
Abstract
Mini abstract
Real-world evidence on the comparative effectiveness and safety of abaloparatide versus teriparatide in women with osteoporosis may help inform treatment decisions. Following 18 months of treatment, abaloparatide was comparable to teriparatide for prevention of nonvertebral fractures, resulted in a 22% risk reduction for hip fractures, and demonstrated similar cardiovascular safety.
Summary
Osteoporotic fracture risk can be reduced with anabolic or antiresorptive medications. In addition to efficacy and safety data from controlled clinical trials, real-world evidence on comparative effectiveness and safety may help inform treatment decisions.
Introduction
The real-world effectiveness of abaloparatide versus teriparatide on nonvertebral fracture (NVF) incidence and cardiovascular safety during the 19-month period after treatment initiation were evaluated (NCT04974723).
Methods
Anonymized US patient claims data from Symphony Health, Integrated Dataverse (IDV)®, May 1, 2017 to July 31, 2019, included women aged ≥ 50 years with ≥ 1 prescription of abaloparatide or teriparatide and no prior anabolic therapy. Most were enrolled in commercial and Medicare health plans. Index was the date of the initial prescription dispensed during the identification period. In 1:1 propensity score matched cohorts, time to first NVF following index date, major adverse cardiovascular events (MACE), and MACE + heart failure (HF) were compared between cohorts using a Cox proportional hazards model.
Results
Propensity score matching yielded 11,616 patients per cohort. Overall median age (interquartile range) was 67 (61, 75) years, and 25.6% had a fracture history. Over 19 months, 335 patients on abaloparatide and 375 on teriparatide had a NVF (hazard ratio [95% confidence interval]: 0.89 [0.77, 1.03]), and 121 and 154 patients, respectively, had a hip fracture [HR (95% CI): 0.78 (0.62, 1.00)]. The MACE and MACE + HF rates were similar between cohorts.
Conclusions
Following 18 months of treatment, abaloparatide was comparable to teriparatide for prevention of NVF and similar cardiovascular safety was demonstrated between cohorts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Background and objectives
Osteoporosis is a skeletal disease characterized by low bone mass, deterioration of bone tissue, and disruption of bone microarchitecture, associated with increased risk of fractures [1]. Approximately 10 million adults in the US have osteoporosis [2]. Two million fractures occur in the USA annually with the estimated lifetime fracture risk of 50% for women 50 years and older [3, 4]. Osteoporotic fractures are associated with functional decline, loss of independence, and reduced health-related quality of life [5, 6]. Furthermore, the economic burden of fractures, including incremental cost of secondary fractures, is significant and on the rise, with hip fractures accounting for the majority of the cost of care [7, 8].
Anabolic drugs, which stimulate new bone formation, and can potentially improve bone microarchitecture, are available as treatment options for individuals with osteoporosis at high risk for fracture [9]. These include patients with a history of osteoporotic fracture, multiple risk factors for fracture, or those with treatment failure on other available therapies. These anabolic drugs include teriparatide [10], a first-in-class anabolic agent that received US Food and Drug Administration (FDA) approval in 2002 and abaloparatide [11], approved by the FDA in 2017. Both drugs are self-administered by daily subcutaneous (SC) injection. Abaloparatide is a novel synthetic analogue of human parathyroid hormone-related peptide [hPTHrP (1–34)], selective for the parathyroid hormone type 1 (PTH1) receptor. Abaloparatide has higher affinity for the RG versus R0 conformation of the PTH1 receptor compared with teriparatide, resulting in more transient receptor signaling consistent with a net anabolic effect [12].
In a large, randomized, phase 3, multicenter, multinational clinical trial (Abaloparatide Comparator Trial in Vertebral Endpoints [ACTIVE]), postmenopausal women with osteoporosis were randomized to 18 months of treatment with abaloparatide 80 μg SC daily, open-label teriparatide 20 μg SC daily, or placebo. Treatment reduced the risk of vertebral (VF), nonvertebral (NVF), clinical, and major osteoporotic fractures versus placebo, independent of baseline risk [13]. The benefits observed with 18 months of abaloparatide during ACTIVE on fracture risk reduction were extended for an additional 2 years with subsequent alendronate treatment in the ACTIVExtend trial, supporting the concept of sequential therapy with an anabolic followed by an antiresorptive agent [14].
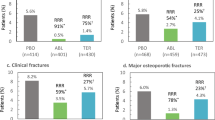
The relative efficacy of abaloparatide compared with other treatment options for fracture risk reduction in women with postmenopausal osteoporosis (PMO) was previously assessed using a network meta-analysis [15]. For VF, abaloparatide had the greatest effect relative to placebo (relative ratio [RR] 0.13; 95% credible interval [CrI]: 0.04, 0.34) compared to teriparatide relative to placebo (RR 0.27; 95% CrI: 0.20, 0.37). For NVF, abaloparatide produced a greater risk reduction versus placebo (RR 0.50; 95% CrI: 0.28, 0.85) compared to teriparatide relative to placebo (RR 0.62; 95% CrI: 0.47, 0.82). Consistent findings have been reported in more recent publications [16, 17].
In addition to clinical outcomes reported from randomized controlled trials (RCT), real-world evidence is important in guiding treatment decisions [18]. An evaluation of real-world effectiveness provides data on a broader population of patients than those who typically meet inclusion/exclusion criteria in randomized controlled trials.
Several approved osteoporosis treatments have been implicated in increasing the risk of cardiovascular (CV) and cerebrovascular events or the composite endpoint of major adverse cardiac events (MACEs) (including myocardial infarction [MI], stroke, and CV death). Hormone therapy and selective estrogen receptor modulators are associated with increased risk of venous thrombosis, and, in some cases, CV disease and stroke [19, 20]. In the pivotal trial investigating the efficacy and safety of odanacatib, the MACE rate was higher with odanacatib versus placebo, with significant differences for risk of stroke [21]. Romosozumab was associated with an increased risk of MACE compared to alendronate, though no difference was seen between romosozumab and placebo [22]. In the ACTIVE trial, the rates of serious cardiac adverse events were similar among the three groups (abaloparatide, placebo, teriparatide), and time to first MACE or MACE plus heart failure event were longer with both abaloparatide and teriparatide compared to placebo [23]. The use of abaloparatide has been associated with transient and reversible increases in heart rate after injection. No published epidemiological studies have examined the CV risk associated with transitory, intermittent increases in heart rate due to an external intervention, as is the case with abaloparatide and teriparatide administration, in the general population and in the target population of postmenopausal women. Because of the postmenopausal population and the potential common etiology or shared risk factors (e.g., age, smoking) between PMO and CV [24], the current study also included an evaluation of new CV events.
The objective of the current study was to evaluate the real-world comparative effectiveness on NVF and comparative CV safety of abaloparatide versus teriparatide during the 19-month period after treatment initiation in propensity score-matched cohorts (NCT04974723).
Methods
Study design
This was a retrospective observational study using anonymized patient claims data from Symphony Health, Integrated Dataverse (IDV)®, May 1, 2017 to July 31, 2019. The database included enhanced hospital data, which are claims and remittance from inpatient hospital setting and proprietary Patient Transactional Dataset claims, prescription data, and mortality data from hospital discharge records. Prescription, medical, and hospital claims, including diagnosis and procedure details, are enhanced with data from electronic medical records, lab centers, patient registries, and pharmacies across the USA and its territories. Data are payer agnostic and provide access to individual-level healthcare claims for more than 317 million US-based commercial and Medicare patients[25].
The index date was defined as the date of initial prescription dispensed for either abaloparatide or teriparatide during the identification period (May 1, 2017 and July 31, 2019) corresponding with the FDA approval of abaloparatide. Patients were assigned to a cohort based on their index anabolic therapy. Data were used as far back as available (May 01, 2012) prior to the index date and included the use of most recent data following treatment initiation (January 31, 2021) (Fig. 1).

Study design and timeline
The pre-index period consists of the 5 years before the index date during which medical and treatment history were available for the patient. The post-index treatment period consists of the 18 months after the index date with the maximum evaluation period of 18 months plus 30-day follow-up (19 months). The evaluation of treatment effectiveness started immediately after treatment initiation and continued for 18 months plus 30-day follow-up after the index date. The evaluation of CV safety outcomes started immediately after treatment initiation and continued while on therapy (until end of treatment) for up to 18 months plus 30-day follow-up.
Study population
The study included women ≥ 50 years of age with ≥ 1 new prescription fill of abaloparatide or teriparatide during the identification period, ≥ 1 claim for a medical or hospital visit, and a pharmacy claim in the 12 months before index date. Patients with a diagnostic claim for Paget’s disease of the bone or malignancy (except for non-melanoma skin cancers, carcinoma in situ of the cervix, ductal carcinoma in situ of breast) at baseline, those with Charlson Comorbidity Index > 10, prior index anabolic therapy, or who switched to a different anabolic treatment after index were excluded.
Propensity score matching
In the absence of randomization, logistic regression-based propensity score matching was used to create the analytic cohorts from all patients meeting the study inclusion/exclusion criteria. A greedy matching algorithm with no replacement was adopted with a caliper width equal to 0.20 times the standard deviation of the logit of the propensity. Cohorts were prospectively specified to match on 73 variables including age, prior fracture history, chronic comorbidities, and prior osteoporosis medications (Appendix A) [26]. The R software MatchIt package [27] was used to find matched pairs. Both prematch and postmatch balance between 2 treatment cohorts were evaluated using standardized difference for each covariate category to ensure that propensity score matching was accepted (i.e., the standardized difference on each covariate between abaloparatide and teriparatide of < 0.10) [28].
Effectiveness and safety endpoints
The primary endpoint was time to first NVF event (hip, pelvis, shoulder [including clavicle and humerus], radius/ulna [including radius and/or ulna and forearm], wrist [including unspecified wrist, wrist/hand, carpal, triquetrum, lunate, capitate, hamate, pisiform, etc.], femur, tibia/fibula, and ankle) within 18 months plus 30-day follow-up after treatment initiation. The secondary endpoints included time to the first composite endpoint of MACE (nonfatal MI, nonfatal stroke, or CV death) with and without heart failure following hospitalization within the 18 months after treatment initiation while on therapy plus 30-day follow-up. The exploratory effectiveness endpoint was time to first hip fracture within 18 months plus 30-day follow-up after treatment initiation. Exploratory safety endpoints included time to first event for MI, stroke, CV death following hospitalization since anabolic treatment initiation, and heart failure while on therapy.
A claim-based validated algorithm with high specificity, which was shown to have over 90% accuracy in previous studies, was used to identify osteoporosis-related fractures [29]. For evaluation of mortality, hospital discharge status was used. Hospital claims do not specify the cause of death nor causal association with a specific medication. We therefore used a previously validated claims-based algorithm to derive hospital CV death (indirect approach 2 described by Xie et al.) [30]. Compared to a previously published fatal MI and stroke method [31], the algorithm we adopted has higher sensitivity while maintaining high specificity improving the net reclassification index. Cardiovascular event was derived from the International Classification of Diseases, 10th Revision, Clinical Modification (ICD-10-CM) codes for MI (I21.x, I22.x), stroke (I61.x-I63.x), and heart failure (I50.x, excluding I50. × 2, I50.8x) consistent with the FDA Mini-Sentinel coding for these events [32].
Statistical analysis
The analysis population consisted of all the patients who met the study inclusion/exclusion criteria who were selected after propensity score matching. The same matched population was used for both effectiveness and safety analyses. The primary analysis of effectiveness based on the time to first NVF event was of the noninferiority of abaloparatide to teriparatide, as measured by the hazard ratio (HR). Noninferiority of abaloparatide to teriparatide was concluded if the upper bound of the 2-sided 95% confidence interval (CI) of the HR between abaloparatide versus teriparatide was < 1.3. Assuming the NVF rate for teriparatide was 3.5% at the end of 18 months [13, 33], a sample size of 8000 matched samples in each treatment cohort was thought to achieve at least 95% power at a 0.05 significance level to estimate the equivalence HR of 1.3 when the actual HR is an equivalence HR of 1.0.
Effectiveness evaluation was conducted using intent-to-treat (ITT) analysis reporting the first fracture event during the 18 months plus 30 days of follow-up from index treatment initiation regardless of when the treatment was discontinued. Comparisons of the time to first fracture between the propensity score matched treatment cohorts were based on a Cox proportional hazards model. P values were obtained from the log-rank test. The HR and 95% CI between the two treatment cohorts were calculated. Duration, in days, from the index date to the last follow-up date was calculated. Comparative effectiveness of therapy was evaluated in a subgroup of patients considered to be at high risk for fracture, including those ≥ 75 years of age, those with prior fracture within 1 year of index date, and those with prior bisphosphonate use within 5 years prior to index date.
The as-treated (AT) analysis was conducted for the safety evaluation, regardless of the anabolic drug gap between two prescription fills. Observation period was for up to 18 months while on treatment plus 30-day follow-up, or until their first CV event or hospital death, whichever came first. Time to first CV event after the index date and within 30 days after the end of treatment was analyzed.
Sensitivity analyses
To evaluate the stability of the propensity score–matched cohorts, the sensitivity analyses on effectiveness and safety endpoints were performed on two additional matching populations using two different calipers (0.15 and 0.3). For effectiveness evaluation, additional sensitivity analyses included anabolic treatment duration (cumulative and consecutive) and treatment response in patients without prior use of denosumab or zoledronic acid. Cumulative and consecutive treatment duration were determined from index date to the last drug supply date regardless of treatment gap (cumulative) and without any gap exceeding 60 days (consecutive). For safety evaluation, sensitivity analysis included an evaluation of outcomes for patients by baseline CV risk factors. To evaluate possible overestimation of new CV events, additional sensitivity analyses excluded diagnosis of CV events in the 183 days preceding the index date according to the sentinel initiative [32].
Results
Matching
Among women ≥ 50 years of age with ≥ 1 new prescription fills of abaloparatide or teriparatide during the identification period (abaloparatide, N = 17,958; teriparatide N = 61,914), 24% in each treatment cohort were ineligible due to not having a medical, hospital visit, or pharmacy claim in the 12 months before index date. Eight percent more in the abaloparatide cohort and 48% of the remaining patients in the teriparatide cohort were excluded because they had a history of anabolic treatment before the index date. A total of 11,617 patients in the abaloparatide cohort and 22,809 patients for teriparatide met all eligibility criteria. Propensity score matching yielded 11,616 patients in each treatment cohort (Table 1). After matching, there was a similar distribution of the propensity score between the two treatment groups, indicating successful matching (Fig. 2). All prespecified variables were well balanced with a standardized mean difference < 0.10 (Table 2).

Distribution of the propensity score before and after propensity score matching
Overall median and interquartile range of age was 67 (61, 75) years old, 25.6% had a history of fracture, and 16.2% had a fracture in the year preceding anabolic treatment initiation (Table 2). On average, abaloparatide and teriparatide patients were diagnosed with osteoporosis 2.8 (± 2.2) years prior to treatment initiation and 45.6% of patients had prior antiresorptive use. The most common comorbid conditions for both cohorts were CV disease (76.7%), arthritis (46.7%), respiratory disease (42.3%), and gastrointestinal disorders (38.4%). The majority (72.4%) had a history of falls, or one or more conditions associated with increased risk for falls.
Exposure
The overall mean duration of abaloparatide and teriparatide exposure was 301.2 and 313.4 days, respectively, with > 45% of patients in both treatment cohorts exposed to treatment > 12 months (Table 3). The mean cumulative duration of abaloparatide and teriparatide exposure was 257.8 and 269.2 days, respectively, with > 33% of patients in both treatment cohorts exposed to treatment > 12 months. The percentage of patients in both treatment cohorts who were exposed to > 12 months of consecutive treatment was > 34%.
Analysis of time to first fracture event
The estimated new NVF rate was comparable for abaloparatide versus teriparatide (2.9% vs 3.2%; HR [95% CI]: 0.89 [0.77, 1.03], P = 0.13), and the risk for hip fractures was reduced 22% (0, 33%) for abaloparatide (new event rate, 1.0% vs 1.3%; HR [95% CI]: 0.78 [0.62, 1.00], P = 0.04) (Table 4 and Fig. 3). Noninferiority for abaloparatide versus teriparatide on time to the first NVF was established since the upper bound of 2-sided 95% CI of the HR between abaloparatide and teriparatide was 1.03 and less than the prespecified 1.3. Outcomes were consistent among all subpopulations in the sensitivity analyses. There was no difference in effectiveness between the two treatment cohorts with variation in matching caliper, for the various durations of treatment, or when excluding patients with prior use of denosumab or zoledronic acid for NVF or hip fractures with one exception (Supplemental Figs. 1a and 1b). For hip fracture sensitivity analyses, when limiting the analyses to patients with > 12 months of consecutive exposure to treatment, the HR (95% CI) was in favor of abaloparatide 0.57 (0.35, 0.94).

a) Time to event of nonvertebral fractures. b) Time to event of hip fractures. CI, confidence interval. aPatients at risk include all patients regardless of when treatment was discontinued, except those who had a fracture event or died
Effectiveness subgroup analyses
Both NVF and hip fractures were higher in the pre-specified subgroups considered to be at high risk for fracture with comparable effectiveness for both treatment cohorts. For patients ≥ 75 years of age (N = 2865), the estimated new NVF event rates were 3.4% for abaloparatide and 4.8% for teriparatide patients (HR [95% CI]: 0.70 [0.54, 0.90]) and hip fracture rates were in favor of abaloparatide versus teriparatide (1.4% vs 2.0%) (HR [95% CI]: 0.69 [0.46, 1.05]). For patients with prior fracture within 1 year of index date (N = 1876), the estimated new events were 6.6% vs 6.1% for NVF (HR [95% CI]: 1.08 [0.84, 1.39]) and 2.2% versus 3.0% for hip fractures (HR [95% CI]: 0.75 [0.50, 1.11]), for abaloparatide versus teriparatide, respectively. Finally, for patients with prior antiresorptive use (N = 5313), the estimated new event rates were 3.2% versus 3.3% for NVF fractures (HR [95% CI]: 0.96 [0.78, 1.18]) and 1.2% versus 1.3% for hip fractures (HR [95% CI]: 0.90 [0.64, 1.26]) for abaloparatide versus teriparatide patients.
Analysis of time to first CV event
The K-M estimated event rates of the composite endpoints of MACE were similar for the abaloparatide (3.0%) versus teriparatide (3.1%) cohorts with comparable risk of new events (HR [95% CI]: 1.00 [0.84, 1.20], P = 0.97). Consistent results were also observed for MACE including heart failure with abaloparatide (6.6%) versus teriparatide (6.4%) with comparable risk of new events (HR [95% CI]: 1.05 [0.93, 1.19], P = 0.41). Results persisted in the sensitivity analyses (Supplemental Figs. 2a, 2b, and 2c).
Discussion
This was the first real-world comparative study of abaloparatide versus teriparatide with the objectives of comparing effectiveness against NVF and CV safety. Propensity matching identified very similar cohorts totaling 23,000 women. Over 19 months of follow-up after the index prescription date, the NVF event rate was numerically lower with abaloparatide versus teriparatide (2.9% vs 3.2%, P = not significant) and the hip fracture event rate was also lower (1.0% with abaloparatide vs 1.3% with teriparatide; P < 0.05). The risks for MACE and MACE + HF were comparable for abaloparatide versus teriparatide cohorts. The efficacy findings were similar in predefined subgroups at particularly high risk for fracture (including women above age 75). The findings suggest a benefit to risk balance for abaloparatide similar to or better than that of teriparatide.
The risk reduction for NVF in this clinical practice setting with abaloparatide versus teriparatide (HR [95% CI]: 0.89 [0.77, 1.03]) was comparable to that reported in the ACTIVE trial (HR [95% CI]: 0.79 [0.43, 1.45]) [13]. Additionally, although ACTIVE and the pivotal teriparatide fracture prevention study were not powered to assess the effects of treatment on hip fracture, the pattern in hip fracture rates in the current study is similar to that reported in previous real-world studies with teriparatide [13, 33, 34].
Although information on bone mineral density (BMD) changes was not available from the claims database in this real-world study, abaloparatide treatment in ACTIVE and two subsequent studies in a subset of participants from ACTIVE increased BMD significantly more at the total hip and femoral neck compared to teriparatide [13, 35, 36]. In both preclinical and clinical studies, teriparatide is associated with increased cortical porosity [37,38,39,40]. Abaloparatide does not increase cortical porosity in preclinical animal models [41, 42]. Consistent with the preclinical data, a post hoc analysis of hip DXA data from ACTIVE using 3D modeling suggests that previously reported differences in areal BMD between abaloparatide and teriparatide may be due to a greater improvement in cortical volumetric BMD of the total hip [36]. Abaloparatide produced greater increases, compared with teriparatide, in cortical volumetric BMD and corresponding biomechanical parameters of the femoral neck, shaft, and trochanter subregions of the hip which might explain, in part, the lower hip fracture risk for abaloparatide versus teriparatide in the current study [35].
The risks for MACE and MACE + HF were comparable for abaloparatide and teriparatide cohorts and results were consistent in the exploratory analyses of individual endpoints of MI, stroke, CV death following hospitalization, and HF. CV event rates reported here in the real-world setting were higher than those reported in the ACTIVE study (MACE: abaloparatide [0.5%] vs teriparatide [0.6%] and MACE + HF: abaloparatide [0.5%] vs teriparatide [0.6%]) [23]. This was expected since a broader population of patients were in the current study with more comorbidities. Furthermore, we may have overestimated the acute event rate since both office visits and all (primary and secondary) hospital diagnosis codes were used to identify CV events, leading to potential redundancy in counting events. A review of the literature, the FDA Adverse Event Reporting System (FAERS) database, and the results from the ACTIVE trial did not identify a signal for serious CV events for either abaloparatide or teriparatide. In fact, in the ACTIVE trial, both abaloparatide and teriparatide groups were associated with a longer time to MACE and MACE + HF than the placebo group.
Lastly, evidence suggests an increased risk for CV events in women with PMO [24]. The inverse relationship between bone density and coronary heart disease risk is supported by reports that postmenopausal women with low BMD values have a greater prevalence and severity of aortic calcification, a predictor of CV disease and mortality [43].
The study results have to be considered within the context of several limitations. First, the data source was administrative claims data, which was not collected for research purposes. Administrative claims data have inherent limitations including coding errors, inconsistencies, outcome misclassifications, or incomplete diagnoses data. Compliance (treatment exposure) cannot be assessed. Cardiovascular events were not adjudicated. In addition, due to data limitations, only mortality recorded on a hospital discharge form was included in the analyses. The current study, however, used a claims-based algorithm with high specificity to identify case-qualifying fractures associated with osteoporosis [29]. The algorithm included the majority of osteoporosis-related fractures but did not include rib fractures, since they are generally difficult to confirm and often do not result in a healthcare encounter. Any misclassification of fractures is likely to be nondifferential between the treatment cohorts compared and should not impact the results. Another limitation of claims-based studies includes potential inaccuracies related to the use of prescription medications. The prescription claim is for the date of fill and not the date of use of the medications, so the assumption was made that these were the same. Detailed clinical data such as BMD values were not available, and unknown confounding factors (e.g., family history, smoking status, alcohol intake) were not adjusted for in propensity score matching. Lastly, in the absence of full medical and treatment history, baseline comorbidity burden in the current population may be underreported. Real-world patient cohorts are likely to include patients with a broader range of comorbidities who would not be eligible to participate in randomized controlled trials [18]. Therefore, caution must be exercised when comparing these results with previous randomized controlled trials.
The current study was observational, and treatments were not assigned. As such, randomization was not possible. Although this was not a randomized study, propensity score matching was used to define the study cohorts and provided confidence that the two treatment groups were comparable in their probability to receive and benefit from treatment. We matched patients on all indicators of disease severity and fracture risk, including prior disease and treatment history for which data were available. Furthermore, we also matched on history of falls, comorbid conditions, and prior osteoporosis medications, or with poor bone quality and strength, and baseline CV risk factors for safety evaluation. This method allowed us to control for known but not unknown confounders. Lastly, there could be residual confounding despite matching.
This observational cohort study examined the comparative effectiveness and CV safety for abaloparatide versus teriparatide. The study design adheres to the guidelines for conduct of comparative effectiveness and safety evaluation with a prespecified protocol and analysis plan, which included consideration of potential biases related to measurement of exposure, outcomes, and confounders [44,45,46]. To address data limitations, the study included new anabolic users, highly specific endpoints, and several sensitivity analyses to test the robustness of findings. Furthermore, comparison of two similar drugs has the advantage to reduce the bias associated with unknown confounders as well as those known confounders not available in claims data (i.e., BMD), given similar market access requirement and place in therapy according to clinical practice guidelines.
In this retrospective real-world database study of patients initiating treatment with abaloparatide or teriparatide, abaloparatide was comparable to teriparatide in the prevention of NVF, resulted in fewer hip fractures and demonstrated similar CV safety. Results of the study are generalizable to the population of managed care enrollees, including commercial and Medicare members. The data are representative of a broad population of patients from multiple payers and are geographically diverse. The study provides additional information on real-world use and outcomes in patients new to abaloparatide outside of the clinical study setting.
Data availability
The source data for this study were licensed from a third party, by Radius Health, Inc (Radius). Although we are not permitted to share the licensed data publicly, the same data used in this study are available for others to license by contracting with the database owners. Radius licensed data from Symphony Health, Integrated Dataverse (IDV)®, May 2012 to January 2021, which included anonymized patient level data from pharmacy claims linked to commercial and Medicare medical claims data. Symphony Health licensing information can be found at www.symphonyhealth.com with the database owners. The authors did not have any special access privileges that other parties who license the data and contract with Symphony would not have.
References
Cosman F, de Beur SJ, LeBoff MS et al (2014) Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int 25(10):2359–2381. https://doi.org/10.1007/s00198-014-2794-2
Wright NC, Looker AC, Saag KG et al (2014) The recent prevalence of osteoporosis and low bone mass in the United Sates based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res 29(11):2520–2526. https://doi.org/10.1002/jbmr.2269
International Osteoporosis Foundation. Key Statistics for North America. https://www.osteoporosis.foundation/facts-statistics/key-statistic-for-north-america. Accessed 29 March 2022.
Osteoporosis Fast Facts. National Osteoporosis Foundation. http://www.bonehealthandosteoporosis.org/wp-content/uploads/2015/12/Osteoporosis-Fast-Facts.pdf. Accessed 29 March 2022.
Gold T, Williams SA, Weiss RJ et al (2019) Impact of fractures on quality of life in patients with osteoporosis: a US cross-sectional survey. J Drug Assess 8(1):175–183. https://doi.org/10.1080/21556660.2019.1677674
Singer A, Exuzides A, Spangler L et al (2015) Burden of illness for osteoporotic fractures compared with other serious diseases among postmenopausal women in the United States. Mayo Clin Proc 90(1):53–62. https://doi.org/10.1016/j.mayocp.2014.09.011
Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A (2007) Incidence and Economic Burden of Osteoporosis-Related Fractures in the United States, 2005–2025. J Bone Miner Res 22(3):465–475. https://doi.org/10.1359/jbmr.061113
Williams SA, Daigle SG, Weiss R et al (2021) Economic Burden of US Osteoporotic Fractures in the US Medicare Population. Ann Pharmacother. https://doi.org/10.1177/1060028020970518
Bilezikian JP, Hattersley G, Fitzpatrick LA et al (2018) Abaloparatide-SC improves trabecular microarchitecture as assessed by trabecular bone score (TBS): a 24-week randomized clinical trial. Osteoporos Int 29(2):323–328
Forteo (2021) Package insert. Lilly USA LLC.
Tymlos (2021) Package insert. Radius Health Inc.
Hattersley G, Dean T, Corbin BA, Bahar H, Gardella TJ (2016) Binding Selectivity of Abaloparatide for PTH-Type-1-Receptor Conformations and Effects on Downstream Signaling. Endocrinol 157(1):141–149. https://doi.org/10.1210/en.2015-1726
Miller PD, Hattersley G, Riis BJ et al (2016) Effects of Abaloparatide vs Placebo on New Vertebral Fractures in Postmenopausal Women with Osteoporosis: A Randomized Clinical Trial. JAMA 316(7):722–733. https://doi.org/10.1001/jama.2016.11136
Bone HG, Cosman F, Miller PD et al (2018) ACTIVExtend: 24 Months of Alendronate After 18 Months of Abaloparatide or Placebo for Postmenopausal Osteoporosis. J Clin Endocrinol Metab 103(8):2949–2957. https://doi.org/10.1210/jc.2018-00163
Reginster J-Y, Bianic F, Campbell R, Martin M, Williams SA, Fitzpatrick LA (2019) Abaloparatide for risk reduction of nonvertebral and vertebral fractures in postmenopausal women with osteoporosis: a network meta-analysis. Osteoporos Int 30(7):1465–1473. https://doi.org/10.1007/s00198-019-04947-2
Barrionuevo P, Kapoor E, Asi N et al (2019) Efficacy of pharmacological therapies for the prevention of fractures in postmenopausal women: a network meta-analysis. J Clin Endocrinol Metab 104(5):1623–1630. https://doi.org/10.1210/jc.2019-00192
Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D (2019) Pharmacological management of osteoporosis in postmenopausal women: An Endocrine Society* clinical practice guideline. J Clin Endocrinol Metab 104(5):1595–1622. https://doi.org/10.1210/jc.2019-00221
Blonde L, Khunti K, Harris SB, Meizinger C, Skolnik NS (2018) Interpretation and Impact of Real-World Clinical Data for the Practicing Clinician. Adv Ther 35(11):1763–1774
Adomaityte J, Farooq M, Qayyum R (2008) Effect of raloxifene therapy on venous thromboembolism in postmenopausal women A meta-analysis. Thromb Haemost 99(2):338–342
Cummings SR, Ensrud K, Delmas PD et al (2010) Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med 362(8):686–696. https://doi.org/10.1056/NEJMoa0808692
McClung MR, O’Donoghue ML, Papapoulos SE et al (2019) Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol 7(12):899–911. https://doi.org/10.1016/S2213-8587(19)30346-8
Evenity (2019) Package insert. Amgen Inc.
Cosman F, Peterson LR, Towler DA, Mitlak B, Wang Y, Cummings SR (2020) Cardiovascular Safety of Abaloparatide in Postmenopausal Women With Osteoporosis: Analysis From the ACTIVE Phase 3 Trial. J Clin Endocrinol Metab 105(11):3384–3395
Tankó LB, Christiansen C, Cox DA, Geiger MJ, McNabb MA, Cummings SR (2005) Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J Bone Miner Res 20(11):1912–1920. https://doi.org/10.1359/JBMR.050711.Erratum.In:JBoneMinerRes.2006Feb;21(2):352
Symphony Health. https://symphonyhealth.com/. Accessed 29 March 2022.
Austin PC (2011) Optimal caliper widths for propensity-score matching when estimating differences in means and differences in proportions in observational studies. Pharm Stat 10(2):150–161. https://doi.org/10.1002/pst.433
Ho DE, Imai K, King G, Stuart EA (2011) MatchIt: Nonparametric Preprocessing for Parametric Causal Inference. J Stat Softw 42(8):1–28. https://doi.org/10.18637/jss.v042.i08
Normand ST, Landrum MB, Guadagnoli E et al (2001) Validating recommendations for coronary angiography following an acute myocardial infarction in the elderly: a matched analysis using propensity scores. J Clin Epidemiol 54:387–398. https://doi.org/10.1016/s0895-4356(00)00321-8
Wright NC, Daigle SG, Melton ME, Delzell ES, Balasubramanian A, Curtis JR (2019) The Design and Validation of a New Algorithm to Identify Incident Fractures in Administrative Claims Data. J Bone Miner Res 34(10):1798–1807. https://doi.org/10.1002/jbmr.3807
Xie F, Colantonio LD, Curtis JR et al (2018) Development of algorithms for identifying fatal cardiovascular disease in Medicare claims. Pharmacoepidemiol Drug Saf 27(7):740–750. https://doi.org/10.1002/pds.4421
Ritchey MD, Loustalot F, Wall HK et al (2017) Million Hearts: Description of the National Surveillance and Modeling Methodology Used to Monitor the Number of Cardiovascular Events Prevented During 2012–2016. J Am Heart Assoc 6(5):e006021
FDA mini-sentinel CV events. FDA's Sentinal System, US FDA.
Neer RM, Arnaud CD, Zanchetta JR et al (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344(19):1434–1441
Silverman S, Langdahl BL, Fujiwara S et al (2019) Reduction of Hip and Other Fractures in Patients Receiving Teriparatide in Real-World Clinical Practice: Integrated Analysis of Four Prospective Observational Studies. Calcif Tissue Int 104(2):193–200
Winzenrieth R, Humbert L, Boxberger JI, Weiss RJ, Wang Y, Kostenuik P (2021) Abaloparatide Effects on Cortical Volumetric BMD and Estimated Strength Indices of Hip Subregions by 3D-DXA in Women with Postmenopausal Osteoporosis. J Clin Densitom. S1094–6950(21)00098–6. Epub ahead of print. https://doi.org/10.1016/j.jocd.2021.11.007.
Winzenrieth R, Ominsky MS, Wang Y, Humbert L, Weiss RJ (2021) Differential effects of abaloparatide and teriparatide on hip cortical volumetric BMD by DXA-based 3D modelling. Osteoporos Int 32(3):575–583. https://doi.org/10.1007/s00198-020-05806-1
Burr DB, Hirano T, Turner CH, Hotchkiss C, Brommage R, Hock JM (2001) Intermittently administered human parathyroid hormone (1–34) treatment increases intracortical bone turnover and porosity without reducing bone strength in the humerus of ovariectomized cynomolgus monkeys. J Bone Miner Res 16(1):157–165. https://doi.org/10.1359/jbmr.2001.16.1.157
Jiang Y, Zhao JJ, Mitlak BH, Wang O, Genant HK, Eriksen EF (2003) Recombinant human parathyroid hormone (1–34) [teriparatide] improves both cortical and cancellous bone structure. J Bone Miner Res 18:1932–1941. https://doi.org/10.1359/jbmr.2003.18.11.1932
MacDonald HM, Nishiyama KK, Hanley DA, Boyd SK (2011) Changes in trabecular and cortical bone microarchitecture at peripheral sites associated with 18 months of teriparatide therapy in postmenopausal women with osteoporosis. Osteoporos Int 22:357–362. https://doi.org/10.1007/s00198-010-1226-1
Sato M, Westmore M, Ma YL et al (2004) Teriparatide [PTH (1–34)] strengthens the proximal femur of ovariectomized nonhuman primates despite increasing porosity. J Bone Miner Res 19(4):623–629. https://doi.org/10.1359/JBMR.040112
Doyle N, Varela A, Haile S et al (2018) Abaloparatide, a novel PTH receptor agonist, increased bone mass and strength in ovariectomized cynomolgus monkeys by increasing bone formation without increasing bone resorption. Osteoporos Int 29(3):685–697
Varela A, Chouinard L, Lesage E, Smith SY, Hattersley G (2017) One year of abaloparatide, a selective activator of the PTH1 receptor, increased bone formation and bone mass in ovariectomized osteopenic rats without increasing bone resorption. J Bone Miner Res 32(1):24–33. https://doi.org/10.1002/jbmr.3003
Samelson EJ, Kiel DP, Broe KE et al (2004) Metacarpal cortical area and risk of coronary heart disease: the Framingham Study. Am J Epidemiol 159(6):589–595. https://doi.org/10.1093/aje/kwh080
European Medicines Agency (2021) Guidelines on registry-based studies. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-registry-based-studies_en-0.pdf. Accessed 29 March 2022.
US Department of Health and Human Services (2021) Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drugs and Biological Products: Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-real-world-data-and-real-world-evidence-support-regulatory-decision-making-drug. Accessed 29 March 2022.
Berger ML, Sox H, Willke RJ et al (2017) Good Practices for Real-World Data Studies of Treatment and/or Comparative Effectiveness: Recommendations from the Joint ISPOR-ISPE Special Task Force on Real-World Evidence in Health Care Decision Making. Pharmacoepidemiol Drug Saf 26(9):1033–1039. https://doi.org/10.1002/pds.4297
Acknowledgements
Funding for this study (NCT04974723 available from www.clinicaltrials.gov) was provided by Radius Health, Inc. (Radius). All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support, Allyson Lehrman, DPM (formatting tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services were provided by AOIC, LLC and were funded by Radius.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The study is fully compliant with the Health Insurance Portability and Accountability Act. The protocol was reviewed and deemed exempt from Institutional Review Board (IRB) oversight using the Department of Health and Human Services regulations found at 45 CFR 46.104 (d)(4) (Advarra IRB; Pro00056338; August 4, 2021).
Consent to participate
A waiver of informed consent was received from Advarra IRB.
Consent for Publication
Not Applicable.
Disclosure
FC has received institutional grants and research support from Amgen; has served as a consultant for Amgen, Radius Health, Inc. (Radius), Enterabio, Haoma, Obseva, and Myovant; has served on the speakers’ bureaus for Amgen and Radius; and has served on advisory boards for Amgen and Radius. She is also co-editor in chief of Osteoporosis International. CC has received lecture fees and honoraria from Amgen, Danone, Eli Lilly, GSK, Medtronic, Merck, Nestlé, Novartis, Pfizer, Roche, Servier, Shire, Takeda, and UCB; YW, BM, SV, and SAW are employees and shareholders of Radius. CC is also the president of International Osteoporosis Foundation, a sponsor of Osteoporosis International.
Conflict of interest
The authors declare that they have no personal conflicts.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cosman, F., Cooper, C., Wang, Y. et al. Comparative effectiveness and cardiovascular safety of abaloparatide and teriparatide in postmenopausal women new to anabolic therapy: A US administrative claims database study. Osteoporos Int 33, 1703–1714 (2022). https://doi.org/10.1007/s00198-022-06413-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-022-06413-y