
Abstract
Purpose
We assessed the effects of targeting low-normal or high-normal arterial carbon dioxide tension (PaCO2) and normoxia or moderate hyperoxia after out-of-hospital cardiac arrest (OHCA) on markers of cerebral and cardiac injury.
Methods
Using a 23 factorial design, we randomly assigned 123 patients resuscitated from OHCA to low-normal (4.5–4.7 kPa) or high-normal (5.8–6.0 kPa) PaCO2 and to normoxia (arterial oxygen tension [PaO2] 10–15 kPa) or moderate hyperoxia (PaO2 20–25 kPa) and to low-normal or high-normal mean arterial pressure during the first 36 h in the intensive care unit. Here we report the results of the low-normal vs. high-normal PaCO2 and normoxia vs. moderate hyperoxia comparisons. The primary endpoint was the serum concentration of neuron-specific enolase (NSE) 48 h after cardiac arrest. Secondary endpoints included S100B protein and cardiac troponin concentrations, continuous electroencephalography (EEG) and near-infrared spectroscopy (NIRS) results and neurologic outcome at 6 months.
Results
In total 120 patients were included in the analyses. There was a clear separation in PaCO2 (p < 0.001) and PaO2 (p < 0.001) between the groups. The median (interquartile range) NSE concentration at 48 h was 18.8 µg/l (13.9–28.3 µg/l) in the low-normal PaCO2 group and 22.5 µg/l (14.2–34.9 µg/l) in the high-normal PaCO2 group, p = 0.400; and 22.3 µg/l (14.8–27.8 µg/l) in the normoxia group and 20.6 µg/l (14.2–34.9 µg/l) in the moderate hyperoxia group, p = 0.594). High-normal PaCO2 and moderate hyperoxia increased NIRS values. There were no differences in other secondary outcomes.
Conclusions
Both high-normal PaCO2 and moderate hyperoxia increased NIRS values, but the NSE concentration was unaffected.
Registration
ClinicalTrials.gov, NCT02698917. Registered on January 26, 2016.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypoxic ischaemic encephalopathy (HIE) is the most common cause of disability and death after out-of-hospital cardiac arrest (OHCA) [1, 2]. The developing neurological damage is initially related to an ischaemia–reperfusion injury and reactive oxygen species (ROS), which may increase the oxidative damage to the brain [3]. This is followed by cerebral hypoperfusion, possibly caused by increased vasoconstriction during the first 72 h of post-resuscitation care, further aggravating the developing HIE [4]. Previous studies have suggested a decrease in cerebral blood flow and an increase in oxygen extraction in cardiac arrest patients [5].
Arterial carbon dioxide tension (PaCO2) is a major determinant of cerebral blood flow (CBF) [6]. In experimental studies, carbon dioxide seems to have anticonvulsive [7], anti-inflammatory and antioxidant properties [8], suggesting a protective role of PaCO2 in the development of HIE. According to a recent meta-analysis of observational data, both hypocapnia and hypercapnia are associated with poor outcomes after cardiac arrest [9]. One randomised controlled trial with 83 patients found, on the contrary, that targeting mild hypercapnia (6.7–7.3 kPa) after cardiac arrest attenuated the increase of neuron-specific enolase (NSE) concentrations over time, suggesting a possible protective effect of mild hypercapnia against neurological injury [10]. Concerns related to higher PaCO2 levels include increased cerebral oedema, respiratory acidosis and impaired right ventricular function, which may all contribute to poor outcomes [11,12,13].
Regarding oxygen, experimental studies have shown that exposure to very high levels of arterial oxygen tension (PaO2) during post-resuscitation care may increase ROS production and exacerbate the developing neurological damage [14]. Indeed, in retrospective and observational human studies, severe hyperoxia (PaO2 > 40 kPa) has been associated with poor outcome after cardiac arrest [15,16,17,18,19]. In contrast, moderate hyperoxia during post-resuscitation intensive care has been associated with better long-term neurological recovery and improved organ function [20]. A retrospective analysis from one large intensive care unit (ICU) registry suggested that the PaO2 associated with the lowest mortality was around 20 kPa [21].
In mechanically ventilated patients, both PaCO2 and PaO2 can be altered via the ventilator settings [22]. Currently there are limited high-quality data on the optimal carbon dioxide and oxygen targets in cardiac arrest patients. Accordingly, we performed a multicentre, randomised pilot trial to assess the feasibility and the effect on the serum concentration of NSE of targeting low-normal or high-normal PaCO2 and normoxia or moderate hyperoxia after OHCA and successful resuscitation. In addition, we investigated the effect of these interventions on other markers of neurological and cardiac injury, electroencephalography (EEG) and cerebral oxygenation. Our hypothesis was that targeting high-normal PaCO2 and moderate hyperoxia would result in lower NSE concentrations at 48 h after cardiac arrest.
Methods
Trial design
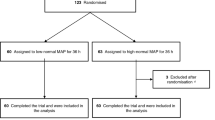
The full details of the Carbon dioxide, Oxygen and Mean arterial pressure After Cardiac Arrest and REsuscitation (COMACARE) study have been previously described [23]. In brief, we conducted a prospective, multicentre, randomised trial with a 23 factorial design. A total of 123 unconscious, mechanically ventilated patients resuscitated from OHCA were randomly assigned to intervention targets of low-normal or high-normal PaCO2, normoxia or moderate hyperoxia and low-normal or high-normal mean arterial pressure (MAP) for the first 36 h in the intensive care unit (ICU). Each patient was randomised into one of eight arms with each arm having a different combination of targets for PaCO2, PaO2 and MAP. In this paper we report the results of the low-normal vs. high-normal PaCO2 and the normoxia vs. moderate hyperoxia comparisons.
Participants
We included adult patients resuscitated from witnessed OHCA of presumed cardiac origin with ventricular fibrillation (VF) or ventricular tachycardia (VT) as the initial rhythm. For details regarding the inclusion and exclusion criteria, the participating centres, the informed consent and the ethical approval, please see the electronic supplemental material (ESM).
Interventions
After ICU admission and randomisation, we directed the treating personnel to target low-normal (4.5–4.7 kPa) or high-normal (5.8–6.0 kPa) PaCO2 by adjusting the minute ventilation (tidal volume and ventilation rate) delivered by the ventilator. Normoxia (10–15 kPa) or moderate hyperoxia (20–25 kPa) were targeted by adjusting FiO2 and PEEP levels on the ventilator. Laminated signs designating the intervention targets were used at the patients’ bedsides and on the ventilators. The ventilator adjustments were guided by arterial blood gas (ABG) analyses (corrected to each patient’s actual temperature) performed at least every 3 h. We used end-tidal carbon dioxide (EtCO2) value as an additional guide in targeting the desired PaCO2 level. In the normoxia group, we used peripheral oxygen saturation (SpO2) as an additional guide, targeting an SpO2 value of 95–98%. A volume-controlled or a pressure-controlled ventilation mode was used according to the treating clinician’s preference. We continued the intervention for 36 h from the ICU admission or until the patient was extubated or ventilation was set to a spontaneous mode, whichever occurred first. All patients received targeted temperature management (TTM) at 33 °C or 36 °C and were sedated according to the treating clinicians’ instructions. All patients received standard care, monitoring and assessments based on the protocol of the ICU, including direct blood pressure monitoring via an arterial catheter.
Outcomes
The primary outcome was the NSE serum concentration at 48 h after cardiac arrest. The main feasibility outcomes were the differences in PaCO2 and PaO2 between the groups targeting low-normal (4.5–4.7 kPa) and high-normal (5.8–6.0 kPa) PaCO2 and normoxia (10–15 kPa) and moderate hyperoxia (20–25 kPa), respectively. The prespecified secondary outcomes were NSE serum concentrations at 24 and 72 h after cardiac arrest; S100B protein (a biomarker of glial injury) serum concentrations at 24, 48 and 72 h after cardiac arrest; cardiac troponin (TnT) plasma concentrations at 24, 48 and 72 h after cardiac arrest; regional frontal cerebral oxygenation (rSO2) measured by continuous near-infrared spectroscopy (NIRS) monitoring during the first 48 h after admission to the ICU; results of continuous EEG monitoring for the first 48 h after admission to the ICU interpreted by an experienced senior neurophysiologist blinded to study group allocation; neurological recovery assessed with Cerebral Performance Category (CPC) at 6 months after cardiac arrest (CPC 1–2 considered a good outcome, and CPC 3–5 a poor outcome) determined by an experienced neurologist blinded to study group allocation; total duration of intensive care; total duration of mechanical ventilation; length of hospital stay; discharge destination and vital status at 30 days after cardiac arrest (dead or alive). Other feasibility outcomes included distribution of values for primary and secondary outcomes, randomised/screened patient ratio, consent rate, data completion rate and recruitment duration. The predefined serious adverse events (SAE) that could be related to the interventions were severe hypercapnia and respiratory acidosis (PaCO2 > 10 kPa and pH < 7.15), unexplained brain oedema on CT scanning and severe ARDS (PaO2/FiO2 ratio of < 100 mmHg).
Data collection, randomisation and statistical methods
The data collection and laboratory methods, randomisation procedure and statistical analysis are described in detail in the ESM.
Results
For the flowcharts demonstrating patient enrolment and group allocations, please see the ESM (Fig. 1a,b). The first patient was randomised on 22 March 2016, and recruitment was completed by 3 November 2017. The 6-month follow-up of the last patient was completed by 3 May 2018. The baseline characteristics and resuscitation factors were comparable between the groups (Table 1). We observed a clear separation during the intervention in PaCO2 between the low-normal and the high-normal PaCO2 groups and in PaO2 between the normoxia and the moderate hyperoxia groups (Fig. 1). The median (interquartile range [IQR]) expiratory tidal volume per body weight and the median (IQR) ventilation rate were 5.8 ml/kg (5.2–6.8 ml/kg) and 12 min−1 (12–14 min−1) in the low-normal PaCO2 group and 5.4 ml/kg (4.8–5.9 ml/kg) and 11 min−1 (10–12 min−1) in the high-normal PaCO2 group, respectively. The median (IQR) FiO2 and PEEP levels were 35% (30–40%) and 7.2 cmH2O (6.2–8.2 cmH2O) in the normoxia group and 50% (45–59%) and 8.2 cmH2O (6.3–10.0 cmH2O) in the moderate hyperoxia group, respectively.

Median (interquartile range) a PaCO2 during the intervention in the low-normal and high-normal PaCO2 groups. b PaO2 during the intervention in the normoxia and moderate hyperoxia groups
We did not find significant differences in the median serum NSE concentration at 48 h after cardiac arrest between the intervention groups (in the low-normal PaCO2 group, 18.8 µg/l [IQR 13.9–28.3 µg/l] and in the high-normal PaCO2 group, 22.5 µg/l [IQR 14.2–34.9 µg/l], p = 0.400; in the normoxia group, 22.3 µg/l [IQR, 14.8–27.8 µg/l] and in the moderate hyperoxia group, 20.6 µg/l [IQR, 14.2–34.9 µg/l], p = 0.594). The NSE, S100B and TnT concentrations were also comparable over time in the low-normal PaCO2 and high-normal PaCO2 groups and the normoxia and moderate hyperoxia groups (Figs. 2, 3, 4).

Baseline, 24 h, 48 h and 72 h median (interquartile range) serum neuron-specific enolase (NSE) concentrations for patients allocated to targeting a low-normal and high-normal PaCO2, b normoxia and moderate hyperoxia

Baseline, 24 h, 48 h and 72 h median (interquartile range) serum S100B concentrations for patients allocated to targeting a low-normal and high-normal PaCO2, b normoxia and moderate hyperoxia

Baseline, 24 h, 48 h and 72 h median (interquartile range) plasma cardiac troponin (TnT) concentrations for patients allocated to targeting a low-normal and high-normal PaCO2, b normoxia and moderate hyperoxia

Median (interquartile range) regional cerebral oxygen saturation (rSO2) during the intervention in the a low-normal and high-normal PaCO2 groups. b normoxia and moderate hyperoxia groups
The median cerebral oxygen saturation (rSO2) was significantly higher in the high-normal PaCO2 group than in the low-normal PaCO2 group, p < 0.001 (Fig. 5a). In addition, the rSO2 was significantly higher in the moderate hyperoxia group than in the normoxia group, p < 0.001 (Fig. 5b). We found no significant differences between any of the groups regarding mortality at 30 days after cardiac arrest, good neurological recovery (CPC 1–2) at 6 months after cardiac arrest, the duration of intensive care or mechanical ventilation, or the frequency of the predefined SAEs (Table 2). The EEG grading at the ICU admission and the end of the intervention was also similar in all groups (Table 3). Regarding the NSE results at 48 h after cardiac arrest or good neurological outcomes at 6 months, we did not find any significant interactions between the PaCO2, PaO2, MAP or TTM targets (ESM Table 4).
Discussion
In this prospective, randomised trial, we found that targeting a specific level of PaCO2 or PaO2 was feasible in comatose, mechanically ventilated patients after cardiac arrest. However, these interventions did not change the concentration of NSE at 48 h after cardiac arrest or any of the measured markers of neurological or cardiac injury. Targeting high-normal PaCO2 or moderate hyperoxia both increased cerebral oxygenation measured by NIRS, but the implications of this are unclear. The preliminary results of the current trial cannot confirm or refute a benefit or harm within the studied carbon dioxide and oxygen ranges.
We chose to use two markers of cerebral injury in this trial. NSE is a cytoplasmic glycolytic enzyme found in neurons and neuroectodermal cells, and S100B is a protein specific to neuroglial cells. Both are released into the cerebrospinal fluid and bloodstream after neuronal damage, and their concentrations during the first 24–72 h after cardiac arrest correlate with the severity of the brain injury and the probability of a poor outcome [24,25,26,27]. We chose the NSE concentration at 48 h as the primary outcome because it is well documented as a surrogate marker of HIE and it has an established role in the multimodal prognostication of the OHCA patients [28]. The similar levels of these surrogate markers in all intervention groups of our study suggest that targeting higher or lower PaCO2 within the normal range or moderately elevated PaO2 instead of normoxia does not markedly affect the development of HIE in post cardiac arrest patients. Compared to previous trials [10, 29], the NSE concentrations in our study were lower and already decreasing at 48 h after cardiac arrest. This is likely explained by our relatively strict inclusion criteria.
It is thought that the development of the HIE begins early after the ROSC and for this reason the interventions aiming at affecting its course should be started as early as possible. For practical reasons, targeting specific levels of PaCO2 and PaO2 is difficult during prehospital care. Thus, we decided to start the interventions immediately after ICU admission at the hospital. Moreover, we think that the delay between the ROSC and the beginning of the interventions was acceptable for most participants (Table 1). However, it is possible that the potential effect of the interventions was decreased as a result of the delay.
This trial was not powered for mortality or neurologic outcome and the results based on a surrogate marker of brain injury cannot exclude benefits or harms from different levels of PaCO2 within the normal range or moderate hyperoxia after cardiac arrest. The results of our study support the safety and feasibility of studying the effects of these interventions in a larger trial. However, because the NSE results were so similar between the intervention groups, it might be more worthwhile to look into the effect of different levels of PaCO2 and PaO2 instead. A larger randomised trial comparing mild hypercapnia (6.7–7.3 kPa) with normocapnia is already taking place (NCT03114033). There is an imminent need for large trials on the long-term effects of different oxygen targets in patients after cardiac arrest and other forms of neurocritical illness [30].
The effect of PaCO2 may differ according to the target temperature during TTM, and hypocapnia may be especially common in patients treated with a target temperature of 33 °C [31]. Cerebrovascular reactivity is maintained during therapeutic hypothermia, and hypocapnia may cause decreased cerebral blood flow and ischaemia in these patients. Regarding oxygen, in the only randomised study performed on the use of oxygen in cardiac arrest patients, an increase in NSE within a subgroup of patients exposed to 100% oxygen and not treated with TTM at 33 °C was seen [32]. This may indicate harmful effects of extreme hyperoxia on the developing HIE especially in patients without the attenuating effect of hypothermia. Indeed, the results of one large registry study suggested no association between oxygen and survival in OHCA patients predominantly treated with hypothermia [17]. We, however, included patients treated with TTM targets of both 33 °C and 36 °C and found no interaction between temperature and neither carbon dioxide nor oxygen targets.
We found that high-normal PaCO2 and moderate hyperoxia resulted in higher cerebral oxygen saturation when compared with low-normal PaCO2 and normoxia, respectively. Higher rSO2 may reflect improved oxygen delivery, and there are some data suggesting that higher rSO2 is related to better outcome after cardiac arrest [33]. Recently, Taccone and colleagues showed a moderate relationship between cerebral perfusion pressure and rSO2 in OHCA patients undergoing TTM [34]. Nevertheless, the relationship between rSO2, CBF and outcome is not fully understood. Combining continuous transcranial Doppler ultrasound with NIRS would likely have provided additional information of the CBF. However, for practical reasons, it was not possible to implement in this multicentre trial.
In experimental studies, CO2 has exhibited potent anticonvulsive properties [7]. In patients with subarachnoid haemorrhage, there appears to be an association between invasively measured low levels of oxygen in brain tissue and periodical epileptic discharges [35]. In this pilot trial, we did not find any association between the targeted carbon dioxide or oxygen level and EEG abnormalities. Considering the relatively small absolute difference in PaCO2 between the groups, this study may not have been powered to exclude an anticonvulsive effect of high-normal PaCO2 or different oxygen levels.
Cardiac arrest and resuscitation result in global ischaemia–reperfusion injury which affects the heart and may lead to myocardial stunning and haemodynamic instability [36]. Moreover, this myocardial ischaemia–reperfusion damage may be aggravated by myocardial infarction because acute coronary syndrome (ACS) is the most common cause of OHCA. Hypercapnia has been related to impaired right ventricular function and acidosis which may negatively affect the recovery of critically ill neurologic patients [12]. The results of two large trials investigating the effect of hyperoxia on myocardial injury in ACS patients were controversial [37, 38]. There are no previous data from randomised trials on the effect of different levels of PaCO2 or moderate hyperoxia on myocardial damage after cardiac arrest and resuscitation. We found that the concentration of TnT was comparable in the two PaCO2 groups as well as in the two PaO2 groups during the first 72 h after cardiac arrest.
Our trial has several strengths. First, the study protocol, including the plan for the statistical analysis, had been previously published [23]. Second, by using relatively strict eligibility criteria, the study was focused on a homogenous group of OHCA patients, thereby reducing the bias caused by differences in baseline characteristics and resuscitation factors. Third, we carried out post-resuscitation intensive care according to the current guidelines for all patients. Fourth, we studied patients in multiple centres in two countries. Fifth, we took frequent ABG samples and used continuous EtCO2 and SpO2 monitoring to ensure that PaCO2 and PaO2 remained at the target level for the whole intervention period. Finally, we recorded frontal rSO2 and EEG continuously during the whole intervention.
This study also has some limitations. First, although we conducted a multicentre trial, most participants were recruited at one hospital. Second, because the PaCO2, PaO2, EtCO2 and SpO2 are routinely monitored variables in the ICU, the study intervention could not be blinded; thus, the treating personnel were aware of the study group allocations. Third, we used a four-channel technique for EEG monitoring. There is a risk that some focal epileptic discharges may have been undetected. Finally, the blood samples taken at the Danish site had to be analysed on-site for logistical reasons. However, to minimise a possible bias resulting from technical issues, the same laboratory kits for NSE and S100B evaluation were used in both Finland and Denmark.
Conclusions
Targeting low-normal or high-normal PaCO2 and normoxia or moderate hyperoxia was feasible in comatose mechanically ventilated patients admitted to the ICU after OHCA and resuscitation. The target levels of PaCO2 or PaO2 did not affect the serum concentration of NSE at 48 h. High-normal PaCO2 and moderate hyperoxia resulted in better cerebral oxygen saturation which may indicate higher CBF and oxygen delivery, but the clinical implications of these findings are unclear.
Other information
Protocol
The protocol of the COMACARE study has been previously published [23].
References
Laver S, Farrow C, Turner D, Nolan J (2004) Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 30:2126–2128. https://doi.org/10.1007/s00134-004-2425-z
Lemiale V, Dumas F, Mongardon N et al (2013) Intensive care unit mortality after cardiac arrest: the relative contribution of shock and brain injury in a large cohort. Intensive Care Med 39:1972–1980. https://doi.org/10.1007/s00134-013-3043-4
Llitjos J-F, Mira J-P, Duranteau J, Cariou A (2016) Hyperoxia toxicity after cardiac arrest: what is the evidence? Ann Intensive Care 6:1–9. https://doi.org/10.1186/s13613-016-0126-8
Buunk G, van der Hoeven JG, Meinders AE (1999) Prognostic significance of the difference between mixed venous and jugular bulb oxygen saturation in comatose patients resuscitated from a cardiac arrest. Resuscitation 41:257–262
Lemiale V, Huet O, Vigué B et al (2008) Changes in cerebral blood flow and oxygen extraction during post-resuscitation syndrome. Resuscitation 76:17–24. https://doi.org/10.1016/j.resuscitation.2007.06.028
Battisti-Charbonney A, Fisher J, Duffin J (2011) The cerebrovascular response to carbon dioxide in humans. J Physiol (Lond) 589:3039–3048. https://doi.org/10.1113/jphysiol.2011.206052
Tolner EA, Hochman DW, Hassinen P et al (2010) Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia 52:104–114. https://doi.org/10.1111/j.1528-1167.2010.02731.x
Shoja MM, Tubbs RS, Shokouhi G et al (2008) The potential role of carbon dioxide in the neuroimmunoendocrine changes following cerebral ischemia. Life Sci 83:381–387. https://doi.org/10.1016/j.lfs.2008.07.007
McKenzie N, Williams TA, Tohira H et al (2017) A systematic review and meta-analysis of the association between arterial carbon dioxide tension and outcomes after cardiac arrest. Resuscitation 111:116–126. https://doi.org/10.1016/j.resuscitation.2016.09.019
Eastwood GM, Schneider AG, Suzuki S et al (2016) Targeted therapeutic mild hypercapnia after cardiac arrest: a phase II multi-centre randomised controlled trial (the CCC trial). Resuscitation 104:83–90. https://doi.org/10.1016/j.resuscitation.2016.03.023
Ganga HV, Kallur KR, Patel NB et al (2013) The impact of severe acidemia on neurologic outcome of cardiac arrest survivors undergoing therapeutic hypothermia. Resuscitation 84:1723–1727. https://doi.org/10.1016/j.resuscitation.2013.07.006
Tiruvoipati R, Pilcher D, Botha J et al (2018) Association of hypercapnia and hypercapnic acidosis with clinical outcomes in mechanically ventilated patients with cerebral injury. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2018.0123
Mekontso Dessap A, Charron C, Devaquet J et al (2009) Impact of acute hypercapnia and augmented positive end-expiratory pressure on right ventricle function in severe acute respiratory distress syndrome. Intensive Care Med 35:1850–1858. https://doi.org/10.1007/s00134-009-1569-2
Pilcher J, Weatherall M, Shirtcliffe P et al (2012) The effect of hyperoxia following cardiac arrest: a systematic review and meta-analysis of animal trials. Resuscitation. 83:417–422. https://doi.org/10.1016/j.resuscitation.2011.12.021
Kilgannon JH, Jones AE, Shapiro NI et al (2010) Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA 303:2165–2171. https://doi.org/10.1001/jama.2010.707
Kilgannon JH, Jones AE, Parrillo JE et al (2011) Relationship between supranormal oxygen tension and outcome after resuscitation from cardiac arrest. Circulation 123:2717–2722. https://doi.org/10.1161/CIRCULATIONAHA.110.001016
Bellomo R, Bailey M, Eastwood GM, Alistair N (2011) Arterial hyperoxia and in-hospital mortality after resuscitation from cardiac arrest. Crit Care 15:R90. https://doi.org/10.1186/cc10090
Janz DR, Hollenbeck RD, Pollock JS et al (2012) Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med 40:3135–3139. https://doi.org/10.1097/CCM.0b013e3182656976
Roberts BW, Kilgannon JH, Hunter BR et al (2018) Association between early hyperoxia exposure after resuscitation from cardiac arrest and neurological disability. Circulation 137:2114–2124. https://doi.org/10.1161/CIRCULATIONAHA.117.032054
Elmer J, Scutella M, Pullalarevu R et al (2014) The association between hyperoxia and patient outcomes after cardiac arrest: analysis of a high-resolution database. Intensive Care Med 41:49–57. https://doi.org/10.1007/s00134-014-3555-6
Vaahersalo J, Bendel S, Reinikainen M et al (2014) Arterial blood gas tensions after resuscitation from out-of-hospital cardiac arrest: associations with long-term neurologic outcome. Crit Care Med 42:1463–1470. https://doi.org/10.1097/CCM.0000000000000228
Nolan JP, Soar J, Cariou A et al (2015) European resuscitation council and European society of intensive Care medicine guidelines for post-resuscitation care 2015: section 5 of the European resuscitation council guidelines for resuscitation 2015. Resuscitation 95:202–222. https://doi.org/10.1016/j.resuscitation.2015.07.018
Jakkula P, Reinikainen M, Hästbacka J et al (2017) Targeting low- or high-normal carbon dioxide, oxygen, and mean arterial pressure after cardiac arrest and resuscitation: study protocol for a randomized pilot trial. Trials 18:1–9. https://doi.org/10.1186/s13063-017-2257-0
Streitberger KJ, Leithner C, Wattenberg M et al (2017) Neuron-specific enolase predicts poor outcome after cardiac arrest and targeted temperature management. Crit Care Med 45:1145–1151. https://doi.org/10.1097/CCM.0000000000002335
Shinozaki K, Shigeto O, Sadahito T, Nakamura M (2009) S-100B and neuron-specific enolase as predictors of neurological outcome in patients after cardiac arrest and return of spontaneous circulation: a systematic review. Crit Care. https://doi.org/10.1186/cc7973
Sandroni C, Cavallaro F, Callaway CW et al (2013) Predictors of poor neurological outcome in adult comatose survivors of cardiac arrest: a systematic review and meta-analysis. Part 1: Patients not treated with therapeutic hypothermia. Resuscitation 84:1310–1323. https://doi.org/10.1016/j.resuscitation.2013.05.013
Calderon LM, Guyette FX, Doshi AA et al (2014) Combining NSE and S100B with clinical examination findings to predict survival after resuscitation from cardiac arrest. Resuscitation 85:1025–1029. https://doi.org/10.1016/j.resuscitation.2014.04.020
Sandroni C, D’Arrigo S, Nolan JP (2018) Prognostication after cardiac arrest. Curr Opin Crit Care. https://doi.org/10.1186/s13054-018-2060-7
Stammet P, Collignon O, Hassager C et al (2015) Neuron-specific enolase as a predictor of death or poor neurological outcome after out-of-hospital cardiac arrest and targeted temperature management at 33 °C and 36 °C. J Am Coll Cardiol 65:2104–2114. https://doi.org/10.1016/j.jacc.2015.03.538
Lång M, Skrifvars MB, Siironen J et al (2018) A pilot study of hyperoxemia on neurological injury, inflammation and oxidative stress. Acta Anaesthesiol Scand 62:801–810. https://doi.org/10.1111/aas.13093
Falkenbach P, Kämäräinen A, Mäkelä A et al (2009) Incidence of iatrogenic dyscarbia during mild therapeutic hypothermia after successful resuscitation from out-of-hospital cardiac arrest. Resuscitation 80:990–993. https://doi.org/10.1016/j.resuscitation.2009.04.044
Kuisma M, Boyd J, Voipio V et al (2006) Comparison of 30 and the 100% inspired oxygen concentrations during early post-resuscitation period: a randomised controlled pilot study. Resuscitation 69:199–206. https://doi.org/10.1016/j.resuscitation.2005.08.010
Storm C, Leithner C, Krannich A et al (2014) Regional cerebral oxygen saturation after cardiac arrest in 60 patients: a prospective outcome study. Resuscitation 85:1037–1041. https://doi.org/10.1016/j.resuscitation.2014.04.021
Taccone FS, Crippa IA, Creteur J, Rasulo F (2018) Estimated cerebral perfusion pressure among post-cardiac arrest survivors. Intensive Care Med 44:966–967. https://doi.org/10.1007/s00134-018-5074-3
Witsch J, Frey H-P, Schmidt JM et al (2017) Electroencephalographic periodic discharges and frequency-dependent brain tissue hypoxia in acute brain injury. JAMA Neurol 74:301–309. https://doi.org/10.1001/jamaneurol.2016.5325
Laurent I, Monchi M, Chiche J-D et al (2002) Reversible myocardial dysfunction in survivors of out-of-hospital cardiac arrest. J Am Coll Cardiol 40:2110–2116
Stub D, Smith K, Bernard S et al (2015) Air versus oxygen in ST-segment elevation myocardial infarction. Circulation 131:1–41. https://doi.org/10.1161/CIRCULATIONAHA.114.014494
Hofmann R, James SK, Jernberg T et al (2017) Oxygen therapy in suspected acute myocardial infarction. N Engl J Med 377:1240–1249. https://doi.org/10.1056/NEJMoa1706222
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. COMACARE STUDY GROUP. Aarhus University Hospital: Thomas Birkelund, Susanne Ilkjaer, Hans Kirkegaard; Central Finland Central Hospital: Raili Laru-Sompa, Anni Pulkkinen, Mikko Reilama, Sinikka Tolmunen; Helsinki University Hospital: Minna Bäcklund, Jonna Heinonen, Johanna Hästbacka, Pekka Jakkula, Nina Lundblom, Marcus Norrgård, Marjatta Okkonen, Ville Pettilä, Markus B Skrifvars, Tarja Suhonen, Marjaana Tiainen, Tuukka Tikka, Marjut Timonen, Jussi Toppila, Miia Valkonen, Erika Wilkman; Jorvi Hospital: Teemu Hult, Tuomas Oksanen; Kuopio University Hospital: Stepani Bendel, Elina Halonen, Sari Rahikainen, Saija Rissanen, Eija Vaskelainen; North Karelia Central Hospital: Tanja Eiserbeck, Sirkku Heino, Helena Jyrkönen, Matti Reinikainen, Johanna Räsänen, Tero Surakka; Päijät-Häme Central Hospital: Talvikki Koskue, Petteri Kujala, Pekka Loisa, Marika Lähde; Tampere University Hospital: Jari Kalliomäki, Sari Karlsson, Atte Kukkurainen, Simo Varila. We thank Tuomas Selander, MSc, biostatistician, for help with the statistical analyses.
Funding
Independent funding support has been received from Helsinki University; Helsinki University Hospital (State funding, Finland); Stiftelsen Dorothea Olivia, Karl Walter och Jarl Walter Perkléns minne; The Laerdal Foundation for Acute Medicine; Medicinska Understödsföreningen Liv och Hälsa; Finska Läkaresällskapet; The Finnish Society of Anaesthesiologists; Orion Research Foundation and Svenska kulturfonden. The funding bodies had no input regarding the design, management, or reporting of the trial.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Members of the COMACARE study group are listed in the Acknowledgements.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jakkula, P., Reinikainen, M., Hästbacka, J. et al. Targeting two different levels of both arterial carbon dioxide and arterial oxygen after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med 44, 2112–2121 (2018). https://doi.org/10.1007/s00134-018-5453-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-018-5453-9