
Abstract
Wild-type Lactococcus lactis strain LAC460 secretes prophage-encoded bacteriocin-like lysin LysL, which kills some Lactococcus strains, but has no lytic effect on the producer. LysL carries two N-terminal enzymatic active domains (EAD), and an unknown C-terminus without homology to known domains. This study aimed to determine whether the C-terminus of LysL carries a cell wall binding domain (CBD) for target specificity of LysL. The C-terminal putative CBD region of LysL was fused with His-tagged green fluorescent protein (HGFPuv). The HGFPuv_CBDlysL gene fusion was ligated into the pASG-IBA4 vector, and introduced into Escherichia coli. The fusion protein was produced and purified with affinity chromatography. To analyse the binding of HGFPuv_CBDLysL to Lactococcus cells, the protein was mixed with LysL-sensitive and LysL-resistant strains, including the LysL-producer LAC460, and the fluorescence of the cells was analysed. As seen in fluorescence microscope, HGFPuv_CBDLysL decorated the cell surface of LysL-sensitive L. cremoris MG1614 with green fluorescence, whereas the resistant L. lactis strains LM0230 and LAC460 remained unfluorescent. The fluorescence plate reader confirmed the microscopy results detecting fluorescence only from four tested LysL-sensitive strains but not from 11 tested LysL-resistant strains. Specific binding of HGFPuv_CBDLysL onto the LysL-sensitive cells but not onto the LysL-resistant strains indicates that the C-terminus of LysL contains specific CBD. In conclusion, this report presents experimental evidence of the presence of a CBD in a lactococcal phage lysin. Moreover, the inability of HGFPuv_CBDLysL to bind to the LysL producer LAC460 may partly explain the host’s resistance to its own prophage lysin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lactococcus lactis is highly valued in the dairy industry for its dual role as a starter in many different fermented products and in aiding food preservation (Lortal and Chapot-Chartier 2005). All lactococcal genomes harbor prophages; genes encoding lysins are present in many of these prophage regions. These lytic enzymes can degrade peptidoglycan in the cell wall, ultimately resulting in cell lysis and cell death (Oliveira et al. 2018).
Phage lysins are generally divided into two groups, namely endolysins and virion-associated lysins (VALs; Abdelrahman et al. 2021). VALs typically exhibit dual functions as both phage structural components and lytic enzymes. VALs are found in different parts of phages, including tails, where they locally degrade the peptidoglycan of target bacteria during phage infection, without lysing the cell. This enzymatic activity facilitates entry of the phage’s genetic material into the host cell (Oliveira et al. 2018). Endolysins, on the other hand, have a role at the end of the life cycle when newly formed phages are released from the host cells. These enzymes work by degrading the peptidoglycan layer of the infected cell from inside, facilitating phage release (Oechslin et al. 2022).
Endolysins targeting Gram-positive bacteria typically consist of one or two enzymatic active domains (EAD) along with a cell wall binding domain (CBD). VALs are typically composed of one or two EADs and are believed to lack a CBD (Chandran et al. 2022). The EAD of an endolysin is usually located in the N-terminus of the lysin. The enzymatic activity is either N-acetylmuramidase or endo-β-N-acetylglucosaminidase, which cleave the sugar backbone of the peptidoglycan, endopeptidase cleaving the peptide segment of the cell wall, or N-acetylmuramyl-l-alanine amidase, hydrolyzing the amide bonds connecting the sugar chain to the peptide (Broendum et al. 2018; Loessner 2005). The activity of the lysin also requires binding to the cell wall; phage endolysins do not work effectively if they are not bound to the cell wall (Loessner et al. 2002). The CBD is usually located in the C-terminus, which is responsible for the enzyme’s selective binding to the cell wall, ultimately defining the specificity of the lysin (Broendum et al. 2018). Several CBDs have been recognized, with LysM standing out as one of the most familiar. LysM is found in motifs and specifically binds to N-acetylglucosamine residues of peptidoglycan (Raya-Tonetti et al. 2021). Another well-established CBD is SH3, which exists in five subgroups and binds to peptidoglycan and facilitates protein–protein interactions (Desvaux et al. 2018). Among Lactococcus phage lysins, CBD types CHW, LysM, PG-1, SH3-5, and SH3b have been identified by sequence analyses (Oliveira et al. 2013). In addition, many lactococcal phage lysins carry uncharacterized regions, which may serve as binding domains. Of these putative anchoring domains, only a few have been experimentally demonstrated to function as CBDs (Plavec et al 2019).
In our previous work, we reported a bacteriocin-like lysin LysL (accession number UCS91464.1), produced by L. lactis strain LAC460 isolated from spontaneously fermented idli batter (Takala et al. 2023). LysL is a 385-aa lysin originating from a defective prophage PLl460-1 in the chromosome of L. lactis LAC460. Figure 1 shows the prediction of the genes in PLl460-1, and the location of the lysL gene in the prophage, according to phage search tool PHASTEST, open reading frame finder ORFfinder, and protein sequence comparison tool BlastP (https://phastest.ca, https://www.ncbi.nlm.nih.gov/orffinder and https://blast.ncbi.nlm.nih.gov/Blast.cgi, respectively). LysL is heat-sensitive, is produced and secreted in late log-phase and early stationary phase, and does not break down the host’s cell wall but kills other Lactococcus strains like a bacteriocin. According to the sequence and conserved domain homologies, LysL possesses EADs of lysozyme and peptidase M23 (approximately aa residues 7-270). However, the function of the C-terminus of about 115 aa is unknown, as no homology to known domains has been detected.

Location of the lysL gene in the defective prophage PLl460-1 and the flanking bacterial genes in L. lactis LAC460 chromosome. The encoded predicted proteins are shown above the genes. Bacterial genes are in light grey, putative phage genes in black, lysL gene in checker board pattern, and the phage lysis cassette genes, including two holins and a putative endolysin, in diagonal stripes. Several phage genes are non-functional, partial, or frameshifted pseudogenes
The aim of this study was to investigate the target specificity of LysL by determining the presence or absence of a CBD in its unknown C-terminus. It was found that the CBDLysL binds only to LysL sensitive Lactococcus sp. strains, but not to LysL resistant strains, which may explain the host’s resistance to its own phage lysin.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Bacterial strains and plasmids used in this study are shown in Table 1, except the Lactococcus sp. strains used only in cell wall polysaccharide (CWPS) genotyping are shown in Table 2 in Results section. E. coli strains were grown in LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl) with shaking (200 rpm) at 37 °C. Ampicillin (150 μg ml−1) was used for selecting transformants. Lactococcus strains were grown in M17 (Oxoid Ltd. Basingstoke, UK) broth supplemented with 0.5% (w/v) glucose (M17G) at 30 °C. Chloramphenicol (10 μg ml−1) was added to the growth media for Lactococcus cremoris LAC275. Solid media were prepared by adding 1.5% agar to the broth media.
DNA techniques
The PCR primers used in this study are presented in the Supplementary Table 1. Phusion High-Fidelity DNA polymerase (Thermo Scientific, Waltham, MA, USA) was used in PCR according to the instructions of the producer. GeneJET PCR purification kit and GeneJET Gel Purification Kit (Thermo Scientific) were used for DNA purification of PCR products and from agarose gel according to the manufacturer’s instructions, respectively. CWPS genotyping of Lactococcus sp. strains was done by multiplex PCR according to Mahony et al. (2013), except the used control primers targeted bacterial 16S rRNA gene (Edwards et al. 1989). Plasmid DNA was isolated from the recombinant cultures with a GeneJET Plasmid Miniprep Kit (Thermo Scientific) as instructed by the manufacturer.
Construction of HGFPuv_CBDlysL expression plasmid and transformation
According to BLAST matches, the EADs in the lysL gene constitute the first 810 bp of the entire length of 1158 bp. Therefore, the rest of the gene was considered as potentially encoding for the putative CBD region, and the last 360 nucleotides were amplified by using L. lactis LAC460 cells as a template with LysL F OE GFP and LysL R primers. The UV light-excited green fluorescent (GFPuv) gene lacking the stop codon but including hexahistidine (6 × His-tag) codons was amplified by PCR using L. cremoris LAC275 cells as template with the His-GFP F and GFP R OE L primers. As the actual Overlap Extension PCR for constructing the HGFPuv_CBDlysL fusion only produced strong bands of wrong size, the HGFPuv and CBDlysL fragments were joined together by polymerization without primers using standard PCR program. The insert HGFPuv_CBDlysL was phosphorylated by T4 polynucleotide kinase (Thermo Scientific) according to manufacturer’s instructions at 37 °C for 30 min, heat inactivated at 65 °C for 20 min, and purified with PCR purification kit. The vector pASG-IBA4 was amplified without its ompA signal sequence by PCR using E. coli ECO809 cells carrying a derivative of pASG-IBA4 plasmid as a template with pASG-IBA4 F and IBA noSS R primers, followed by purifying the vector fragment with a PCR purification kit. The insert and the vector fragments were ligated using T4 DNA ligase (Thermo Scientific) overnight at room temperature, as instructed by the manufacturer.
The ligation mixture was then electroporated into E. coli DH5α (Zabarovsky and Winberg 1990). Cells were spread onto selective plates (LB Amp150) and incubated at 37 °C overnight. Obtained transformant colonies were screened by PCR with His-GFP F and LysL R primers. The plasmid was then extracted from the correct clones and sequenced to confirm the correct construct. The recombinant E. coli DH5α carrying the correct pASG-IBA4-HGFPuv_CBDlysL plasmid pLEB836 was stored as E. coli ECO854.
Western blot and purification of HGFPuv_CBDLysL
HGFPuv_CBDLysL was purified with affinity chromatography by an outsourced protein purification service (Protein Service core facility of the Tampere University). Briefly, 200 ml of E. coli ECO854 culture in LB Amp150 was induced with 0.5 μg ml−1 tetracycline after OD600 reached about 0.5. The cells were harvested by centrifugation (5000×g, 10 min) after 20 h of continued incubation. The cell pellet was resuspended in 50 ml of PBS (pH 7.2) and treated four times with an Emulsiflex C3 high-pressure homogenizer (Avestin Inc., Ottawa, Canada). The content was then centrifuged (30,000×g, 4 °C, 20 min) and the supernatant was used as a cell lysate sample.
Western blotting was performed essentially as described by Towbin et al. (1979). SDS-PAGE of the cell lysate proteins was carried out as described by Laemmli (1970) excluding the staining and destaining steps. Briefly, the cell lysate was mixed in a 5:1 ratio with Tris–Glycine SDS-PAGE loading buffer (Bio-Rad Laboratories, Hercules, CA, USA) and heated at 95°C for 5 min. Then, 5 µl of the sample was loaded into a gradient SDS-PAGE gel (4–20%, Bio-Rad Laboratories) and electrophoresed at 200 V for 30 min at room temperature. Electrotransfer of the protein bands from SDS gel to a cellulose nitrate membrane was done under the voltage 120 V at 4 °C for 40 min. The membrane was then exposed to blocking solution (10% (w/v) bovine serum albumin) for 1 h with mild agitation followed by washing three times with TBS-T buffer (150 mM NaCl, 25 mM Tris, 0.1% Tween 20, pH 7.5) for 5 min. The membrane was then probed with primary mouse anti-His.H8 diluted 1:10,000 in 5% milk-TBST (Thermo Scientific) at 4 °C overnight with mild agitation followed by washing three times with TBST buffer for 5 min. The membrane was then incubated with horse anti-mouse IgG(H + L) Peroxidase (Vector Laboratories, Newark, CA, USA) at 1:20 000 dilution in TBST for 1 h at room temperature. Afterward, the membrane was washed three times with TBST buffer for 5 min each time. Protein bands were visualized using the WesternBright ECL HRP substrate (Advansta, San Jose, CA, USA) following the manufacturer’s instructions. Imaging was performed using the ChemiDoc MP Imaging System (Bio-Rad Laboratories).
For purification of HGFPuv_CBDLysL, the cell lysate was obtained from 900 ml of tetracycline-induced culture of E. coli ECO854 as described above. 10 mM imidazole was added to the pellet lysate sample to prevent nonspecific binding, then the sample was bound to HisPur™ Ni–NTA agarose (Thermo Scientific) in a batch mode at 4 °C for 1 h. The bound protein was washed with 10 column volumes with Wash Buffer (PBS, 250 mM NaCl, 50 mM imidazole, pH 7.2) before stepwise elution with 5 column volumes of Elution Buffer (20 mM Tris–HCl, 500 mM NaCl, 250 mM imidazole, pH 7.5). NanoDrop One (Thermo Scientific) was used for measuring protein concentration of the samples at A280.
Cell wall binding assays
Binding of HGFPuv_CBDLysL onto Lactococcus cells was determined by the binding assay described previously (Loessner et al. 2002). The indicator strains (Table 2) were cultured (10 ml) overnight, harvested by centrifugation (5000×g, 10 min), and resuspended in the same volume of PBS-T buffer (pH 7.4, 0.1% Tween 80). 600 µl of cell suspensions were mixed with 5 µl of the purified HGFPuv_CBDLysL (1.73 mg/ml) and incubated at room temperature for 15 min. Cells were pelleted (5000×g, 5 min), washed with 1 ml of PBS-T, resuspended in 600 µl of PBS-T, and applied to a microplate. Fluorescence was measured with Infinite M200 plate reader (Tecan, Männedorf, Switzerland) with excitation at 395 nm and emission at 508 nm. Part of the cell suspensions of strains L. cremoris MG1614, L. lactis LM0230 and L. lactis LAC460 were also used for visualization with a Zeiss Axioscope 2 Plus fluorescence microscope using Axiocam 305 color camera and Carl Zeiss microscope software 2.6 pre (Carl Zeiss Microscopy, Jena, Germany) with a WTGFP filter (502 nm emission).
Results
Cloning and expression of HGFPuv_CBDLysL in E. coli
With the purpose of producing GFPuv-fused CBD of LysL, HGFPuv_CBDLysL was cloned in E. coli DH5α. When exposed to UV light, the cell pellet of the tetracycline-induced E. coli ECO854 culture showed green fluorescence, whereas the parallel non-induced cell pellet did not (result not shown). This indicates that the cloning and expression of HGFPuv_CBDLysL in E. coli was successful and that the HGFPuv in the fusion protein is functional.
Western blot and purification of HGFPuv_CBDLysL
Western blotting was performed prior to purification to verify the presence of LysL(CBD) and the N-terminal 6 × His-tag in the fusion protein in cell lysate of E. coli ECO854. As the molecular weight of HGFPuv is approximately 28 kDa and CBD_LysL about 14 kDa, the distinct band above 40 kDa on the blotted membrane represents HGFPuv_CBDLysL, recognized by the anti-His antibody (Fig. 2). Purification of HGFPuv_CBDLysL was performed using 6 × His-tag and HisPur™ Ni–NTA chromatography. From the 900 ml of the initial culture of E. coli ECO854, the total protein yield was 3.06 mg with the concentration of 1.73 mg/mL.

Western blot of the tetracyclin-induced cell lysate of E. coli ECO854. Lane 1, HGFPuv_CBDLysL; M, PageRuler Broad Range Protein Ladder (Thermo Scientific)
Binding of HGFPuv_CBDLysL protein onto Lactococcus cells
The purified HGFPuv_CBDLysL was used to examine the specific binding of CBDLysL to the cell wall of the Lactococcus strains. As shown in the microscope images (Fig. 3), green fluorescence was only observed on the L. cremoris MG1614 cell surface, demonstrating that CBD of LysL binds to the cell wall of the LysL-sensitive Lactococcus strain. No green decoration was observed on the cells of the LysL-resistant L. lactis LM0230 or the LysL producer strain L. lactis LAC460.

Phase contrast and fluorescence microscopy images of L. cremoris MG1614 (a), L. lactis LM0230 (b), and L. lactis LAC460 (c) cells mixed with HGFPuv_CBDLysL protein. Only L. cremoris MG1614 cells were decorated with green fluorescence
In addition, binding of the purified HGFP_CBDLysL fusion protein was examined with Lactococcus strains representing different CWPS genotypes. The strains’ CWPS genotype was determined by multiplex PCR, LysL sensitivity was tested on indicator plates, and the binding of HGFP_CBDLysL was measured with a fluorescence plate reader. The results are shown in Table 2, and the HGFP_CBDLysL binding to a few selected strains is also shown in Fig. 4. HGFP_CBDLysL only bound to the LysL sensitive strains, and it did not bind to LysL resistant strains. The LysL sensitive strains represented either the CWPS genotype C or an unknown genotype. The LysL resistant strains were found from all CWPS genotypes, showing no clear correlation between the CWPS genotype and the binding of CBDLysL.

Fluorescence of Lactococcus strains mixed with purified HGFPuv_CBDLysL protein measured by a fluorescence plate reader. Blank, cells without added HGFPuv_CBDLysL protein, i.e., background fluorescence from the cells. RFU relative fluorescence unit. Error bars represent standard deviation of two duplicates
Domain prediction
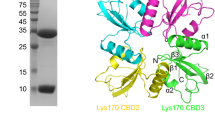
The results of the binding assay suggest that there is a region that binds the protein onto the cell surface of LysL-sensitive bacteria within the 119 C-terminal amino acids in LysL (Fig. 5a). However, unlike the two N-terminal EADs in LysL, lysozyme and peptidase, the NCBI Conserved Domain search tool did not recognize any structural domain in the C-terminus of LysL. Therefore, we sought to elucidate the three-dimensional structure of LysL by using Alphafold2. Three distinct domains in the 3-D structure of LysL were revealed (Fig. 5b). The two N-terminal structures are likely the lysozyme and peptidase domains predicted by the Conserved Domains search. The third separate structure in the unknown C-terminus is presumably the CBD, demonstrating that the cell binding region forms a distinct structural domain in LysL. The precise location and the length of the CBD, the 58 C-terminal residues forming a seemingly independent structure after a long linker, is still only an estimate based on the 3-D prediction.

Predicted domains and structure of LysL. a Amino acid sequence. The underlined sequence is the 119-aa C-terminal region fused with GFP. b structure prediction by Alphafold2. The three distinct domains of lysozyme, peptidase, and cell binding domain are marked in both sequence and 3-D structure with cyan, magenta, and green, respectively. The linkers between the domains are shown in black. Lysozyme and peptidase positions were predicted by NCBI Conserved Domains search tool. The location of CBD is an estimate based on 3-D structure
Discussion
This study sought to investigate the cell binding domain of the bacteriocin-like lysin LysL from Lactococcus lactis LAC460. The CBD was fused with GFPuv and the fusion protein was mixed with different Lactococcus cells to detect the possible binding of the CBD onto the cell surface. The HGFPuv_CBDLysL protein bound to LysL-sensitive strains, but not to LysL-resistant strains, including the LysL-producer L. lactis LAC460, as observed with fluorescence microscopy and as measured with a plate reader. The results confirmed that the C-terminus of LysL carries a CBD with strain-specific binding property that determines the target specificity of the LysL lysin. Based on protein BLAST comparisons, identical or similar (78–79% identity) C-terminal domains seem to be linked with M23 peptidases. Identical CBDs are only found in LysL-like enzymes carrying a lysozyme and peptidase domains, but similar (putative) CBDs are also present in enzymes with only M23 peptidase domain. However, these M23 peptidases show only around 50% aa-sequence identities to the peptidase domain of LysL.
The inability of HGFPuv_CBDLysL to bind to L. lactis LAC460 may be the reason why LysL does not lyse the host’s own cell wall. It has been suggested that bacteria domesticate prophages for their own benefit, for example to improve secretion, gene transfer, and defense (Bobay et al. 2014). This work provides an example of domestication of a prophage lysin by L. lactis strain. However, the receptor for the CBDLysL and the reason why L. lactis LAC460 contains remnants of prophages that do not work against the host, remain to be investigated.
There are not many studies about experimental evidence of the function of lactococcal phage lysin CBDs. Roces et al. (2016) have characterized the LysM domain of the endolysin LysTP712, and Plavec et al (2019) have tested a few putative CBDs, including unchacterized domains, for surface display of proteins on L. lactis cells. In addition to phage lysin CBDs, also other cell binding proteins, e.g. lactococcal autolysin AcmA carrying LysM repeats, have been exploited for displaying proteins on the Lactococcus cell surface (Zahirović et al. 2022). Endolysin CBDs of some other genera have also been studied. For instance, the binding of the CBDs of Listeria monocytogenes and lactobacilli phage endolysins has been studied by the same approach as in this work, namely constructing CBD fusions with fluorescent protein and testing protein binding to different bacteria (Dorosky et al. 2023; Schmelcher et al. 2011). In the aforementioned study by Dorosky et al (2023), a method to differentiate specific lactobacilli from a mixture was developed based on CBD binding.
Studies on the receptors of CBD targets are very limited; only a few ligands are known. Of the known CBD targets, the involvement of wall teichoic acids in the binding of L. monocytogenes phage lysins to CBDs have been shown (Eugster and Loessner 2012; Eugster et al. 2011). Research by Mahony et al. in 2013 revealed that the host range of 936-type phages correlates with the cell wall polysaccharide (CWPS) types of the targeted L. lactis bacteria. Among the 11 lactococcal 936-type phages studied, most infected strains with MG/SK CWPS (genotype C), others with IL/KF CWPS (genotype B), and a small portion demonstrated infectivity towards both. Even though CWPS has been shown to be a major factor determining phage sensitivity in Lactococcus spp. (Ainsworth et al 2014), it seems not to correlate with LysL sensitivity, as both LysL sensitive and resistant strains were found from different CWPS genotype groups (Table 2). Consequently, it seems unlikely that the CBDLysL would use CWPS as the binding target. However, additional investigation is required for a better understanding of the specific target to which CBDLysL binds.
It is not fully certain whether LysL is a VAL or endolysin. VALs are often claimed to lack a CBD, although there is an exception reported where the VAL of Staphylococcus aureus phage P68 carries a CBD (Takác and Bläsi 2005). Regarding EADs, M23 peptidase present in LysL is rarely an EAD in endolysins, while being frequently found in VAL catalytic active domains (Oliveira et al. 2018). Otherwise the LysL protein structure resembles a conventional endolysin. However, the location of the lysL gene in the genome is not typical for an endolysin gene. Endolysin and holin genes are usually located adjacently in the lysis cassette of prophages (Summer et al. 2007; Shin et al. 2014). However, this is not always the case, as for instance in the ΦC2 prophage of Clostridioides difficile the endolysin gene is downstream of holin gene after abiF gene, which confers phage resistance to bacteria (Goh et al. 2007). In the case of lysL gene, there is no adjacent holin gene, but there are two holin genes about 800 bp downstream of lysL (Fig. 1). However, immediately downstream of the holin genes, there is another gene encoding a lysin with an N-terminal amidase as an apparent EAD and with an unknown C-terminus for a putative CBD, making this lysin gene to look like a typical endolysin. Still, some phages carry two endolysins, e.g. Lactococcus phage KSY1 and E. coli phage swi2 (Chopin et al. 2007; Sui et al. 2021). These endolysin genes are also neighboured by holin genes in the phage genomes. However, lysL gene is located in an incomplete prophage PLl460-1, where it is unknown which parts of the prophage have been rearranged. Hence, the set and the order of the genes may have earlier been different in PLl460-1. Phages can evolve by recombinations, causing gene exchanges (Oechslin et al 2022). It is therefore possible that the origin of lysl gene is not prophage PLl460-1, or that even larger part of the prophage region is a result of recombinations and genomic rearrangements. Based on BLAST nucleotide sequence comparisons, identical (≥ 99%) prophage PLl460-1 fragment in the same location in the chromosome (Fig. 1), is found from several other L. lactis strains, for instance the strains SRCM103457, JXNPKM 1305, and D53 (accession numbers CP035757.1, JAKIVG010000004.1, and WKFC01000004.1). Thus, the mutations, rearrangements and possible recombinations in the prophage fragment causing gene exchanges have most likely happened earlier in an ancestor strain, and not in LAC460.
References
Abdelrahman F, Easwaran M, Daramola OI, Ragab S, Lynch S, Oduselu TJ et al (2021) Phage-encoded endolysins. Antibiotics 10:124. https://doi.org/10.3390/antibiotics10020124
Ainsworth S, Sadovskaya I, Vinogradov E, Courtin P, Guerardel Y, Mahony J et al (2014) Differences in lactococcal cell wall polysaccharide structure are major determining factors in bacteriophage sensitivity. Mbio 5:e00880-e1814. https://doi.org/10.1128/mBio.00880-14
Bobay LM, Touchon M, Rocha EPC (2014) Pervasive domestication of defective prophages by bacteria. PNAS 111:12127–12132. https://doi.org/10.1073/pnas.1405336111
Broendum SS, Ashley MB, Sheena M (2018) Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol Microbiol 110:879–896. https://doi.org/10.1111/mmi.14134
Chandran C, Tham HY, Abdul RR, Lim SHE, Yusoff K, Song AA (2022) Lactococcus lactis secreting phage lysins as a potential antimicrobial against multi-drug resistant Staphylococcus aureus. PeerJ 10:e12648. https://doi.org/10.7717/peerj.12648
Chopin A, Chopin MC, Moillo-Batt A, Langella P (1984) Two plasmid-determined restriction and modification systems in Streptococcus lactis. Plasmid 11:260–263. https://doi.org/10.1016/0147-619x(84)90033-7
Chopin A, Deveau H, Ehrlich SD, Moineau S, Chopin M (2007) KSY1, a lactococcal phage with a T7-like transcription. Virol 365:1–9. https://doi.org/10.1016/j.virol.2007.03.044
Colombo A (2016) Reformulation and quality evaluation by microbiological analyses of idli, a fermented Indian products. MSc thesis, Università Degli Studi di Milano, Italy, and University of Helsinki, Finland
Desvaux M, Candela T, Serror P (2018) Surfaceome and proteosurfaceome in parietal monoderm bacteria: focus on protein cell-surface display. Front Microbiol 9:100. https://doi.org/10.3389/fmicb.2018.00100
Dorosky RJ, Lola SL, Brown HA, Schreier JE, Dreher-Lesnick SM, Stibitz S (2023) Characterization of lactobacilli phage endolysins and their functional domains-potential live biotherapeutic testing reagents. J Viruses 15:1986. https://doi.org/10.3390/v15101986
Edwards U, Rogall T, Blöcker H, Emde M, Böttger EC (1989) Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16s ribosomal RNA. Nucleic Acids Res 17(19):7843–7853. https://doi.org/10.1093/nar/17.19.7843
Efstathiou JD, McKay LL (1977) Inorganic salts resistance associated with a lactose-fermenting plasmid in Streptococcus lactis. J Bacteriol 130:257–265. https://doi.org/10.1128/jb.130.1.257-265.1977
Eugster MR, Loessner MJ (2012) Wall teichoic acids restrict access of bacteriophage endolysin Ply118, Ply511, and Plyp40 cell wall binding domains to the Listeria monocytogenes peptidoglycan. J Bacteriol 194:6498–6506. https://doi.org/10.1128/JB.00808-12
Eugster MR, Haug MC, Huwiler SGB, Loessner MJ (2011) The cell wall binding domain of Listeria bacteriophage endolysin PlyP35 recognizes terminal GlcNAc residues in cell wall teichoic acid. Mol Microbiol 81:1419–1432. https://doi.org/10.1111/j.1365-2958.2011.07774.x
Gasson MJ (1983) Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol 154:1–9. https://doi.org/10.1128/jb.154.1.1-9.1983
Goh S, Ong PF, Song KP, Riley TV, Chang BJ (2007) The complete genome sequence of Clostridium difficile phage phiC2 and comparisons to phiCD119 and inducible prophages of CD630. Microbiol (reading) 153:676–685. https://doi.org/10.1099/mic.0.2006/002436-0
Hakovirta J, Reunanen J, Saris PEJ (2006) Bioassay for nisin in milk, processed cheese, salad dressings, canned tomatoes, and liquid egg products. AEM 72:1001–1005. https://doi.org/10.1128/AEM.72.2.1001-1005.2006
Hanahan D (1985) Techniques for transformation of Escherichia coli. In: Glover DM (ed) DNA cloning: a practical approach. IRL Press, Virginia, pp 109–135
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. https://doi.org/10.1038/227680a0
Loessner MJ (2005) Bacteriophage endolysins–current state of research and applications. COMICR 8:480–487. https://doi.org/10.1016/j.mib.2005.06.002
Loessner MJ, Kramer K, Ebel F, Scherer S (2002) C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol 44:335–349. https://doi.org/10.1046/j.1365-2958.2002.02889.x
Lortal S, Chapot-Chartier MP (2005) Role, mechanisms and control of lactic acid bacteria lysis in cheese. Int Dairy J 15:857–871. https://doi.org/10.1016/j.idairyj.2004.08.024
Mahony J, Kot W, Murphy J, Ainsworth S, Neve H, Hansen LHK et al (2013) Investigation of the relationship between lactococcal host cell wall polysaccharide genotype and 936 phage receptor binding protein phylogeny. AEM 79:4385–4392. https://doi.org/10.1128/AEM.00653-13
Oechslin F, Zhu X, Dion MB, Shi R, Moineau S (2022) Phage endolysins are adapted to specific hosts and are evolutionarily dynamic. PLoS Biol 20:e3001740. https://doi.org/10.1371/journal.pbio.3001740
Oliveira H, Melo LD, Santos SB, Nóbrega FL, Ferreira EC, Cerca N et al (2013) Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 87:4558–4570. https://doi.org/10.1128/JVI.03277-12
Oliveira H, São-José C, Azeredo J (2018) Phage-derived peptidoglycan degrading enzymes: challenges and future prospects for in vivo therapy. J Viruses 10:292. https://doi.org/10.3390/v10060292
Plavec TV, Štrukelj B, Berlec A (2019) Screening for new surface anchoring domains for Lactococcus lactis. Front Microbiol 10:1879. https://doi.org/10.3389/fmicb.2019.01879
Raya-Tonetti F, Müller M, Sacur J, Kitazawa H, Villena J, Vizoso-Pinto MG (2021) Novel LysM motifs for antigen display on lactobacilli for mucosal immunization. Sci Rep 11:21691. https://doi.org/10.1038/s41598-021-01087-8
Schmelcher M, Tchang VS, Loessner MJ (2011) Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol 4:651–662. https://doi.org/10.1111/j.1751-7915.2011.00263.x
Shin H, Lee JH, Yoon H, Kang DH, Ryu S (2014) Genomic investigation of lysogen formation and host lysis systems of the Salmonella temperate bacteriophage SPN9CC. AEM 80:374–384. https://doi.org/10.1128/AEM.02279-13
Sorokina D (2015) Enkasiini-bakteriosiinin tuotto Escherichia coli-bakteerissa. MSc thesis, University of Helsinki. Finland
Steen MT, Chung YJ, Hansen JN (1991) Characterization of the nisin gene as part of a polycistronic operon in the chromosome of Lactococcus lactis ATCC 11454. Appl Environ Microbiol 57:1181–1188. https://doi.org/10.1128/aem.57.4.1181-1188.1991
Sui B, Qi X, Wang X, Ren H, Liu W, Zhang C (2021) Characterization of a novel bacteriophage swi2 harboring two lysins can naturally lyse Escherichia coli. Front Microbiol 12:670799. https://doi.org/10.3389/fmicb.2021.670799
Summer EJ, Berry J, Tran TA, Niu L, Struck DK, Young R (2007) Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J Mol Biol 373:1098–1112. https://doi.org/10.1016/j.jmb.2007.08.045
Takác M, Bläsi U (2005) Phage P68 virion-associated protein 17 displays activity against clinical isolates of Staphylococcus aureus. Antimicrob Agents Chemother 49:2934–2940. https://doi.org/10.1128/AAC.49.7.2934-2940
Takala TM, Mokhtari S, Ahonen SL, Wan X, Saris PEJ (2023) Wild-type Lactococcus lactis producing bacteriocin-like prophage lysins. Front Microbiol 14:1219723. https://doi.org/10.3389/fmicb.2023.1219723
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. PNSA 76:4350–4354. https://doi.org/10.1073/PNAS.76.9.4350
Wan X, Takala TM, Qiao M, Saris PEJ (2021) Complete genome sequence of nisin-producing Lactococcus lactis subsp. lactis N8. Microbiol Resour Announc 10:e01147-e1220. https://doi.org/10.1128/MRA.01147-20
Zabarovsky ER, Winberg G (1990) High efficiency electroporation of ligated DNA into bacteria. Nucleic Acids Res 18:5912. https://doi.org/10.1093/nar/18.19.5912
Zahirović A, Plavec TV, Berlec A (2022) Dual functionalized Lactococcus lactis shows tumor antigen targeting and cytokine binding in vitro. Front Bioeng Biotechnol 10:822823. https://doi.org/10.3389/fbioe.2022.822823
Acknowledgements
The authors acknowledge the Biocenter Finland and Tampere facility of Protein Services (PS) for their services.
Funding
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital). The Niemi-Säätiö (application numbers: 20210081 and 20220051) and The Finnish Cultural Foundation (27.02.2022) are also acknowledged for awarding personal working grants to Samira Mokhtari.
Author information
Authors and Affiliations
Contributions
Timo Takala and Per Saris designed the research. Samira Mokhtari, Yanru Li, and Timo Takala performed the experiments except the outsourced Western blot and protein purification. Samira Mokhtari wrote the manuscript draft and all the co-authors contributed to revising it.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mokhtari, S., Li, Y., Saris, P.E.J. et al. Analysis of the cell wall binding domain in bacteriocin-like lysin LysL from Lactococcus lactis LAC460. Arch Microbiol 206, 336 (2024). https://doi.org/10.1007/s00203-024-04066-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-024-04066-5