
Abstract
Microbial mats are often found in intertidal areas experiencing a large range of salinities. This study investigated the effect of changing salinities on nitrogenase activity and on the composition of the active diazotrophic community (nifH transcript libraries) of three types of microbial mats situated along a littoral gradient. All three mat types exhibited highest nitrogenase activity at salinities close to ambient seawater or lower. The response to lower or higher salinity was strongest in mats higher up in the littoral zone. Changes in nitrogenase activity as the result of exposure to different salinities were accompanied by changes in the active diazotrophic community. The two stations higher up in the littoral zone showed nifH expression by Cyanobacteria (Oscillatoriales and Chroococcales) and Proteobacteria (Gammaproteobacteria and Deltaproteobacteria). At these stations, a decrease in the relative contribution of Cyanobacteria to the nifH transcript libraries was observed at increasing salinity coinciding with a decrease in nitrogenase activity. The station at the low water mark showed low cyanobacterial contribution to nifH transcript libraries at all salinities but an increase in deltaproteobacterial nifH transcripts under hypersaline conditions. In conclusion, increased salinities caused decreased nitrogenase activity and were accompanied by a lower proportion of cyanobacterial nifH transcripts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coastal ecosystems, including salt marshes, mangroves, wetlands, estuaries and bays, are often characterized by highly fluctuating environmental conditions, mainly due to the tidal cycle. One important factor in intertidal areas is salinity. Salinities range from almost freshwater, for example, through runoff or upwelling groundwater, to hypersaline conditions due to evaporation, for example, when seawater is confined to shallow areas with a negative water balance. These intertidal areas are colonized by a variety of microorganisms. Salt stress may limit the diversity of any group of organisms, including microorganisms, probably for reasons of impaired bioenergetics (Oren 1999). In the light of global climate change, an increasing number of habitats could be subjected to increased salinities due to rising sea levels. It is therefore of interest to evaluate the consequences of elevated salinities on the communities of these habitats and the ecosystem services provided by these communities.
Studies on the diversity of Bacteria and Archaea as well as their distribution along a salinity gradient in a coastal solar saltern showed a diverse community consisting of Proteobacteria, Cyanobacteria, high GC-Gram-positive Bacteria and members of the Cytophaga-Flavobacterium-Bacteroides group (CFB) (Benlloch et al. 2002). The diversity of different clusters decreased with increasing salinity, but a considerable degree of microdiversity within these remaining clusters persisted. Similar observations were made for a salinity gradient in a soda lake where the 16S rRNA gene sequences of members of a variety of different bacterial groups, for example, Cyanobacteria, Proteobacteria, Actinobacteria and Clostridia were retrieved (Foti et al. 2008). Again, the lowest bacterial diversity was found at the highest salinities. The change of bacterial communities along a salinity gradient has also been studied in other environments. Free-living bacterioplankton in two Chesapeake Bay estuaries showed a dominance of Alphaproteobacteria in the saltwater regions, members of the Cytophaga-Flavobacterium cluster in the turbidity maximum and Betaproteobacteria in the freshwater regions, while Gammaproteobacteria abundances exhibited only sporadic peaks (Bouvier and del Giorgio 2002). Possible factors for these compositional changes were partly associated with but not exclusively driven by salinity. On a global scale, salinity has been identified as an important environmental determinant of microbial community composition (Lozupone and Knight 2007). The effect of increased salinities on microbial activities and community composition has been studied for freshwater sediments (Edmonds et al. 2009). Despite the changes in activity, the microbial community composition remained largely unaltered. It was therefore argued that the changes in metabolic activity were driven by shifts in gene expression and regulation rather than by changes in the composition of the microbial community (Edmonds et al. 2009).
In addition to salinity changes, coastal intertidal sandy sediments are often characterized by low nutrient concentrations and different degrees of wave and wind energy. On some intertidal sand flats, these harsh conditions largely exclude grazing organisms and thus allow for the development of microbial mats. These small-scale ecosystems are vertically stratified microbial communities, usually built by Cyanobacteria. Especially coastal tidal flats with low slopes and fine sandy sediment serve as excellent habitats for microbial mats (Stal 2000). Cyanobacteria are oxygenic photoautotrophs with low nutritional requirements. Many of them are also capable of fixing N2, which provides them with a distinctive advantage that allows them to colonize nutrient-poor and nitrogen-depleted environments. The ability to fix N2 is not limited to Cyanobacteria and occurs among a variety of other members of the Bacteria as well as in some Archaea. In addition to Cyanobacteria, microbial mats may accommodate a variety of other functional groups of microorganisms, for example, anoxygenic phototrophs, sulfate-reducing bacteria and sulfur-oxidizing bacteria. Especially the members of the Proteobacteria were identified as potential diazotrophs based on the possession of nifH, the gene encoding for dinitrogenase reductase, one of the two components of nitrogenase, the enzyme complex catalyzing the reduction of N2 to NH3. Gammaproteobacteria and Deltaproteobacteria were most frequently encountered in different types of microbial mats (e.g., Zehr et al. 1995; Bauer et al. 2008). Especially within the Deltaproteobacteria, the role of sulfate-reducing bacteria in microbial mat N2 fixation has been discussed (Steppe and Paerl 2002). The high bacterial diversity of microbial mats has also been reported for mats in high salt environments and includes Cyanobacteria, Chloroflexi, Bacteroidetes and a variety of Proteobacteria (Fourçans et al. 2004; Ley et al. 2006). Hypersaline microbial mats are found in different regions of the world (Severin and Stal 2010a), and high salinities, therefore, do not seem to prevent the existence of these microbial communities. Nevertheless, metabolic processes such as CO2 and N2 fixation can be severely inhibited by salt stress. In cultures of diazotrophic Cyanobacteria, increased salinities greatly decreased nitrogenase activity (e.g., Fernandes et al. 1993; Fu and Bell 2003). But also microbial mat community showed lower CO2 and N2 fixation rates at higher salinities (Pinckney et al. 1995).
Hitherto, no experimental study showed the effect of salinity on N2 fixation and the associated shifts in the expression of nitrogenase genes in coastal microbial mats. We hypothesized that changes in salinity result in shifts within the active diazotrophic community and therefore possibly also in altered nitrogenase activity. This study aimed at testing this hypothesis by exposing coastal microbial mats to an experimental series of salinities and subsequently measuring nitrogenase activity and determining the active diazotrophic community, that is, the fraction of the bacterial community expressing nifH.
Materials and methods
Sampling
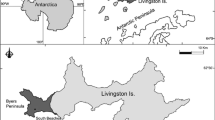
The study site was located on the Dutch barrier island Schiermonnikoog. The geographic coordinates of the study site were N 53°29′ and E 6°08′. Microbial mats were found on the sandy beach covering the north bank of the island facing the North Sea. Areas of the beach are currently turning into a salt marsh, resulting in mats partly overgrown by higher plants. Due to this succession and the gradually changing influence of the North Sea, different mat types developed along the littoral gradient.
Three sites within this area were chosen based on the microscopic observations of the cyanobacterial community composition as well as their situation along the littoral gradient. Station I was located near the dunes and influenced by both seawater and freshwater (rain and groundwater). This area was only irregularly inundated by the sea. The mats found at Station I revealed high cyanobacterial species diversity containing both heterocystous and non-heterocystous filamentous Cyanobacteria as well as unicellular species. Station II is situated near the low water mark. Due to the tidal inundation, seawater was far more important than the occasional rain showers, and this distinguished it from Station I. The Cyanobacteria at Station II were mostly non-heterocystous forms, predominantly Lyngbya aestuarii. Occasionally, heterocystous Cyanobacteria have been observed, but these organisms did not seem to be a structural part of this community. Station III was located between Stations I and II and therefore represented an area influenced by seawater and fresh water depending on the tidal amplitude. At Station III, higher plants were partly overgrowing the established mats. Based on the microscopic observation, Microcoleus chthonoplastes was identified as the dominant cyanobacterial component. A variety of other non-heterocystous as well as heterocystous species were found as well.
For the incubations, mat samples of each location were collected by using disposable 10 ml-syringes of which the needle connector was cut off to obtain a corer with a diameter of 1.5 cm. The top 2–3 mm of the mat was sampled and transferred in the incubation vials. After finishing the nitrogenase activity measurements, sediment and biomass were retrieved by filtering the samples over a glass microfiber filter (Whatman GF/F, nominal pore size 0.7 μm, Whatman plc, Kent, United Kingdom). Each sample was divided into four parts, and each part was transferred into a separate cryovial (Simport Plastics, Beloeil, Qc J3G 4S5, Canada) and immediately frozen in liquid nitrogen.
Incubations and nitrogenase activity measurements
The intact mat samples were incubated in 30-ml serum flasks which were sealed with a butyl rubber stopper (Rubber B.V., 1211 JG Hilversum, The Netherlands). To each of the samples, 2 ml of medium with the following salinities was added. Filtered North Sea water (Whatman GF/F glass microfiber filter, nominal pore size of 0.7 μm) with a salinity of 33 PSU from the location served as the control (‘natural’ salinity). Seawater salinity was determined on every sampling occasion using a refractometer (ATAGO CO., LTD., Tokyo 173-0001, Japan). Tap water (produced from the island’s own groundwater) was used as the 0 PSU medium (‘freshwater’), and a 1:1 dilution of filtered seawater with tap water provided the 16.5 PSU medium (‘half the natural salinity’). Salinities of 66 PSU and 165 PSU (‘twice the natural salinity’ and ‘fivefold the natural salinity’) were obtained by adding NaCl to the filtered seawater. Samples of all three mat types were incubated in duplicate. The entire incubation lasted 72 h. Measurements of nitrogenase activity were taken at the starting of the experiment and after 12, 24, 60 and 72 h. At each of the time points, the vials were closed, and 5 ml acetylene (Messer B.V., 4782 PM Moerdijk, The Netherlands) was injected, the gas phase was mixed by pumping with the syringe, and 5 ml of the headspace was removed to maintain normal pressure. Incubations lasted 3 h after which 5 ml of the headspace gas was transferred into evacuated and sealed 5-ml crimp top vials (Chrompack-Varian, Inc., 4338 PL Middelburg, The Netherlands). Between the incubations, the vials were left open to permit gas exchange with the atmosphere. Ethylene concentrations were measured by using a gas chromatograph (Chrompack CP 9001) equipped with a flame ionization detector. The temperatures of injector, detector and oven were 90, 120 and 55°C, respectively. Helium served as a carrier gas (Westfalen Gassen Nederland BV, 7418 EW Deventer, The Netherlands) at a flow rate of 10 ml min−1. The supply rates of H2 and clinical air (Westfalen Gassen Nederland BV, 7418 EW Deventer, The Netherlands) for the flame ionization detector were 30 and 300 ml min−1, respectively. The column was a 25 m-long wide-bore silica fuse column (inner diameter of 0.53 mm), packed with Poropak U (Chrompack-Varian, Inc., 4338 PL Middelburg, The Netherlands).
Nucleic acid extraction
RNA was extracted from triplicate samples at the end of the incubations using the Qiagen RNeasy Mini-kit (Qiagen GmbH, 40724 Hilden, Germany) following the manufacturer’s instructions but replacing the enzyme-digest by a bead-beating step (0.1-mm glass beads, maximum speed for 2 min) (Sartorius AG, 37075 Göttingen, Germany). Quality and quantity of the extracted RNA were checked on a 1% agarose gel. The triplicates were combined and treated with DNase (Deoxyribonuclease I, Invitrogen Corporation, Carlsbad, CA 92008, USA), following the manufacturer’s instructions. The lack of genomic DNA in the RNA extracts was confirmed by negative PCR results using the primers described below and under the conditions described in the next paragraph. DNA-free RNA was immediately used for reversed transcription using random primers (Superscript II Reverse Transcriptase and Random Primers, Invitrogen Corporation, Carlsbad, CA 92008, USA). The resulting cDNA was checked on a 1% agarose gel and stored at −20°C. RNA extraction, and therefore, all subsequent steps failed for the Station III at freshwater conditions. The entire procedure and all subsequent steps were repeated, and the duplicates were analyzed separately in order to investigate within-sample variability (Fig. 2; Supplement Tables 2 and 3) as well as in combination in order to achieve comprehensive results (Fig. 1; Tables 1, 2, 3 and Supplement Table 1).

Nitrogenase activity at five time points within the 72-h incubation period as measured by acetylene reduction assay (ARA, normalized to m2) at the different salinities. Standard deviation is indicated by error bars. The horizontal bar at the top depicts the time of sampling and measurement (white bar for daytime and black bar for nighttime measurement)
PCR, cloning and sequencing
For the amplification of nifH transcripts, a nested PCR with the internal primer pair nifH 1 (5′ TGY GAY CCN AAR GCN GA 3′) and nifH 2 (5′ ADN GCC ATC ATY TCN C 3′) (Zehr and McReynolds 1989) and the external primers nifH 3 (5′ ATR TTR TTN GCN GCR TA 3′) nifH 4 (5′ TTY TAY GGN AAR GGN GG 3′) was performed (Zani et al. 2000). For the first amplification, each 25-μl PCR reaction mix contained 25 pmol of each primer, 0.2 mM dNTPs, 1× reaction buffer, 0.625 U Qiagen HotStar Taq (Qiagen GmbH, 40724 Hilden, Germany), as well as 10–15 ng DNA. For the nested reaction, 2 μl of the PCR product from the first reaction was used as template. Cycling conditions for both PCR steps included 94°C for 15 min, 35 cycles of 94°C for 1 min, 54°C (first reaction) or 57°C (nested reaction) for 1 and 72 min for 1 min, followed by an extension period of 72°C for 10 min. PCR products were checked on a 1% agarose gel.
The fresh PCR product was cloned using the TOPO TA Cloning Kit for Sequencing (Invitrogen Corporation, Carlsbad, CA 92008, USA), following the manufacturer’s instructions. Only white transformants were used for amplification with the M13-primer pair (F: 5′ GTA AAA CGA CGG CCA G 3′ and R: 5′ CAG GAA ACA GCT ATG AC 3′) and checked by gel electrophoresis. Cycling conditions were 95°C for 2 min, followed by 40 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 2 min, and a final extension step at 72°C for 19 min. PCR products were purified with the Sephadex G-50 Superfine powder (GE Healthcare Bio-Sciences AB, 751 84 Uppsala, Sweden) and 45 μl Millipore MultiScreen plates (Millipore Corporation, Billerica, MA 01821, USA). After determining the quantity of the purified PCR product, a sequencing reaction was performed using the BigDye Terminator chemistry (BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems, Inc., Foster City, CA 94404, USA) and the M13 reverse primer.
Sequence analysis
Sequences were aligned in BioEdit (Ibis Biosciences, Carlsbad, CA 92008, USA) using the ClustalW Multiple Alignment function, corrected by manual inspection and analyzed for similarity in BLASTn (Basic Local Alignment Search Tool, National Center for Biotechnology Information, 8600 Rockville Pike, Bethesda, USA). Nucleotide sequences were also translated into amino acid sequences to check the correct alignment.
Alpha- and beta-diversity of the active diazotrophic community at all three stations was estimated by the DOTUR and SONS software programs (Schloss and Handelsman 2005, 2006), based on the alignment files of the nifH transcript libraries.
All sequences are deposited at the NCBI GenBank database under the accession numbers GU192465 to GU193156 and JF896875 to JF897602.
Results
Nitrogenase activity
Nitrogenase activity (NA) was measured at different time points during the incubation period of 72 h and for five salinities ranging from 0 to 165 PSU (Fig. 1).
Highest NA was always recorded at half the natural salinity (16.5 PSU) or natural salinity (33 PSU), and all stations showed decreased NA at two- and fivefold the natural salinity (66 PSU and 165 PSU, respectively). At Station I, the response to changing salinities was stronger when NA was measured during the day, especially after 24 h of incubation. At Station II, differences between NA at the five salinities were less pronounced than at the other two stations. Highest NA was recorded at half the natural salinity with the most distinct response recorded at the beginning of the incubation and after 24 h. Station III showed the most pronounced differences in NA at different salinities after 60 h of incubation.
Composition of the active diazotrophic community
Diversity of nifH transcripts, based on the Chao1 richness estimator, varied between the stations and for the different salinities (Table 1). We used a 99% similarity cutoff level to include the microdiversity of nifH. Chao1 diversity richness estimator based on the nifH transcripts predicted highest diversity for Stations I and III at natural and fivefold salinity. At Station II, highest diversity was predicted for natural salinities but decreased again for the highest salinity. Differences in the estimated diversity of the active diazotrophic community at different salinities were highest for Station II.
The active diazotrophic community, as assessed by the nifH transcripts present at the end of the incubations at three of the salinities, showed differences between the salinities at one station as well as between the different stations (Fig. 2). However, the results of the two rounds of sequence analysis per sample differed. These differences were more pronounced for Stations II and III. For Station I, the parallel results showed a higher similarity and are therefore described in more detail. At all stations, most sequences belonged to Cyanobacteria and Proteobacteria. A list of phylotypes identified as the closest relatives to the retrieved nifH sequences is given in Supplement Table 1. Table 2 lists the increase in the relative contributions of major bacterial groups to the nifH transcript libraries.

Relative contributions of major bacterial groups to the nifH transcript libraries for Stations I, II and III at three different salinities. a and b denote the two separate sequence analyses
At Station I, Oscillatoriales-related sequences dominated at freshwater conditions whereas the members of the Chroococcales prevailed at natural salinity (Fig. 2). At the highest salinity, both groups as well as some sequences belonging heterocystous Cyanobacteria (Nostocales) were found, but the contribution of Cyanobacteria to the nifH transcript libraries decreased markedly with increasing salinity (Table 2). Hence, the percentage of proteobacterial sequences in the nifH transcript libraries increased with increasing salinity. At natural salinity, Gammaproteobacteria (Chromatiales) prevailed, whereas Deltaproteobacteria (Desulfomonadales) dominated at the highest salinity.
At Station II, Gammaproteobacteria (Chromatiales) and Deltaproteobacteria (Desulfomonadales) were prominent in the nifH transcript libraries at all salinities (Fig. 2). Among the cyanobacterial nifH transcripts, no heterocystous Cyanobacteria were found. Chroococcales dominated at higher salinities. No clear trend in the relative contributions of major bacterial groups to the nifH transcript libraries could be identified (Table 2).
At Station III, no good-quality RNA could be extracted from the incubation at 0 PSU, and therefore, no analysis of the active diazotrophic community at freshwater conditions was possible. The relative contribution of nifH transcripts belonging to Cyanobacteria (Oscillatoriales and Chroococcales) decreased with increasing salinity. Proteobacterial nifH transcripts, predominantly belonging to Gammaproteobacteria (Chromatiales) and Deltaproteobacteria (Desulfovibrionales), prevailed (Fig. 2). With increasing salinity, there was an increase in deltaproteobacterial nifH transcripts (Table 2).
We also used non-parametric richness diversity estimators to estimate the similarity of the active diazotrophic community of each station at the three different salinities. We compared the fraction of shared OTUs between the salinities and the fraction of sequences belonging to shared OTUs (Table 3). Based on this overlap of OTUs, the active diazotrophic communities at all stations were more similar to each other at natural and fivefold the natural salinity than under freshwater and natural conditions.
Discussion
We analyzed the response of N2 fixation and diazotrophic community composition to the changes in the ambient salinity in three microbial mats. Studies of this kind are rare and have been limited to hypersaline microbial mats (Pinckney et al. 1995; Yannarell et al. 2006).
The difference in nitrogenase activity (NA) rates per station and salinity observed for the different time points results from the fact that measurements were taken both at day and at night. The highly variable daily NA pattern which has been observed for these and other diazotrophic microbial mats (Omoregie et al. 2004a, b; Steppe and Paerl 2005; Severin and Stal 2008) results in different NA rates depending on the time of the measurement, even if environmental conditions, for example, temperature, are kept constant in laboratory experiments (e.g., Severin and Stal 2008).
At Station I, the mat highest up in the littoral zone, highest NA at salinities ranging from freshwater to ambient seawater salinity demonstrates the adaptation to conditions likely to occur at this station but also the intolerance of the present diazotrophic community with regard to elevated salinities. The shift within the active diazotrophic community was more pronounced for the transition from freshwater to natural salinity than from natural to fivefold the natural salinity. This illustrates that the diazotrophic fraction of the community best adapted to each of these salinities was most active and ensured high NA within the natural salinity range at this station. Such plasticity is not expected for high salinities that occur only episodically, and this is consistent with the observation of decreasing NA at high salinities. The active diazotrophic community which is presumably responsible for high NA under freshwater and seawater conditions showed a remarkable change within the most common groups, Cyanobacteria and Proteobacteria. Members of the structurally dominant Oscillatoriales seemed to be better adapted to lower salinities, whereas the members of the Chroococcales contributed the major part of cyanobacterial nifH transcripts at ambient salinity. Hypersaline microbial mats in different environments contain filamentous as well as unicellular forms (e.g., Fourçans et al. 2004; Yannarell et al. 2006), but there is some evidence that unicellular Cyanobacteria become more important at elevated salinities, probably due to the ability to synthesize quaternary ammonium compounds that serve as compatible solutes that maintain the cell’s osmotic pressure (e.g., Garcia-Pichel et al. 1998 and references therein). In the mats investigated in this study, gammaproteobacterial and deltaproteobacterial nifH transcripts dominated the clone libraries at higher salinities. Proteobacteria are common member of intertidal microbial mat communities and have been shown to contribute substantially to the nifH gene pool (Olson et al. 1999; Bauer et al. 2008) and to the part of the diazotrophic community that is actually expressing nifH (Steppe and Paerl 2002, 2005). Their prevalence in the nifH transcript libraries at hypersaline conditions is also in line with their presence and activity in hypersaline microbial mats (Omoregie et al. 2004a, b). Nevertheless, it has been shown that higher salinities correlate with lower NA in these types of mats as well (Pinckney et al. 1995). This is generally attributed to bioenergetic aspects (for a review see Oren 1999). The synthesis and/or uptake of compatible solutes to balance the osmotic pressure of the environment as well as transporting sodium ions out of the cytoplasm are energetically expensive processes, which might restrict other energy-demanding metabolic processes such as the fixation of N2. It is therefore plausible that the supply of energy constrains N2 fixation and could be the main reason for a decrease in NA under increased salinities.
At Station II, we observed smaller changes in NA at the different salinities. Since this station was situated close to the low water mark and therefore regularly inundated by seawater, a selection for a halotolerant diazotrophic community due to more frequent changes in salinity during the tidal cycle could have taken place. Differences in the community composition of the three mat types are likely to be attributed to their position along the tidal gradient and the associated differences in salinity (e.g., Bolhuis and Stal 2011). The observation that highest NA occurred at half the natural salinity rather than natural salinity is probably due to the activity of diazotrophs with a broad halotolerance. At lower salinities, these organisms spend less energy for the synthesis of quaternary ammonium compounds that serve as compatible solutes and hence can devote more energy and reducing equivalents to fuel nitrogenase activity. NA in general was slightly lower at Station II than at Stations I and III, likely caused by the low contribution of Cyanobacteria to the nifH transcript libraries, that is, to the active diazotrophic community, at all salinities.
Station III showed no initial response to altered salinity. This might have been caused by the stronger cohesiveness of this mat compared to the other two mat types. It therefore might have taken longer to reach equilibrium in this mat type. Subsequently, the response to altered salinity was similar for all time points in this mat, regardless of the time of the day of the measurement. This could indicate i) a similar active diazotrophic community at day and night or ii) a similar performance with respect to NA by different active diazotrophic communities. An exception was the NA measured after 60 h of incubation. The reason for that is unknown. We were unable to analyze the active diazotrophs at freshwater conditions, but the community shift from natural salinity to hypersaline conditions demonstrated a more or less stable contribution of major bacterial groups to the nifH transcript libraries.
The presumably active diazotrophic community as inferred from the nifH transcript libraries was reproducible for Station I but not for Stations II and III (Fig. 2). This can most likely be attributed to the fact that our sampling effort with respect to the amount of sequences retrieved in both of the analysis rounds was not sufficient, as indicated by rarefaction curves (not shown). Deviating sets of sequences retrieved for the same sample at the two instances result in different images of the active diazotrophic community (Fig. 2) and made it difficult to identify trends in the change of community composition with changing salinities at Stations II and III (Table 2). Based on the deviating sets of sequences, also the separate analyses of alpha- and beta-diversity of these sequences differed (Supplement Tables 2 and 3). When thought appropriate, we presented the results of the combined analyses to achieve a more comprehensive image of the diversity in the three mat types, but the above-mentioned differences should be kept in mind.
The diversity of nifH-OTUs at 99% similarity predicted by the Chao1 diversity indicator was highest at natural salinities for two of the three stations. It might be expected that diversity of an active community is higher at natural conditions as opposed to occasionally occurring extreme environmental conditions which are likely to render some parts of this community inactive. At Station I, however, diversity was slightly higher at fivefold than at natural salinities. This could account for an increase in microdiversity. We chose the cutoff level of 99% sequence similarity to account for possible Taq errors but allow the consideration of microdiverse clusters as observed in the majority of marine microbial communities (Acinas et al. 2004; Klepac-Ceraj et al. 2004; Haverkamp et al. 2008). A decrease in overall diversity but an increase in microdiversity with increasing salinity has also been observed for the bacterial community of a solar saltern (Benlloch et al. 2002) and a soda lake (Foti et al. 2008). This phenomenon might hence also apply for the active diazotrophic community of microbial mats.
In conclusion, mats naturally exposed to smaller and/or more frequent salinity changes showed a more variable active diazotrophic community and a stronger response to decreased or increased salinities with regard to NA than mats situated in a zone with a larger range in natural salinity. In general, the diazotrophic community responded better to salinities that likely occur in nature than to those beyond the normal salinity range. We could show that for one of the stations expression of nifH that accompanied low NA at higher salinities was dominated by members of the Proteobacteria, mainly Gammaproteobacteria and Deltaproteobacteria. However, their contribution to whole mat NA remains unresolved, because gene expression does not necessarily coincide with activity (Severin and Stal 2010b). In all three microbial mats, salinity was an important factor shaping the active diazotrophic community and consequently also NA.
References
Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF (2004) Fine scale phylogenetic architecture of a complex bacterial community. Nature 430:551–554
Bauer K, Diez B, Lugomela C, Seppälä S, Borg AJ, Bergman B (2008) Variability in benthic diazotrophy and cyanobacterial diversity in a tropical intertidal lagoon. FEMS Microbiol Ecol 63:205–221
Benlloch S, López-López A, Casamayor EO, Øvreas L, Goddard V, Daae FL, Smerdon G, Massana R, Joint I, Thingstad F, Pedros-Alio C, Rodriguez-Valera F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol 4:349–360
Bolhuis H, Stal LJ (2011) Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J 5:1701–1712
Bouvier TC, del Giorgio PA (2002) Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol Oceanogr 47:453–470
Edmonds JW, Weston NB, Joye SB, Mou XZ, Moran MA (2009) Microbial community response to seawater amendment in low-salinity tidal sediments. Microb Ecol 58:558–568
Fernandes TA, Iyer V, Apte K (1993) Differential responses of nitrogen-fixing cyanobacteria to salinity and osmotic stresses. Appl Environ Microbiol 59:899–904
Foti MJ, Sorokin DY, Zacharova EE, Pimenov NV, Kuenen JG, Muyzer G (2008) Bacterial diversity and activity along a salinity gradient in soda lakes of the Kulunda Steppe (Altai, Russia). Extremophiles 12:133–145
Fourçans A, de Oteyza TG, Wieland A, Solé A, Diestra E, van Bleijswijk J, Grimalt JO, Kühl M, Esteve I, Muyzer G, Caumette P, Duran R (2004) Characterization of functional bacterial groups in a hypersaline microbial mat community (Salins-de-Giraud, Camargue, France). FEMS Microbiol Ecol 51:55–70
Fu FX, Bell PRF (2003) Effect of salinity on growth, pigmentation, N2 fixation and alkaline phosphatase activity of cultured Trichodesmium sp. Mar Ecol Prog Ser 257:69–76
Garcia-Pichel F, Nübel U, Muyzer G (1998) The phylogeny of unicellular, extremely halotolerant cyanobacteria. Arch Microbiol 169:469–482
Haverkamp T, Acinas SG, Doeleman M, Stomp M, Huisman J, Stal LJ (2008) Diversity and phylogeny of Baltic Sea picocyanobacteria inferred from their ITS and phycobiliprotein operons. Environ Microbiol 10:174–188
Klepac-Ceraj V, Bahr M, Crump BC, Teske AP, Hobbie JE, Polz MF (2004) High overall diversity and dominance of microdiverse relationships in salt marsh sulphate-reducing bacteria. Environ Microbiol 6:686–698
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM, Maresca JA, Bryant DA, Sogin ML, Pace NR (2006) Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72:3685–3695
Lozupone CA, Knight R (2007) Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104:11436–11440
Olson JB, Litaker RW, Paerl HW (1999) Ubiquity of heterotrophic diazotrophs in marine microbial mats. Aquat Microb Ecol 19:29–36
Omoregie EO, Crumbliss LL, Bebout BM, Zehr JP (2004a) Comparison of diazotroph community structure in Lyngbya sp and Microcoleus chthonoplastes dominated microbial mats from Guerrero Negro, Baja, Mexico. FEMS Microbiol Ecol 47:305–318
Omoregie EO, Crumbliss LL, Bebout BM, Zehr JP (2004b) Determination of nitrogen-fixing phylotypes in Lyngbya sp and Microcoleus chthonoplastes cyanobacterial mats from Guerrero Negro, Baja California, Mexico. Appl Environ Microbiol 70:2119–2128
Oren A (1999) Bioenergetic aspects of halophilism. Microbiol Mol Biol Rev 63:334–348
Pinckney J, Paerl HW, Bebout BM (1995) Salinity control of benthic microbial mat community production in a Bahamian hypersaline lagoon. J Exp Mar Biol Ecol 187:223–237
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schloss PD, Handelsman J (2006) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72:6773–6779
Severin I, Stal LJ (2008) Light dependency of nitrogen fixation in a coastal cyanobacterial mat. ISME J 2:1077–1088
Severin I, Stal LJ (2010a) Microbial mats: diazotrophic microbial mats. In: Seckbach J, Oren A (eds) Cellular origin, life in extreme habitats and astrobiology. Springer, Berlin/Heidelberg, pp 323–339
Severin I, Stal LJ (2010b) NifH expression by five groups of phototrophs compared to nitrogenase activity in coastal microbial mats. FEMS Microbiol Ecol 73:55–67
Stal LJ (2000) Mats and stromatolites. In: Whitton A, Potts M (eds) The ecology of Cyanobacteria: their diversity in time and space. Kluwer, Dordrecht, pp 61–120
Steppe TF, Paerl HW (2002) Potential N2 fixation by sulfate-reducing bacteria in a marine intertidal microbial mat. Aquat Microb Ecol 28:1–12
Steppe TF, Paerl HW (2005) Nitrogenase activity and nifH expression in a marine intertidal microbial mat. Microb Ecol 49:315–324
Yannarell AC, Steppe TF, Paerl HW (2006) Genetic variance in the composition of two functional groups (diazotrophs and cyanobacteria) from a hypersaline microbial mat. Appl Environ Microbiol 72:1207–1217
Zani S, Mellon MT, Collier JL, Zehr JP (2000) Expression of nifH genes in natural microbial assemblages in Lake George, New York, detected by reverse transcriptase PCR. Appl Environ Microbiol 66:3119–3124
Zehr JP, McReynolds LA (1989) Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl Environ Microbiol 55:2522–2526
Zehr JP, Mellon M, Braun S, Litaker W, Steppe TF, Paerl HW (1995) Diversity of heterotrophic nitrogen fixation genes in a marine cyanobacterial mat. Appl Environ Microbiol 61:2527–2532
Acknowledgments
This work was financially supported by the Netherlands Research Council (NWO) contract ALW 815.01.003 and the Schure-Beijerinck-Popping fund (KNAW). This is publication 5188 of the Netherlands Institute of Ecology (NIOO-KNAW).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Joerg Overmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Severin, I., Confurius-Guns, V. & Stal, L.J. Effect of salinity on nitrogenase activity and composition of the active diazotrophic community in intertidal microbial mats. Arch Microbiol 194, 483–491 (2012). https://doi.org/10.1007/s00203-011-0787-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-011-0787-5