gene 325:187–200
Bovee TFH, Helsdingen RJR, Hamers ARM, van Duursen MBM, Nielen MWF, Hoogenboom RLAP (2007) A new highly specific and robust yeast androgen bioassay for the detection of agonists and antagonists. Anal Bioanal Chem 389:1549–1558. https://doi.org/10.1007/s00216-007-1559-6
Dancik Y, Troutman JA, Jaworska J (2015) Estimation of in vivo dose of dermally applied chemicals leading to estrogen/androgen receptor-mediated toxicity from in vitro data—Illustration with four reproductive toxicants. Reprod Toxicol 55:50–63. https://doi.org/10.1016/j.reprotox.2015.01.002
EFSA (2004) Opinion of the Scientific Panel on food additives, flavourings, processing aids and materials in contact with food (AFC) related to 2,2-bis(4-hydroxyphenyl)propane bis(2,3-epoxypropyl)ether (Bisphenol A diglycidyl ether, BADGE). REF. No 13510 and 39700. EFSA J 2:86. https://doi.org/10.2903/j.efsa.2004.86
EFSA (2015) Scientific opinion on the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J 13:3978. https://doi.org/10.2903/j.efsa.2015.3978
Fabian E, Gomes C, Birk B, Williford T, Hernandez TR, Haase C, Zbranek R, van Ravenzwaay B, Landsiedel R (2019) In vitro-to-in vivo extrapolation (IVIVE) by PBTK modeling for animal-free risk assessment approaches of potential endocrine-disrupting compounds. Arch Toxicol 93:401–416. https://doi.org/10.1007/s00204-018-2372-z
Fic A, Žegura B, Gramec D, Mašič LP (2014) Estrogenic and androgenic activities of TBBA and TBMEPH, metabolites of novel brominated flame retardants, and selected bisphenols, using the XenoScreen XL YES/YAS assay. Chemosphere 112:362–369. https://doi.org/10.1016/J.CHEMOSPHERE.2014.04.080
Gramec Skledar D, Troberg J, Lavdas J, Peterlin Mašič L, Finel M (2015) Differences in the glucuronidation of bisphenols F and S between two homologous human UGT enzymes, 1A9 and 1A10. Xenobiotica 45:511–519. https://doi.org/10.3109/00498254.2014.999140
Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C (2007) The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact 168:66–73. https://doi.org/10.1016/j.cbi.2006.12.003
Jones H, Rowland-Yeo K (2013) Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT pharmacometrics Syst Pharmacol 2:e63. https://doi.org/10.1038/psp.2013.41
Kilford PJ, Gertz M, Houston JB, Galetin A (2008) Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos 36:1194–1197. https://doi.org/10.1124/dmd.108.020834
Kuester SRK, Sipes IG (2007) Prediction of metabolic clearance of bisphenol A (4,4-dihydroxy- 2,2-diphenylpropane) using cryopreserved human hepatocytes. Drug Metab Dispos 35:1910–1915. https://doi.org/10.1124/dmd.107.014787
Kurebayashi H, Okudaira K, Ohno Y (2010) Species difference of metabolic clearance of bisphenol A using cryopreserved hepatocytes from rats, monkeys and humans. Toxicol Lett 198:210–215. https://doi.org/10.1016/J.TOXLET.2010.06.017
Louisse J, Beekmann K, Rietjens IMCM (2017) Use of physiologically based kinetic modeling-based reverse dosimetry to predict in vivo toxicity from in vitro data. Chem Res Toxicol 30:114–125. https://doi.org/10.1021/acs.chemrestox.6b00302
Meinl W, Ebert B, Glatt H, Lampen A (2008) Sulfotransferase forms expressed in human intestinal Caco-2 and TC7 cells at varying stages of differentiation and role in benzo[a]pyrene metabolism. Drug Metab Dispos 36:276–283. https://doi.org/10.1124/dmd.107.018036
OECD (2018) Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. OECD Series on Testing and Assessment, No. 150, OECD Publishing, Paris. https://doi.org/10.1787/9789264304741-en
Peters SA (2008) Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet 47:261–275. https://doi.org/10.2165/00003088-200847040-00004
Peters SA (2012) Physiological Model for Absorption. Physiologically-based pharmacokinetic (PBPK) modeling and simulations. Wiley, Hoboken, pp 43–88
Poulin P, Theil F-P (2002) Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 91:1358–1370. https://doi.org/10.1002/jps.10128
Rochester JR, Bolden AL (2015) Bisphenol S and F: a systematic review and comparison of the hormonal activity of bisphenol A substitutes. Environ Health Perspect. https://doi.org/10.1289/ehp.1408989
Rodgers T, Rowland M (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. https://doi.org/10.1002/jps.20502
Rosenmai AK, Dybdahl M, Pedersen M, Alice van Vugt-Lussenburg BM, Wedebye EB, Taxvig C, Vinggaard AM (2014) Are structural analogues to bisphenol A safe alternatives? Toxicol Sci 139:35–47. https://doi.org/10.1093/toxsci/kfu030
Simcyp (2016) Simcyp prediction tools-fu. https://members.simcyp.com/account/tools/. Accessed May 2019
Strikwold M, Spenkelink B, de Haan LHJ, Woutersen RA, Punt A, Rietjens IMCM (2017) Integrating in vitro data and physiologically based kinetic (PBK) modelling to assess the in vivo potential developmental toxicity of a series of phenols. Arch Toxicol 91:2119–2133. https://doi.org/10.1007/s00204-016-1881-x
Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, Fleisher D, Lee KD, Amidon GL (2002) Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res 19:1400–1416. https://doi.org/10.1023/A:1020483911355
Teeguarden JG, Twaddle NC, Churchwell MI, Yang X, Fisher JW, Seryak LM, Doerge DR (2015) 24-h human urine and serum profiles of bisphenol A: evidence against sublingual absorption following ingestion in soup. Toxicol Appl Pharmacol 288:131–142. https://doi.org/10.1016/J.TAAP.2015.01.009
Thayer KA, Doerge DR, Hunt D, Schurman SH, Twaddle NC, Churchwell MI, Garantziotis S, Kissling GE, Easterling MR, Bucher JR, Birnbaum LS (2015) Pharmacokinetics of bisphenol A in humans following a single oral administration. Environ Int 83:107–115. https://doi.org/10.1016/J.ENVINT.2015.06.008
van Leeuwen SP, Bovee TF, Awchi M, Klijnstra MD, Hamers AR, Hoogenboom RL, Portier L, Gerssen A (2019) BPA, BADGE and analogues: a new multi-analyte LC–ESI-MS/MS method for their determination and their in vitro (anti)estrogenic and (anti)androgenic properties. Chemosphere 221:246–253. https://doi.org/10.1016/J.CHEMOSPHERE.2018.12.189
Wetmore BA, Wambaugh JF, Allen B, Ferguson SS, Sochaski MA, Setzer RW, Houck KA, Strope CL, Cantwell K, Judson RS, LeCluyse E, Clewell HJ, Thomas RS, Andersen ME (2015) Incorporating high-throughput exposure predictions with dosimetry-adjusted in vitro bioactivity to inform chemical toxicity testing. Toxicol Sci 148:121–136. https://doi.org/10.1093/toxsci/kfv171
Yang X, Doerge DR, Teeguarden JG, Fisher JW (2015) Development of a physiologically based pharmacokinetic model for assessment of human exposure to bisphenol A. Toxicol Appl Pharmacol 289(3):442–456. https://doi.org/10.1016/j.taap.2015.10.016
Yoon M, Campbell JL, Andersen ME, Clewell HJ (2012) Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit Rev Toxicol 42:633–652. https://doi.org/10.3109/10408444.2012.692115
Zanelli U, Caradonna NP, Hallifax D, Turlizzi E, Houston JB (2011) Comparison of cryopreserved HepaRG cells with cryopreserved human hepatocytes for prediction of clearance for 26 drugs. Drug Metab Dispos 40:104–110. https://doi.org/10.1124/dmd.111.042309
Zhang M, van Ravenzwaay B, Fabian E, Rietjens IMCM, Louisse J (2018) Towards a generic physiologically based kinetic model to predict in vivo uterotrophic responses in rats by reverse dosimetry of in vitro estrogenicity data. Arch Toxicol 92:1075–1088. https://doi.org/10.1007/s00204-017-2140-5
Acknowledgements
This work was financed by the Netherlands Food and Consumer Product Safety Authority.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Punt, A., Aartse, A., Bovee, T.F.H. et al. Quantitative in vitro-to-in vivo extrapolation (QIVIVE) of estrogenic and anti-androgenic potencies of BPA and BADGE analogues. Arch Toxicol 93, 1941–1953 (2019). https://doi.org/10.1007/s00204-019-02479-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02479-6