
Abstract
The aryl hydrocarbon receptor (AhR) is a nuclear receptor that facilitates a wide transcriptional response and causes a variety of adaptive and maladaptive physiological functions. Such functions are entirely dependent on the type of ligand activating it, and therefore, the nuances in the activation of this receptor at the single-cell level have become a research interest for different pharmacological and toxicological applications. Here, we investigate the activation of the AhR by diverse classes of compounds in a Hepa1c1c7-based murine hepatoma cell line. The exogenous compounds analyzed produced different levels of ultrasensitivity in AhR activation as measured by XRE-coupled EGFP production and analyzed by both flow cytometric and computational simulation techniques. Interestingly, simulation experiments reported herein were able to reproduce and quantitate the natural single-cell stochasticity inherent to mammalian cell lines as well as the ligand-specific differences in ultrasensitivity. Classical AhR modulators 2,3,7,8-tetrachlorodibenzodioxin (10− 1–105 pM), PCB-126 (10− 1–107 pM), and benzo[a]pyrene (10− 1–107 pM) produced the greatest levels of single-cell ultrasensitivity and most maximal responses, while consumption-based ligands indole-3-carbinol (103–109 pM), 3,3′-diindolylmethane (103–108 pM), and cannabidiol (103–108 pM) caused low-level AhR activation in more purely graded single-cell fashions. All compounds were tested and analyzed over a 24 h period for consistency. The comparative quantitative results for each compound are presented within. This study aids in defining the disparity between different types of AhR modulators that produce distinctly different physiological outcomes. In addition, the simulation tool developed for this study can be used in future studies to predict the quantitative effects of diverse types of AhR ligands in the context of pharmacological therapies or toxicological concerns.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aryl hydrocarbon receptor (AhR) is a nuclear receptor known classically for its dioxin-dependent activation and subsequent toxicological effects (Landers and Bunce 1991). The triggered activation of AhR requires the intracellular presence of a high-affinity ligand, many signaling events that allow for its translocation into the nucleus, and additional protein complexes that enable binding to DNA for a wide transcriptional response of mainly drug-metabolizing enzyme genes (Abel and Haarmann-Stemmann 2010). Classically identified ligands for this receptor include a variety of polycyclic aromatic hydrocarbons (PAHs) such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other members of the dioxin family (Landers and Bunce 1991); 3,3′,4,4′,5-pentachlorobiphenyl (PCB-126) and other polychlorinated biphenyls (PCBs) (Chubb et al. 2004); and large nonpolar pyrene derivatives such as benzo[a]pyrene (BaP) (Whitlock 1999). The AhR-mediated responses elicited by these ligands are all deleterious in nature and primarily result in immunotoxicity, skin lesions known as chloracne, hepatotoxicity, and various forms of tumor promotion (Denison et al. 2011; Wei et al. 2016).
While early studies focusing on the AhR all provide strong evidence of its toxicological responses, recent studies have reoriented our understanding of this complex receptor and solidified it as a potential therapeutic target in modulating the progression of several inflammatory diseases and cancers (Beischlag et al. 2008; Kiss and Diefenbach 2012; Kolluri et al. 2017; Safe et al. 2017). As one potential benefit, it has been shown that sustained activation of the AhR by TCDD can suppress the adaptive immune response and reduce Leishmania major burdens in mice (Bowers et al. 2006). While this parasitic resistance is a therapeutic outcome of TCDD exposure, classical toxicological data suggest that exposure to this compound will result in many unintended health hazards and render it unsuitable as a treatment for any infection or disease.
In light of these data, current research has turned to the activation of AhR using more natural endogenous and exogenous ligands. Indole-3-carbinol (13C) is one such exogenous ligand identified in cruciferous vegetables that allows for potent AhR activation upon consumption (Rogan 2006). This receptor activation is mediated through direct binding as well as through binding of I3C’s major metabolite 3,3′-diindolylmethane (DIM) (Bradlow and Zeligs 2010; Patel et al. 2012; Rogan 2006). These compounds fall within a class of substituted indoles that predominately originate from dietary sources and all variably activate the AhR (Stepankova et al. 2018). It is known that I3C and related indoles can reduce oxidative stress in cell-culture models (Fan et al. 2009) in addition to regulating carbohydrate metabolism in mouse models (Poornima and Mirunalini 2014). AhR activation by such ligands is critical for preventing the metabolic and genetic dysfunction seen in mammalian metabolic syndrome (Natividad et al. 2018). As perhaps, the most impactful pharmacological aspect of these indole compounds, their activation of the AhR regulates intestinal immune cells and drives a powerfully therapeutic anti-inflammatory response in the rodent gut (Murray and Perdew 2017). Mice treated with I3C displayed a significant increase in beneficial intraepithelial lymphocytes and a drastic reduction in intestinal inflammation and damage (Li et al. 2011). Several laboratories have further elucidated additional gastrointestinal benefits, showing I3C- and DIM-mediated prevention of intestinal carcinogenesis in mice (Kawajiri et al. 2009, Su et al. 2017). These studies have revealed a compelling AhR-based reason to advocate for liberal cruciferous vegetable intake and functional nutrition (Hooper 2011; Hubbard et al. 2017). Investigators have also identified an endogenously sourced indole ligand that potently activates the human AhR: 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) (Song et al. 2002; Henry et al. 2006). Much like the other indole ligands, ITE has been shown to upregulate specific regulatory T cells and reduce the intestinal inflammation that drives mammalian colitis in an AhR-dependent manner (Goettel et al. 2016; Abron et al. 2018).
Non-classical ligands lacking an indole moiety can also trigger AhR activity (Denison and Nagy 2003). Cannabidiol, derived as a primary cannabinoid constituent from the Cannabis plant genus, can potently induce CYP1A1 through AhR-mediated signaling (Yamaori et al. 2015). Natural phenolic compounds such as curcumin have identified AhR activity (Ciolino et al. 1998), and many plant-derived tetraterpenoids such as apocarotenal, canthaxanthin, and astaxanthin have been identified as AhR agonists as well mainly by their CYP1A1 and CYP1A2 induction (Gradelet et al. 1996a, b). These compounds, while less understood for their AhR modulation than the aforementioned indole compounds, may also provide insight into the differential effects of consumption-based AhR activation.
The AhR is activated by a structurally diverse spectrum of compounds (Bock 2016; Denison and Nagy 2003). The variable response profile of the AhR has been thoroughly reviewed (Denison et al. 2011; Murray et al. 2014), where any given AhR-mediated physiological outcome is highly dependent upon the ligand that stimulates it. Other receptors have different signaling profiles and thus dichotomous physiological outcomes for a spectrum of agonists as well, which has been extensively documented as “biased agonism” (Wisler et al. 2014); examples of such receptors are the opioid receptors (Pradhan et al. 2012), serotonin 5-HT2A receptor (Shan et al. 2012), glucagon-like-peptide-1 receptor (Wootten et al. 2013), and the glucocorticoid receptor (Hadley et al. 2011).
While differences in AhR-mediated transcriptional responses have been well documented between different cell types (Maier et al. 2007), there are no documented differences in immediate expressional changes within the same cell type by different ligand types (Henry et al. 2010). While gene expression profile changes appear to be the similar for AhR ligands, physiological outcomes between TCDD and ITE are vastly different in biological systems like the placenta (Wu et al. 2014). Ligand-intrinsic and dose-dependent differences in AhR activity have also been well documented for the fate of differentiating CD4+ T cells, where different amounts of different AhR ligands will facilitate the production of different sets and quantities of lymphocytic markers (Ehrlich et al. 2018).
It is assumed that there are a variety of complex factors that allow for different ligands to activate the same receptor and produce dichotomous responses. Such factors may include different direct binding domains on the intracellular receptor, dynamic activation nuances (e.g., via direct binding or indirect phosphorylation events), alterations in metabolic activity, pharmacokinetics of a given ligand, and different intrinsic affinities for the receptor and resulting activation durations. In addition, receptors and signaling proteins are sometimes rapidly stimulated within a narrow stimulus window, giving rise to ultrasensitive or “switch-like” behavior (Ferrell and Machleder 1998), and indeed, we have previously demonstrated that the AhR is activated by the classical ligand PCB-126 in a variably ultrasensitive manner (Broccardo et al. 2004, 2005; French et al. 2004). Ultrasensitivity dynamics have been unpacked extensively at the molecular level, and the mathematics explaining this phenomenon have been keenly approximated for computational modeling studies (Ferrell and Ha 2014). Ultrasensitive response motifs manifest as a result of a variety of molecular processes including cooperative binding, dimerization sequences, and multistep signaling processes (Zhang et al. 2013). For the AhR, the protein modification and translocation that occurs after ligand binding appears to form the basis of this switch-like activation; however, the rapid activity of this bound complex is highly ligand-dependent.
Because of these observations and in light of recent non-classical ligand experimentation, we hypothesized that the ligand-specific disparity between AhR-mediated responses can be in part explained by the nuances in each ligand’s ability to directly bind and ultrasensitively activate this nuclear receptor. To define these nuances, we offer here an experimental approach that integrates a high-throughput single-cell in vitro assay and a robust mathematical simulation method to understand the graded and switch-like dynamics of this nuclear receptor activation, and predict the impact of specific exogenous stimuli on this molecular target. The AhR modulators used in this study consist of three classical aryl hydrocarbon ligands and three non-traditional exogenous ligands, as displayed in Fig. 1.

Chemical structures of the six compounds used in this study that act differentially to modulate AhR activity
Materials and methods
Chemicals and reagents
TCDD (≥ 98% pure) was purchased from Cambridge Isotopes (Andover, MA, USA). PCB-126 (≥ 99% pure) was obtained from Accustandard (New Haven, CT, USA) and confirmed by GCMS to be pure and free of other congeners. BaP (≥ 96% pure), I3C (≥ 96% pure), and DIM (≥ 98% pure) were purchased from Sigma–Aldrich (St. Louis, MO, USA). CBD (≥ 99% pure) was purchased from Cerilliant Corporation (Round Rock, TX, USA). For treatments, all compounds were diluted in sterile-filtered DMSO; treatments and vehicle groups contained less than or equal to 0.1% v/v DMSO. Geneticin (G418) used for selective media was obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Cell-culture model and flow cytometry
H1G1.1C3-GFP cells, the generous gift of Dr. Michael Denison at University of California Davis, were used as an AhR reporter line as described previously (Nagy et al. 2002). Briefly, this is a stably transfected mouse hepatoma (Hepa1c1c7) cell line with an enhanced green fluorescent protein (EGFP) reporter gene linked to the aryl hydrocarbon response element, referred to here as the xenobiotic response element (XRE). Cells were maintained in αMEM containing 10% fetal bovine serum in a humidified environment of 5% CO2 at 37 °C. Because it was observed by Nagy et al. (2002) that these cells require constant selective pressure in order for the EGFP gene to be strongly expressed, cells were placed in selective media (αMEM containing 10% fetal bovine serum and 968 mg/L G418) 48 h prior to treatments. Cells were plated into 24-well plates and allowed 24 h to adhere, where they were subsequently treated for 24 h with varying doses of each of the 6 treatment compounds. Cells were treated with TCDD (7-point scheme, 10− 1–105 pM), PCB-126 (9-point scheme, 10− 1–107 pM), BaP (8-point scheme, 10− 1–106 pM), I3C (7-point scheme, 103–109 pM), DIM (6-point scheme, 103–108 pM), or CBD (6-point scheme, 103–108 pM). After each treatment duration, cells were trypsinized (0.025% trypsin/EDTA) and resuspended in media at 106 cells/mL for flow cytometric analyses. Cells suspensions were read by a Beckman Coulter CyAnADP flow cytometer equipped with a 488 nm laser for excitation and FITC channel (530/30 bandpass filter) for EGFP intensity readouts. FCS files were captured for each treatment group using a Summit Software System (v4.3; Cytomation, Fort Collins, CO, USA), and files were further analyzed using FlowJo Software (v10.5.0; FlowJo LLC, Ashland, OR, USA) for mean fluorescence intensity (MFI) values and bifurcation-gated cell percentages. All samples were run in biological triplicate.
Computational simulation
The simulation approach employed herein utilizes the AnyLogic multimethod simulation software (v8.0; The AnyLogic Company, Chicago, IL, USA), integrating agent-based, discrete event, and system-dynamics modeling techniques. Our group has been a pioneer in the use of this platform for exploration of cellular responses as well as physiologically based pharmacokinetics (Hoffman et al. 2017; Hoffman and Hanneman 2017). Primarily, the simulation interface was built with a population of hepatocyte agents (cell number adjustable), each with an intrinsic equation network defining AhR activation. This equation network uses the tissue-level exposure of each compound of interest (assumed first-order diffusion and first-order elimination) within a standard Hill equation that defines a cell’s AhR response fraction. The mathematics and equation sets defining biological responses used here have been previously documented (Ferrell and Ha 2014). The main components of this equation are the Hill coefficient and threshold (EC50) value (as shown in Fig. 2a), both of which have been systematically optimized to fit to the previous experimental data sets. Validations and corrections of these simulation optimizations were further performed using our own experimental data. The hill coefficient is optimized based on histogram readouts generated by the flow cytometer as well as MFI values that define activation levels; as cells are activated experimentally, their response shape, and thus Hill coefficient, remain the same within a cellular population for each given ligand. For the threshold value, however, cellular variance exists, as some cells will respond to different amounts of ligands; because of this, the threshold is defined as a distribution for each cellular population respective to each specific ligand. Upon simulation startup, each cell is instructed by the program to pull threshold values from a theoretical distribution. Optimization experiments performed within the simulation for these value distributions all consist of 50,000 randomly sampled iterations each consisting of 1000-cell populations to reach the most appropriate values and ranges. Each treatment scenario was further analyzed by Monte Carlo analyses of 100 sampling iterations each to elucidate the level of variance in the stochastic simulation data. All other details on simulation builds are elaborated on in “Results” and files used may be openly accessed via the AnyLogic Cloud online platform. Representative 1000 pM TCDD simulation experiment can be run directly in web browser at: https://tinyurl.com/y9yzt8dd. Files (.alp format) can be downloaded from this same site.

Optimized AhR activation assay using the XRE-EGFP-coupled hepatoma cell line. a Gating scheme for each sample that was used to select hepatoma cell populations for analysis. Intact cells were first separated from debris and noise based on appropriate size (FS forward scatter) and granularity (SS side scatter); single-cell events were then separated from doublet events. b Dose-dependent variation in EGFP-based fluorescence intensity (captured via FITC channel) seen in cell populations after 24 h of different TCDD exposures (101–104 pM). c 7-point dose–response analysis (10− 1–105 pM TCDD) for response fraction and activated cell counts, measured as corrected mean fluorescence intensity (MFI) and bifurcated FITC+ cells, respectively. d Fluorescence imaging of the transfected hepatoma cells exposed to either DMSO vehicle or 1000 pM TCDD, as shown in the original documentation of these cells by Nagy et al. (2002)
Data visualization and further mathematical analyses
Graphical representations and non-linear curve analyses of data were produced using both AnyLogic simulation software and GraphPad Prism (v7.0; GraphPad Software Inc.; La Jolla, CA, USA). One-phase decay analyses were conducted to further characterize Weibull-based threshold distributions and elucidate median values. Simulated fluorescence histograms were fitted accordingly using either a smoothed fourth-order polynomial analysis or Gaussian analysis for biphasic (highly ultrasensitive) or monophasic (graded) plots, respectively. Sigmoidal (variable slope; four parameter) analyses and concomitant EC50 value generation were conducted for all dose–response curves.
Results
AhR activation assay
The flow cytometric assay used here has been optimized by our laboratory and is suitable for investigating ultrasensitivity dynamics for functional activation of the AhR. This assay directly measures the level of AhR-dependent protein product (EGFP) in individual H1G1.1C3-GFP cells. Collecting these single-cell and population-based metrics were critical for use in the subsequent simulation approach.
We first identified the hepatocyte population of interest gathered by the cytometer by gating for appropriate size and complexity seen in the cells (measured as forward and side scatter) as well as by gating out false doublet or triplet events as clumped cells can sometimes make their way through the point of observation (Fig. 2a). Each treatment group’s cell population was excited by the 488 nm laser and their level of EGFP-based fluorescence was observed, which for increasing TCDD treatments, showed mainly two distinct states: inactive or fully active. This is defined by the bimodal distributions in the fluorescence intensity histograms shown (Fig. 2b). These mean fluorescence intensity (MFI) data were quantified at the population level and plotted against the varying doses of TCDD used, resulting in a steep sigmoidal or ultrasensitive curve (Fig. 2c, left panel). Mean fluorescence intensity values were corrected for autofluorescence and normalized to reach a response fraction. A bifurcation gating scheme was also employed to divide the percent of cells that exist within the inactive state (FITC−) or the fully active state (FITC+), which was possible to analyze, because there were no states observed in between in response to TCDD exposure (Fig. 2c, right panel). A visual representation of this TCDD-dependent switch-like response in H1G1.1C3-GFP cells was previously gathered by Nagy et al. (2002), where the cells were treated with DMSO vehicle or 103 pM TCDD (Fig. 2d).
Ultrasensitivity dynamics elucidated via computational simulation
We designed a simulation for this study that is consistent with the behavior of individual cells as they respond to AhR modulators, in either slowly graded or highly ultrasensitive manners. The foundational construct of our simulation is an agent-based approach, where “cell agents” exist as a population and each contain a stochastic equation-based response network that is dependent upon intracellular exposure inputs (Fig. 3a). TCDD was the prototypical compound used for the initial simulation build, and its ligand-specific Hill coefficient of 7 and threshold distribution (also displayed in Fig. 3a) were systematically optimized and defined by simulation experiments. The graphical representation of the threshold distribution elicited by TCDD is displayed in Fig. 3b. After a simulation run at 103 TCDD exposure, the responses of 100 sampled cells were collected and overlaid, displaying the stochastic nature of the simulation interface (Fig. 3c, left panel); these data sets were then compounded to reveal the population-based response (Fig. 3c, right panel). The population-based response was collected for experiments containing 200 or 1000 cell agents, where the data sets change only marginally, showing that these are enough agents in the simulation to display the compounded effects of the distribution metrics. Variance of the programmed stochasticity was further quantified by Monte Carlo analysis (100 iterations per group), and the results are displayed as Gaussian distribution subplots (Fig. 3c, right panel). Cells were then phenotyped based on their response fraction values (inactive, 25%, 50%, 75%, and ≥ 90% or fully active), and cell percentages dominated either the inactive or fully active states in response to 103 TCDD (Fig. 3d), correlating appropriately to our experimental data. After the 24 h runtime in the simulation, histograms of response fractions for different TCDD doses were collected (Fig. 3e), paralleling the experimental histograms shown in Fig. 2b. As final analyses in the simulation design, dose–response curves were generated for all TCDD doses after 24 h of exposure. Response fractions and activated cell percentages were recorded and overlaid on top of experimental data, as shown in Fig. 3f, g, respectively. The similarity between the experimental and simulation data sets highlights the validity of this approach for studying the ligand-dependent AhR response.

Computational constructs and simulation results for the TCDD-mediated AhR response in immortalized hepatocytes. a Simplified schematic of the simulation interface and primary mathematics. Circles represent cellular agents in the simulation, where each is designed to respond probabilistically based on their threshold magnitude pulled from an identified distribution. The identified vales and ranges that correspond to TCDD are enumerated. b Simulation-generated threshold distribution for TCDD, sampled from 1000 cellular agents. c Single-cell (left panel, 100 sampled curves from runtime) and compounded population (right panel) simulation results following 1000 pM TCDD. Population results were iterated 100 times each in a Monte Carlo analysis to determine variance from the stochastic simulations; resulting Gaussian curves with output data are seen in the subplot for 200 and 1000 cell agents. d Levels of activation within the simulated cell population following 1000 pM TCDD. Cells were phenotyped based on their response fraction values. e Dose-dependent variation in simulated fluorescence intensity seen in cell populations after 24 h of different TCDD exposures (101–104 pM). f Experimental and simulated dose–response analyses overlaid for response fraction (10− 1–105 pM TCDD). g Experimental and simulated dose–response analyses overlaid for activated cell counts (10− 1–105 pM TCDD)
Dose–response and simulation analyses performed for each test compound
Following the preliminary TCDD optimization and analysis, the flow cytometric and simulation analyses were performed for all classical and consumption-based ligands. PCB-126 was exposed to the hepatoma cells at a range of 10− 1–107 pM. Experimental histograms of fluorescence intensity after 24 h of PCB-126 treatment are displayed in Fig. 4a, showing the switch from inactive to fully active states. Current flow cytometric data (top panel) and historical data from our laboratory (published as Broccardo et al. 2004; bottom panel) are shown for PCB-126 in both the mutated and traditional Hepa1c1c7 cells, respectively. Historical data were used to further confirm the actions of PCB-126 qualitatively, but optimized simulation data for this ligand were gathered using newly obtained flow cytometric data for consistency throughout this study. Simulation experiments revealed a Hill coefficient of 2 and a threshold distribution similar in nature and shape to that of TCDD, but different in magnitude (Fig. 4b, top panel); in addition, since all compounds were normalized to maximal induction TCDD data, it was found experimentally that PCB-126 hit a maximum response fraction of 0.72 which was included in the simulation makeup. Response fraction simulation data from 100 sampled cell agents after 103 pM of PCB-126 were collected and overlaid to display individual stochastic activations (Fig. 4b, bottom panel). Simulated fluorescence intensity histograms were overlaid for relevant PCB-126 doses (Fig. 4c, top panel), and simulation-generated time-course compounded population results were overlaid for the same doses (Fig. 4c, bottom panel). Full dose–response curves were then generated for response fraction values (Fig. 4d, top panel) and activated cell percentages (Fig. 4d, bottom panel), overlaid on top of experimental flow cytometry data. The full experimental and simulation analyses were performed for BaP as well (not shown for brevity), resulting in a Hill coefficient of 3, a threshold distribution median of 1231 pM, and experimental EC50 of 491.4 pM.

Quantitative analysis of AhR activation by PCB-126. a Dose-dependent variation in EGFP-based fluorescence intensity (captured via FITC channel) seen in cell populations after 24 h of different PCB-126 exposures (101–106 pM). The lower panel displays historical flow cytometric histogram data from Broccardo et al. (2004), of PCB-126-mediated AhR activation following 24 h of treatment using CYP1A1 antibodies. b Hepatocyte population characteristics with respect to PCB-126-mediated AhR response. The optimized threshold distribution with mathematical setup is displayed (top panel), and single-cell simulation results are overlaid for 100 sampled cells following 1,000 pM PCB-126 (bottom panel). c For varying doses of PCB-126 (101–106 pM), response fraction histograms are overlaid (top panel), and time-course evaluations for the same doses are overlaid (bottom panel). d Comparison of PCB-126 dose–response simulation data to experimental flow data (10− 1–106 pM); response fraction (top panel) and cell percentages (bottom panel) were evaluated
Consumption-based ligands all underwent the same experimental and simulation analyses, and the highlights of these results are displayed for I3C (Fig. 5a), DIM (Fig. 5b), and CBD (Fig. 5c). Shown for each of these ligands are (i) the generated threshold distribution and equation framework employed in simulation experiments, (ii) the overlaid histograms of simulated fluorescence intensity for all relevant doses within the experimental curve, and (iii) the overlaid full dose–response curves generated experimentally and via simulation. Representative dose–response histograms generated via flow cytometry experiments are shown for each compound as well.

Analyses of consumption-based ligands. a I3C, b DIM, and c CBD computational analyses and experimental flow data. For each consumption-based ligand, the optimized threshold distribution with mathematical setup is displayed (left panels), simulated fluorescence histograms are overlaid for all dose points (middle panels), and comparative experimental-to-simulation dose–response plot is shown (right panels). Representative experimental histograms of measured fluorescence intensity for each compound are seen in the dose–response subplots
Comparative analyses between AhR modulators
The experimental and simulation-generated response activation values were finally compared for all the classical AhR ligands and contemporary consumption-based ligands. The experimental EC50 and simulated threshold distribution medians were markedly higher for the consumption-based ligands than for the classical AhR ligands, whereas the simulated Hill coefficients revealed for each of the consumption-based ligands were markedly lower. When plotted as Hill coefficients against either threshold distribution medians or against experimental EC50 values, the resulting graph can be divided into quadrants defining the type of AhR modulation, and thus predicting regions with distinct physiological outcomes (Fig. 6a). These outcomes of differential AhR activation are summarized in Fig. 6b, where small amounts of potent classical ligands are required to elicit highly ultrasensitive responses with a propensity for deleterious outcomes. In contrast, this summary diagram shows how much larger quantities of contemporary AhR-modulating ligands result in much less steepness in their activation curves, which may in part be responsible for their mild transcriptional conditioning and harmless or even therapeutic outcomes, with respect to AhR activity alone.

Final comparisons between all AhR modulators of interest. a Hill coefficient values plotted against respective threshold distribution medians (left axis) and experimentally measured EC50 values (right axis). Classical/toxicological and contemporary/dietary/therapeutic ligands are clustered in different quadrants outlining graphical regions with differential physiological outcomes. b Conclusive disparity between different ligand-specific ultrasensitivity dynamics and how they correlate with cellular and physiological outcomes
Discussion
This study is highly innovative, because it greatly advances our understanding of how different AhR ligands can elicit drastically different and sometimes opposite physiological outcomes. While there are many theoretical components accounting for the disparity observed in the literature, our results view the kinetic and dynamic relationships that exist through an intricate quantitative lens. We have previously identified ultrasensitivity dynamics within AhR-mediated responses in liver cell lines and primary hepatocytes (Broccardo et al. 2004, 2005; French et al. 2004). These previous analyses of the AhR response have utilized population-based mathematics that have been derived for these types of substrate-dependent curves and phosphorylation systems (Ferrell and Ha 2014). Due to the advancement of adaptable agent-based simulation technology, our current study now reveals specific nuances to quantifying each individual cell that mounts the response and to further characterizing different types of ligands as AhR modulators.
While we have generated an appropriate quantitative model for cell distribution-based Hill dynamics relative to the AhR, different ultrasensitivity mathematics born out of either positive feedback (Ferrell and Xiong 2001) or negative cooperativity (Ha and Ferrell 2016) are yet to be fully understood for this particular receptor. Our study indicates that potent prototypical ligands like TCDD allow for a more bistable system with reinforced signal transduction and long-lasting effects. Alternative ligands like DIM or CBD may result in weaker cooperativity at the nuclear dimerization point and thus more graded and short-lived responses, as it is possible to produce AhR dimers with quantifiably lower affinities for DNA binding (Corrada et al. 2016; Wu et al. 2013).
The nuances in activating this receptor are also purportedly attributed to differing recruitment biomolecules in the cytosol giving rise to varying levels of receptor translocation and transcriptional activity. In specific cases with ligands like omeprazole, receptor activation is completely dependent upon particular AhR protein residues and co-activating kinases, whereas traditional toxicological ligands like TCDD elicit no such dependency (Powis et al. 2011). Investigators have also noted distinct differences in the binding fingerprints of TCDD versus ITE and other indole-based ligands toward murine AhR residues (Dolciami et al. 2018), finding that particular moieties within contemporary ligands are best for targeting the AhR with less stable molecular confirmations for therapy. These limiting factors in AhR activation overall show reduced transcriptional magnitudes and shortened activity durations (Powis et al. 2011). Such factors may be at play with many contemporary consumption-based ligands such as those observed in the current study. As a critical observation, when the endogenous indole ITE is compared to TCDD exposure in mouse lung fibroblasts, immediate expressional changes are seemingly analogous (Henry et al. 2010). Looking at these data from acute exposures and considering the limiting factors for low affinity ligands, it appears that the AhR response disparity is fully observed in a more subacute or even chronic fashion when probed with subtler levels of ultrasensitivity.
In contrast to the intestinal and hepatic differences noted for varying AhR ligand types (Bock 2016), different AhR ligands causing similar physiological responses have also been observed for very specific tissues like the placenta. Following treatment with TCDD or ITE, a modulation in the angiogenic response of the placenta is seen due to differential AhR roles between placental endothelial cells (Li et al. 2015, 2017). While TCDD and ITE both appear to have suppressive angiogenic responses in the placenta, it has also been reported that the expressional changes and final vascular remodeling elicited by both are vastly different (Wu et al. 2014). These observations lead to the speculation that AhR modulators of any kind may have a more hypervariable physiological response within tissues that are highly estrogen responsive, as there is much signaling crosstalk and shared ligands between the AhR and estrogen receptor (ER) family (Saito et al. 2014). While the AhR may be an emerging therapeutic target for alleviating the progression of intestinal diseases and inflammatory responses, it appears that there are estrogen-dependent caveats to stimulating this pathway. Known AhR modulators TCDD and ITE can potentially harm a growing fetus via stunting placental growth (Li et al. 2015, 2017). DIM has been shown to allow for the proliferation of breast cancer cells (Marques et al. 2014). Because of these reports, future work into therapeutic AhR modulators should carefully take into account direct and indirect estrogenic potential to eliminate any unintended effects.
Throughout this study and future experimental analyses, it is important to consider experimental dosing schemes and their relevancies to environmental and dietary exposures. Because many studies for AhR ligands are performed in vitro and in vivo, exposure assessments and physiologically based pharmacokinetic analyses are required to understand the correlation between the two. For the toxicological ligands addressed in this study, the dose ranges applied are identified intracellular ranges that are known to activate the AhR and were once environmentally relevant, but measured internal blood doses for average and at risk populations have since declined far below their activation thresholds following years of remediation for TCDD (Pavuk et al. 2014; Warner et al. 2014) and most PCB congeners (Magoni et al. 2016). Interestingly, lung perfusate doses up to 104 pM BaP can still be reached and maintained for cigarette smokers (Campbell et al. 2016). Internal doses for contemporary consumption-based ligands are user-dependent, and investigators must rely on established PBPK studies for internal exposure estimation. Following 100 mg/kg I3C prodrug oral dosing in mice, >106 pM serum ranges can be reached for several hours (Moussata et al. 2014). Following 250 mg/kg crystalline DIM oral dosing in mice, >107 pM serum ranges can be reached for several hours (Anderton et al. 2004). Following 10 mg/kg CBD oral dosing in rats, >105 pM serum ranges can be reached for several hours (Hložek et al. 2017). While these pharmacokinetic dosing schemes may be relatively high for human supplements (e.g., 10 mg/kg CBD in an average human would be a 600 mg CBD oral dose), the internal doses reached are all relevant to the activation ranges elucidated.
AhR activation by the appropriate amount of a particular ligand can elicit therapeutic responses, as it has been seen for carbohydrate metabolism in mice (Poornima and Mirunalini 2014), gastric cancer reduction (Kawajiri et al. 2009; Su et al. 2017), and alleviated intestinal inflammation (Li et al. 2011). The current study greatly aids in the understanding of why a potent AhR ligand can result in hepatic oxidative stress and hepatocellular carcinoma, while a weak activator like I3C can result in enhanced hepatic and metabolic processes. The growing body of the literature on therapeutic effects, however, warrants more differential ultrasensitivity quantification for other cell types such as intestinal lymphocytes and endothelial cells.
Conclusion
In conclusion, we have provided a novel integrated experimental simulation approach that allows us to understand and visualize the dynamics of the AhR response for a variety of classical toxicological ligands as well as consumption-based constituents. Using these newly developed tools, we have been able to quantify important constants respective to each ligand that affords us the opportunity to draw a correlation to the phenotypic AhR-dependent response each ligand will mount. This analysis along with future studies will greatly aid in quantifying and targeting ligand-specific AhR responses as a therapeutic modality.
References
Abel J, Haarmann-Stemmann T (2010) An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol Chem 391(11):1235–1248
Abron JD, Singh NP, Mishra MK, Price RL, Nagarkatti M, Nagarkatti PS, Singh UP (2018) An endogenous aryl hydrocarbon receptor (AhR) ligand, ITE induces regulatory T cells (Tregs) and ameliorates experimental colitis. Am J Physiol Gastrointest Liver Physiol 315(2):G220–G230
Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, Mager DE (2004) Physiological modeling of formulated and crystalline 3,3′-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos 32(6):632–638
Beischlag TV, Morales JL, Hollingshead BD, Perdew GH (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr 18(3):207–250
Bock K (2016) Human and rodent aryl hydrocarbon receptor (AHR): from mediator of dioxin toxicity to physiologic AHR functions and therapeutic options. Biol Chem 398(4):455–464
Bowers OJ, Sommersted KB, Sowell RT, Boling GE, Hanneman WH, Titus RG, Dekrey GK (2006) 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) reduces Leishmania major burdens in C57BL/6 mice. Am J Trop Med Hyg 75(4):749–752
Bradlow H, Zeligs M (2010) Diindolylmethane (DIM) spontaneously forms from indole-3-carbinol (I3C) during cell culture experiments. In Vivo 24(4):387–391
Broccardo CJ, Billings RE, Chubb LS, Andersen ME, Hanneman WH (2004) Single cell analysis of switch-like induction of CYP1A1 in liver cell lines. Toxicol Sci 78(2):287–294
Broccardo CJ, Billings RE, Andersen ME, Hanneman WH (2005) Probing the control elements of the CYP1A1 switching module in H4IIE hepatoma cells. Toxicol Sci 88(1):82–94
Campbell J, Franzen A, Van Landingham C, Lumpkin M, Crowell S, Meredith C, Loccisano A, Gentry R, Clewell H (2016) Predicting lung dosimetry of inhaled particleborne benzo[a]pyrene using physiologically based pharmacokinetic modeling. Inhal Toxicol 28(11):520–535
Chubb LS, Andersen ME, Broccardo CJ, Legare ME, Billings RE, Dean CE, Hanneman WH (2004) Regional induction of CYP1A1 in rat liver following treatment with mixtures of PCB 126 and PCB 153. Toxicol Pathol 32(4):467–473
Ciolino HP, Daschner PJ, Wang TTY, Yeh GC (1998) Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem Pharmacol 56:197–206
Corrada D, Soshilov AA, Denison MS, Bonati L (2016) Deciphering dimerization modes of PAS domains: computational and experimental analyses of the AhR:ARNT complex reveal new insights into the mechanisms of AhR transformation. PLoS Comput Biol 12(6):e1004981
Denison MS, Nagy SR (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43:309–334
Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B (2011) Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci 124(1):1–22
Dolciami D, Gargaro M, Cerra B, Scalisi G, Bagnoli L, Servillo G, Fazia MAD, Puccetti P, Quintana FJ, Fallarino F, Macchiarulo A (2018) Binding mode and structure–activity relationships of ITE as an aryl hydrocarbon receptor (AhR) agonist. ChemMedChem 13(3):270–279
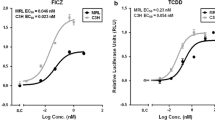
Ehrlich A, Pennington J, Bisson W, Kolluri S, Kerkvliet N (2018) TCDD, FICZ, and other high affinity AhR Ligands dose-dependently determine the fate of CD4+ T cell differentiation. Toxicol Sci 161(2):310–320
Fan S, Meng Q, Saha T, Sarkar FH, Rosen EM (2009) Low concentrations of diindolylmethane, a metabolite of indole-3-carbinol, protects against oxidative stress in a BRCA1-dependent manner. Can Res 69(15):6083–6091
Ferrell JE, Machleder EM (1998) The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science 280(5365):895–898
Ferrell JE, Xiong W (2001) Bistability in cell signaling: How to make continuous processes discontinuous, and reversible processes irreversible. Chaos 11(1):227–236
Ferrell JE, Ha SH (2014) Ultrasensitivity part I: michaelian responses and zero-order ultrasensitivity. Trends Biochem Sci 39(10):496–503
French CT, Hanneman WH, Chubb LS, Billings RE, Andersen ME (2004) Induction of CYP1A1 in primary rat hepatocytes by 3,3′,4,4′,5-pentachlorobiphenyl: evidence for a switch circuit element. Toxicol Sci 78(2):276–286
Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, Griffith AE, Patel B, Shouval DS, Weiner HL, Snapper SB, Quintana FJ (2016) AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep 17(5):1318–1329
Gradelet S, Leclerc J, Siess M-H, Astorg PO (1996a) B-Apo-8′-carotenal, but not β-carotene, is a strong inducer of liver cytochromes P4501A1 and 1A2 in rat. Xenobiotica 26:909–919
Gradelet S, Astorg P, Leclerc J, Cheva- lier J, Vernevaut M-F, Siess M-H (1996b) Effects of canthaxanthin, astaxanthin, lycopene and lutein on liver xenobiotic-metabolizing enzymes in the rat. Xenobiotica 6:49–63
Ha SH, Ferrell JE (2016) Thresholds and ultrasensitivity from negative cooperativity. Science 352(6288):990–993
Hadley K, Louw A, Hapgood J (2011) Differential nuclear localisation and promoter occupancy play a role in glucocorticoid receptor ligand-specific transcriptional responses. Steroids 76(10–11):1176–1184
Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA (2006) A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys 450(1):67–77
Henry EC, Welle SL, Gasiewicz TA (2010) TCDD and a putative endogenous AhR ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol Sci 114(1):90–100
Hložek T, Uttl L, Kadeřábek L, Balíková M, Lhotková E, Horsley RR, Nováková P, Šíchová K, Štefková K, Tylš F, Kuchař M, Páleníček T (2017) Pharmacokinetic and behavioural profile of THC, CBD, and THC + CBD combination after pulmonary, oral, and subcutaneous administration in rats and confirmation of conversion in vivo of CBD to THC. Eur Neuropsychopharmacol 27(12):1223–1237
Hoffman TE, Hanneman WH (2017) Physiologically-based pharmacokinetic analysis of benzoic acid in rats, guinea pigs and humans: Implications for dietary exposures and interspecies uncertainty. Comput Toxicol 3:19–32
Hoffman TE, Barnett KJ, Wallis L, Hanneman WH (2017) A multimethod computational simulation approach for investigating mitochondrial dynamics and dysfunction in degenerative aging. Aging Cell 16(6):1244–1255
Hooper L (2011) You AhR what you eat: Linking diet and immunity. Cell 147(3):489–491
Hubbard T, Murray I, Nichols R, Cassel K, Podolsky M, Kuzu G, Tian Y, Smith P, Kennett MJ, Patterson AD, Perdew G (2017) Dietary broccoli impacts microbial community structure and attenuates chemically induced colitis in mice in an Ah receptor dependent manner. J Funct Foods 37:685–698
Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS, Sakaki T, Hirokawa T, Akiyama T, Kurosumi M, Poellinger L, Kato S, Fujii-Kuriyama Y (2009) Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin /+ mice with natural ligands. Proc Natl Acad Sci USA 106(32):13481–13486
Kiss EA, Diefenbach A (2012) Role of the aryl hydrocarbon receptor in controlling maintenance and functional programs of RORγt+ innate lymphoid cells and intraepithelial lymphocytes. Front Immunol 3:124
Kolluri S, Jin U, Safe S (2017) Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch Toxicol 91(7):2497–2513
Landers JP, Bunce NJ (1991) The Ah receptor and the mechanism of dioxin toxicity. Biochem J 276(2):273–287
Li Y, Innocentin S, Withers D, Roberts N, Gallagher A, Grigorieva E, Wilhelm C, Veldhoen M (2011) Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147(3):629–640
Li Y, Wang K, Zou QY, Magness RR, Zheng J (2015) 2,3,7,8-Tetrachlorodibenzo-p-dioxin differentially suppresses angiogenic responses in human placental vein and artery endothelial cells. Toxicology 336:70–78
Li Y, Wang K, Zou QY, Jiang YZ, Zhou C, Zheng J (2017) ITE suppresses angiogenic responses in human artery and vein endothelial cells: differential roles of AhR. Reprod Toxicol 74:181–188
Magoni M, Donato F, Speziani F, Leonardi L, Orizio G, Scarcella C, Gaia A, Apostoli P (2016) Substantial decline of polychlorinated biphenyls serum levels 10 years after public health interventions in a population living near a contaminated site in Northern Italy. Environ Int 95:69–78
Maier MS, Legare ME, Hanneman WH (2007) The aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl induces distinct patterns of gene expression between hepatoma and glioma cells: chromatin remodeling as a mechanism for selective effects. Neurotoxicology 28(3):594–612
Marques M, Laflamme L, Benassou I, Cissokho C, Guillemette B, Gaudreau L (2014) Low levels of 3,3′-diindolylmethane activate estrogen receptor α and induce proliferation of breast cancer cells in the absence of estradiol. BMC Cancer 14:524
Moussata J, Wang Z, Wang J (2014) Development and validation of an HPLC method for the simultaneous quantification of indole-3-carbinol acetate, indole-3-carbinol, and 3,3′-diindolylmethane in mouse plasma, liver, and kidney tissues. J Chromatogr 958:1–9
Murray IA, Perdew GH (2017) Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr Opin Toxicol 2:15–23
Murray IA, Patterson AD, Perdew GH (2014) Ah receptor ligands in cancer: friend and foe. Nat Rev Cancer 14(12):801–814
Nagy SR, Sanborn JR, Hammock BD, Denison MS (2002) Development of a green fluorescent protein-based cell bioassay for the rapid and inexpensive detection and characterization of Ah receptor agonists. Toxicol Sci 65(2):200–210
Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, Michel M-L, Chong-Nguyen C, Roussel R, Straube M, Jegou S, McQuitty C, Gall ML, da Costa G, Lecornet E, Michaudel C, Modoux M, Glodt J, Bridonneau C, Sovran B, Dupraz L, Bado A, Richard ML, Langella P, Hansel B, Launay J-M, Xavier RJ, Duboc H, Sokol H (2018) Impaired Aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metabolism S1550-4131(18):30444
Patel AR, Spencer SD, Chougule MB, Safe S, Singh M (2012) Pharmacokinetic evaluation and in vitro–in vivo correlation (IVIVC) of novel methylene-substituted 3,3′diindolylmethane (DIM). Eur J Pharm Sci 46(1–2):8–16
Pavuk M, Patterson DG Jr, Turner WE (2014) Serum concentrations of TCDD and other dioxin-like compounds in US Air Force veterans of Operation Ranch Hand. Chemosphere 102:18–23
Poornima J, Mirunalini S (2014) Regulation of carbohydrate metabolism by indole-3-carbinol and its metabolite 3,3′-diindolylmethane in high-fat diet-induced C57BL/6J mice. Mol Cell Biochem 385(1–2):7–15
Powis M, Celius T, Matthews J (2011) Differential ligand-dependent activation and a role for Y322 in aryl hydrocarbon receptor-mediated regulation of gene expression. Biochem Biophys Res Commun 410(4):859–865
Pradhan AA, Smith ML, Kieffer BL, Evans CJ (2012) Ligand-directed signalling within the opioid receptor family. Br J Pharmacol 167(5):960–969
Rogan E (2006) The natural chemopreventive compound indole-3-carbinol: State of the science. In Vivo 20(2):221–228
Safe S, Cheng Y, Jin UH (2017) The aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr Opin Toxicol 2:24–29
Saito R, Miki Y, Hata S, Takagi K, Iida S, Oba Y, Ishida T, Suzuki T, Ohuchi N, Sasano H (2014) Aryl hydrocarbon receptor in breast cancer—a newly defined prognostic marker. Hormones Cancer 5(1):11–21
Shan J, Khelashvili G, Mondal S, Mehler EL, Weinstein H (2012) Ligand-dependent conformations and dynamics of the serotonin 5-HT2Areceptor determine its activation and membrane-driven oligomerization properties. PLoS Comput Biol 8(4):e1002473
Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF (2002) A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA 99(23):14694–14699
Stepankova M, Bartonkova I, Jiskrova E, Vrzal R, Mani S, Kortagere S, Dvorak Z (2018) Methylindoles and methoxyindoles are agonists and antagonists of human aryl hydrocarbon receptor AhR. Mol Pharmacol 93(6):631–644
Su M, Qian C, Hu Y, Lu W, Huang R, Chen M, Chen J (2017) Inhibitory effect of the low-toxic exogenous aryl hydrocarbon receptor modulator 3′3-diindolylmethane on gastric cancer in mice. Oncol Lett 14(6):8100–8105
Warner M, Mocarelli P, Brambilla P, Wesselink A, Patterson DG, Turner WE, Eskenazi B (2014) Serum TCDD and TEQ concentrations among seveso women, twenty years after the explosion. J Eposure Sci Environ Epidemiol 24(6):588–594
Wei Y, Zhao L, He W, Yang J, Geng C, Chen Y, Liu T, Chen H, Li Y (2016) Benzo[a]pyrene promotes gastric cancer cell proliferation and metastasis likely through the Aryl hydrocarbon receptor and ERK-dependent induction of MMP9 and c-myc. Int J Oncol 49(5):2055–2063
Whitlock J (1999) Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol 39:103–125
Wisler JW, Xiao K, Thomsen ARB, Lefkowitz RJ (2014) Recent developments in biased agonism. Curr Opin Cell Biol 0:18–24
Wootten D, Savage E, Willard F, Bueno A, Sloop K, Christopoulos A, Sexton P (2013) Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol Pharmacol 83(4):822–834
Wu D, Potluri N, Kim Y, Rastinejad F (2013) Structure and dimerization properties of the aryl hydrocarbon receptor PAS-A domain. Mol Cell Biol 33(21):4346–4356
Wu Y, Chen X, Zhou Q, He Q, Kang J, Zheng J, Wang K, Duan T (2014) ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS ONE 9(1):e86549
Yamaori S, Kinugasa Y, Jiang R, Takeda S, Yamamoto I, Watanabe K (2015) Cannabidiol induces expression of human cytochrome P450 1A1 that is possibly mediated through aryl hydrocarbon receptor signaling in HepG2 cells. Life Sci 136:87–93
Zhang Q, Bhattacharya S, Andersen ME (2013) Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open Biol 3(4):130031
Acknowledgements
This research was supported by the Center for Environmental Medicine at Colorado State University. The authors would like to thank Dr. Michael Denison at UC Davis for providing the H1G1.1C3-GFP cells. The authors also thank Dr. Christopher Allen for his assistance with the flow cytometry work.
Author information
Authors and Affiliations
Contributions
TEH, ERA, KFC, and VSG performed the in vitro work. TEH, ERA, KFC, and WHH analyzed data sets and prepared figures. TEH, ERA, KFC, and VSG regularly maintained experimental equipment and cultures. TEH, LEW, and WHH provided computational simulation expertise. WHH provided experimental equipment and reagents. TEH, ERA, KFC, VSG, LEW, and WHH aided in the preparation of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hoffman, T.E., Acerbo, E.R., Carranza, K.F. et al. Ultrasensitivity dynamics of diverse aryl hydrocarbon receptor modulators in a hepatoma cell line. Arch Toxicol 93, 635–647 (2019). https://doi.org/10.1007/s00204-018-2380-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2380-z