
Abstract
Carbonate-bearing fluorapatite rocks occur at over 30 globally distributed carbonatite complexes and represent a substantial potential supply of phosphorus for the fertiliser industry. However, the process(es) involved in forming carbonate-bearing fluorapatite at some carbonatites remain equivocal, with both hydrothermal and weathering mechanisms inferred. In this contribution, we compare the paragenesis and trace element contents of carbonate-bearing fluorapatite rocks from the Kovdor, Sokli, Bukusu, Catalão I and Glenover carbonatites in order to further understand their origin, as well as to comment upon the concentration of elements that may be deleterious to fertiliser production. The paragenesis of apatite from each deposit is broadly equivalent, comprising residual magmatic grains overgrown by several different stages of carbonate-bearing fluorapatite. The first forms epitactic overgrowths on residual magmatic grains, followed by the formation of massive apatite which, in turn, is cross-cut by late euhedral and colloform apatite generations. Compositionally, the paragenetic sequence corresponds to a substantial decrease in the concentration of rare earth elements (REE), Sr, Na and Th, with an increase in U and Cd. The carbonate-bearing fluorapatite exhibits a negative Ce anomaly, attributed to oxic conditions in a surficial environment and, in combination with the textural and compositional commonality, supports a weathering origin for these rocks. Carbonate-bearing fluorapatite has Th contents which are several orders of magnitude lower than magmatic apatite grains, potentially making such apatite a more environmentally attractive feedstock for the fertiliser industry. Uranium and cadmium contents are higher in carbonate-bearing fluorapatite than magmatic carbonatite apatite, but are much lower than most marine phosphorites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phosphorus is essential to all life and is a limiting nutrient for plant growth (Cordell and White 2014). It is one of three macro-nutrients in fertiliser, but is a non-renewable resource principally mined from phosphorites, carbonatites and alkaline igneous rocks (Northolt et al. 1989). As of 2015, carbonatites and alkaline rocks account for ~10% of global phosphate production and make up ~5% of the world’s phosphorus reserves (Pufahl and Groat 2017). Extraction from igneous sources is predominantly from large, low-grade (5–15% P2O5) but easily beneficiated intrusions (e.g. Siilinjärvi, Finland; Khibiny, Russia), where apatite (Ca5[PO4]3[OH,Cl,F]) crystallised as a magmatic phase. However, non-magmatically derived carbonate-bearing fluorapatite rocks can also occur in association with carbonatite and these rocks are actively mined for their phosphate content at, for example, Catalão I (Brazil), and have been mined out at the Cargill (Canada) and Glenover (South Africa) carbonatites.
Carbonate-bearing fluorapatite rocks (also termed ‘staffelite rocks’ or ‘francolite rocks’) are known from dozens of carbonatite complexes (Fig. 1). They are principally composed of massive carbonate-bearing fluorapatite, which accommodates the carbonate ion through substitution into the apatite Cl/F/OH site (type A substitution) or phosphate site (type B; Fleet 2015). In comparison with igneous apatite deposits, these rocks can reach very high grades. Martison, Canada, for example, has an indicated resource of 54.3 Mt at 23.4% P2O5 (Horner et al. 2016), compared with igneous apatite mined at Siilinjärvi with a grade of 3–6% P2O5 (Puustinen and Kauppinen 1989). In addition, in areas where phosphorites are scarce and commercial fertiliser is expensive, such as sub-Saharan Africa, direct application of local carbonate-bearing fluorapatite can deliver increased crop yields (Appleton 1994, 2002; Van Straaten 2002; Sanchez 2002). In these cases, substitution of carbonate into the apatite structure is particularly beneficial as the reactivity and agronomic potential of apatite dramatically increases with higher carbonate content (Lehr and McClellan 1972).

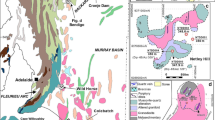
Locations of carbonatites where secondary carbonate-bearing fluorapatite has been described, grouped by genetic interpretation. Key references for each locality are as follows: 1McConnell and Gruner (1940), 2Burton (1986), 3Fletcher and Litherland (1981), 4Litherland et al. (1986), 5Walter et al. (1995a, 6b), 7Girard et al. (1993), 8Sage (1988b, 91991), 10Potapoff (1989), 11Horner et al. (2016), 12Sage (1988a), 13Sandvik and Erdosh (1977), 14Pressacco (2001), 15Erdosh (1979), 16Laval et al. (1988), 17Boulingui (1997), 18Brasseur et al. (1961), 19Decrée et al. (2015, 202016), 21Witt et al. (2019), 22Verwoerd (1986), 23Graupner et al. (2018), 24Prins (1973), 25Fernandes (1978, 261989), 27Mgonde (1994), 28Egerov (1991), 29Lapin and Lyagushkin (2014), 30Kravchenko and Pokrovsky (1995), 31Lapin et al. (2016), 32Slukin (1994), 33Chebotarev et al. (2017), 34Jayawardena (1989), 35Dahanayake and Subasinghe (1988, 361989a, 37b), 38Tazaki et al. (1986, 391987) , 40Lottermoser (1990), 41Soubiès et al. (1991), 42Ferrari et al. (2001). Bold localities with larger location markers correspond to those included in this study. Blow-out maps show the simplified bedrock geology of the main study sites, sample locations and, when available/present, the location of apatite-rich regoliths. Regolith covers the majority of the Bukusu and Catalão I complexes. The position of pits, rivers and towns are also highlighted for reference. Maps redrawn from Vartiainen and Paarma (1979), Vartiainen et al. (1990) [Sokli]; Ivanyuk et al. (2002), Mikhailova et al. (2016) [Kovdor]; Davies (1947), Baldock (1971) [Bukusu]; Verwoerd (1967) [Glenover]; and Oliveira et al. (2017) [Catalão]. Geological maps are projected using an auxiliary sphere Mercator projection, WGS1984 geoid. Note that insufficient data were available to georeference the Glenover map, and a coordinate position is given as only an approximation
Despite the importance of carbonatite-related carbonate-bearing fluorapatite rocks as a source of phosphate, there is little consensus on how they form. Two principal models are favoured: (1) a hydrothermal origin, from a late-stage, carbonatite-derived fluid (Kapustin 1983; Krasnova 2003; Verwoerd 1986; Walter et al. 1995a; Broom-Fendley et al. 2016; Chakhmouradian et al. 2017) or, (2) formation through supergene, low-temperature, dissolution and reprecipitation of apatite (Davies 1947; Verwoerd 1967; Tazaki et al. 1987; Vartiainen 1989; Vartiainen et al. 1990; Walter et al. 1995b; Richardson and Birkett 1996; Lapin and Lyagushkin 2014; Decrée et al. 2016). A third possibility combines both models as the presence of deep weathering does not necessarily preclude an earlier hydrothermal overprint ( Toledo et al. 2004). Microbiologically mediated dissolution-reprecipitation has also been suggested (Dahanayake and Subasinghe 1989a, b; Subasinghe 2013; Lazareva et al. 2015; Ponomarchuk et al. 2020), although the evidence, at present, appears circumstantial, a biological factor does warrant further investigation.
The competing hypotheses outlined above invoke contrasting mineralisation environments, but the geological evidence supporting each model is equivocal. For instance, brecciation, which is commonly associated with carbonate-bearing fluorapatite deposits, can be interpreted as a high-temperature hydraulic fracturing/explosive degassing process (e.g. at Kovdor: Kapustin 1983; Krasnova 2003), or low-temperature karstic collapse (e.g. Glenover; Verwoerd 1966). Commonly, the genesis of a deposit is determined by comparison with better-understood localities. For example, deposits considered to have formed by palaeo-tropical weathering processes, which are currently located in arctic climates (e.g. Sokli, Finland, and Martison, Canada; Vartiainen 1989; Sage 1991), bear similarity to deposits formed through modern tropical weathering in Brazil (e.g. at Catalão I, Toledo et al. 2004). However, extensive petrographic and geochemical analyses of many of these deposits are not commonly reported in detail, and compositional data for carbonate-bearing fluorapatite is typically limited to major elements from electron probe microanalysis (EPMA) and lacks CO3 contents. This scarcity of geochemical data limits the robustness of inter-deposit comparison as means of interpreting rock genesis.
The paucity of minor and trace element data is also problematic for evaluating the concentration of beneficial and deleterious elements in carbonate-bearing fluorapatite. Some trace elements, such as Mg, S, Se, Mo, Zn, Cu and Cr, are micronutrients which increase crop yield and crop health (Alloway 2008; Pufahl and Groat 2017), while the REE and U could be saleable co-products from apatite processing (Ihlen et al. 2014; Ulrich et al. 2014; Emsbo et al. 2015; Chen and Graedel 2015; Tulsidas et al. 2019). However, phosphate deposits can also contain notable concentrations of approximately 16 elements that are hazardous to human health (e.g. Cd, U, Th; Van Kauwenbergh 1997). While bulk-rock trace element analyses have been undertaken on carbonate-bearing fluorapatite rocks (Zanin and Zamirailova 2009), no study, to our knowledge, has evaluated how the concentration of these key trace elements varies on a mineral scale.
In this contribution, we present the first extensive comparison of in situ trace- and volatile-element data from carbonate-bearing fluorapatite, supported by additional textural data, using examples from Kovdor (Russia), Sokli (Finland), Glenover (South Africa), Bukusu (Uganda) and Catalão I (Brazil). We aim to (a) expand upon existing evidence for the genesis of carbonate-bearing fluorapatite rocks, (b) comment on the potential of these rocks to supply REE and other commodities as a by-product and (c) comment on the concentration of potentially deleterious trace elements for fertiliser production (e.g. Cd, U, Th).
Carbonate-bearing fluorapatite occurrences in carbonatites
Occurrences of carbonate-bearing fluorapatite in carbonatites are often unpublished or are presented in publications which may be of limited circulation or difficult to obtain. Moreover, a myriad of terms are used for carbonate-bearing fluorapatite rocks from carbonatites, including ‘carbonate-fluorapatite’, ‘staffelite’, ‘dahlite’, ‘collophane’ or ‘francolite’ rocks, as well as ‘weathered carbonatite’ and (erroneously) ‘phosphorite’, making direct comparisons challenging. Several of these terms are widely used but are ambiguous, poorly defined or are prefixed by minerals which are discredited by the International Mineralogical Association. For example, the most commonly used term, francolite, is defined as a carbonate-bearing fluorapatite with greater than 1% fluorine and ‘an appreciable amount of CO2’ (Sandell et al. 1939). However, McClellan and Van Kauwenbergh (1990) highlight that while most carbonate-bearing fluorapatite contains high fluorine contents (>3.77%), fluorine contents of <1% F can also occur in such apatite. Moreover, the term is commonly applied even where compositional data are not available, based on historical interpretations or textural data. An alternative term, ‘carbonate-fluorapatite’, has been discredited by the International Mineralogical Association as carbonate substitution is typically a subordinate component and ‘carbonate-fluorapatite’ lacks a distinct XRD pattern (Pasero et al. 2010). The term ‘carbonate-bearing fluorapatite’ is recommended instead and is used herein. However, we note that magmatic apatite from carbonatites, which is not the focus of this study, also contains a small amount of structurally bound CO3 (Prins 1973). Consequently, the term ‘carbonate-bearing fluorapatite’ is preferentially used in this manuscript to describe fluorapatite with a measurable amount of carbonate substitution formed after the cessation of magmatic crystallisation. Where an alternative term has previously been used to name a local rock type (e.g. ‘apatite francolite regolith’), this is also included for consistency.
The challenges outlined above mean that detailed accounts of carbonate-bearing fluorapatite rocks are uncommon and can be difficult to obtain. While it is not the intention of this contribution to provide a thorough review of all examples, Fig. 1 indicates where carbonate-bearing fluorapatite has been described at a carbonatite and includes key references for each locality. It is evident that carbonate-bearing fluorapatite rocks are globally distributed and are not solely localised to areas of modern tropical weathering.
In carbonatites, carbonate-bearing fluorapatite typically forms the matrix of a breccia cementing clasts of altered carbonatite, fenite or fragments of other locally derived alkaline rocks and minerals. It occurs in two geological environments: (1) as a regolith covering the carbonatite and (2) as localised breccia units within a carbonatite complex. Regolith-hosted examples are commonly interpreted as a weathering product of the underlying carbonatite host rock, and these are highlighted in Fig. 1. Such an interpretation is relatively straightforward, especially when the weathering is ongoing, as the weathering zone typically has a fixed depth, covers a large part of the complex, lacks associated carbonate minerals which have been removed through dissolution and contains residually enriched magmatic apatite grains and other minerals which are resistant to chemical weathering, such as pyrochlore. Many of these localities are also located in equatorial areas, where deep weathering profiles are common (Fig. 1). Where carbonate-bearing fluorapatite is localised to a breccia unit, its genesis is less clear and two interpretations are common: (a) karstic collapse—in a (palaeo)-weathering environment or (b) a hydrothermal vent. In both instances, mixed interpretations are common (Fig. 1).
Study sites and sample selection
Five, globally distributed, study sites were selected for this study in order to assess a representative suite of carbonate-bearing fluorapatite deposits. Bukusu and Catalão I are representative of sites where modern weathering processes are active, and samples were acquired from the weathering profiles at each of these locations. At Sokli, Kovdor and Glenover, the role of modern weathering is less apparent and, as outlined above, their genesis is less clear.
All of the study sites have some form of associated economic activity. Sokli and Bukusu are mineral resources at different stages of development, the carbonate-bearing fluorapatite rock at Kovdor is presently stockpiled while igneous apatite is mined as a co-product from the Zheleznyi pit, Glenover had a mineral reserve but was mined out between 1950 and 1970, and phosphate has been mined by at least two different companies at Catalão I for the past 30 years.
Sokli, Finland
The Sokli carbonatite complex is located approximately 145 km NE of Kemijärvi and is part of the Devonian Kola Alkaline Province. It is ~18 km2 in size, is ellipsoidal and composed of concentric rings (towards the centre) of fenite, metacarbonatite and a core comprising phoscorite, calcite and dolomite carbonatites, cross-cut by late Sr- REE and Ba-bearing veins (Vartiainen and Paarma 1979; Vartiainen 1980; O’Brien and Hyvönen 2015; Fig. 1). The complex is covered by 5–10 m of glacial till (Vartiainen and Paarma 1979), with the underlying bedrock heavily weathered to a typical depth of 5–30 m, but to a maximum of 107 m (Vartiainen 1989; Seppo Gehör, pers comm 2019).
Phosphate mineralisation is hosted in the weathered rock and principally occurs in two localities: Loitso, in the north-western part of the complex, and Pierkuli, along the eastern contact between carbonatite and fenite (Vartiainen 1989; Vartiainen et al. 1990; Fig. 1). The phosphate mineralisation is divided into three main types termed: (1) apatite francolite regolith, (2) apatite silicate residue and (3) apatite residue above ring dykes (Vartiainen 1989). The apatite francolite regolith is the most extensive ore type, and the only one which contains substantial quantities of carbonate-bearing fluorapatite. The latter two types are smaller in extent and are composed of eluvial apatite grains from the dissolution of carbonate with little/no development of secondary minerals.
The apatite francolite regolith is unconsolidated, loose and heterogeneous (Fig. 2a) and comprises several sub-types, as defined by Vartiainen (1989), including hard, granular, soft, mica-rich, Fe-rich and Mn-rich varieties. These are principally defined on the abundance of residual magmatic minerals and neo-formed carbonate-bearing fluorapatite, goethite and Mn oxides. Hard and granular sub-types are cemented by carbonate-bearing fluorapatite, to different degrees. The samples analysed for this study fit the description of the hard apatite francolite regolith subtype (e.g. Fig. 2b) and were acquired from near to the test pit in the Loitso area (Fig. 1). These rocks are creamy-white in colour, hard, porous and predominantly composed of massive apatite, with no clear fragments of carbonatite visible on the hand-specimen scale.

Examples of carbonate-bearing fluorapatite rocks from the studied localities. a Excavated unconsolidated, loose, apatite francolite regolith, and b sampled ‘hard apatite francolite regolith subtype’ (terminology after Vartiainen 1989) from Sokli. c Carbonate-bearing fluorapatite at Kovdor, with cream-white colloform apatite forming on top of a groundmass of altered mica and iron oxides. d Example of a clast in breccia from Bukusu, predominantly composed of apatite mixed with Fe oxides. e Contact between ferruginous apatite-rich breccia and apatite-bearing pyroxenite at the Glenover open pit, interpreted as a karstic collapse structure (Verwoerd 1967; van der Walt et al. 2012). Profile height is approximately 20 m. f Sample taken from waste pit at Glenover, representative of the apatite-rich breccia. Despite the brown colouration, the rock is predominantly made up of apatite, with darker areas formed of massive apatite with Fe impurities and lighter areas comprising late euhedral apatite in a pseudo-colloform habit. g Profile of weathered carbonatite at Catalão I, showing (A) barren material / laterite, (B) saprolite and (C) unweathered rock. Bench height is 5 m. The location of sample BRR1 is indicated with a close-up in (h), demonstrating the apatite-rich (cream/white in colour) composition
Kovdor, Russia
Kovdor is located ~50 km ESE of Sokli and is also part of the Kola Alkaline Province. It is ~42 km2 in size and principally composed of alkali-ultramafic rocks (olivinite, pyroxenite), surrounded by a fenite aureole. Carbonatite and phoscorite make up a much smaller component of the complex, limited to a concentrically zoned plug in the southwestern part of the intrusion, surrounded by calcite carbonatite dykes (Ivanyuk et al. 2002; Krasnova et al. 2004).
The francolite deposit at Kovdor is located to the south of the Zheleznyi iron ore mine, which currently exploits the phoscorite for magnetite, apatite and baddeleyite (Fig. 1). The carbonate-bearing fluorapatite mineralisation forms structurally complex vein-like bodies of approximately 15–300 m wide, occurring in a semi-circular zone of about 3.5 km in diameter. The structure of these carbonate-bearing fluorapatite units roughly matches the surrounding carbonatite bodies, suggesting an association between the two rock types (Lapin and Lyagushkin 2014). The carbonate-bearing fluorapatite units form in funnel-shaped depressions, typically pinching out at a depth of 70–100 m, but locally extending to ~200 m. The contact between unaltered and carbonate-bearing fluorapatite rocks is commonly sharp but disintegrated (Lapin and Lyagushkin 2014). Notably, some of the deposits are not connected with the surface (Krasnova 2003).
Krasnova (2003) identifies three cross-cutting types of carbonate-bearing fluorapatite rocks at Kovdor: (1) loose rocks of carbonate-bearing fluorapatite and vermiculite, cross-cut by (2) massive brecciated rocks which are locally cross-cut by stringers of psilomelane, which in-turn is associated with (3) psilomelane-vermiculite ‘brecciaform’ rocks. Breccia clasts in all these instances comprise residual fragments of carbonatite and other country rocks (Krasnova 2003; Lapin and Lyagushkin 2014). Three samples of material from Kovdor were acquired for this study, all of which predominantly fit the description of the loose carbonate-bearing fluorapatite and vermiculite rocks of Krasnova (2003). They were sourced from approximately 3–4 m depth, from a site of excavation to the south of the Zheleznyi mine (Fig. 1). The samples are soft and friable and composed of residual mica and magnetite, cemented by a groundmass of massive apatite and cross-cut by colloform apatite veins (Fig. 2c).
Bukusu, Uganda
Bukusu is the largest Cenozoic carbonatite complex in south east Uganda, spanning 50 km2. It consists of rings of syenite, ijolite and pyroxenite, with veins of magnetite-apatite-phlogopite rocks and carbonatite, centred on a central hill of vent agglomerate (Davies 1956; Baldock 1971; Fig. 1). Outcrop, however, is poor due to the formation of up to 45 m of laterite (Davies 1947, 1956).
Phosphate rock occurs on Busumbu ridge, to the south of the central vent (Davies 1947, 1956; Fig. 1). These rocks form in the regolith above unweathered carbonatite and magnetite-apatite-phlogopite rocks. The phosphate rock is heterogeneous and consists of a mixture of hard phosphatic rock in a mass of soft limonitic material. A single sample of the hard phosphatic rock was investigated for this study, sourced from the British Geological Survey collections (Fig. 2d). Little information is available about the environment from which this sample was collected, except that it is a surface sample. The hard phosphatic rock is grey–brown, friable and concretionary. It is composed of spongey, silvery-white carbonate-bearing fluorapatite locally forming stellate or fibrous aggregates around cores of colourless apatite crystals and, to a lesser extent, mica or magnetite.
Glenover, South Africa
The Glenover carbonatite complex is located approximately 170 km NNW of Johannesburg. It is approximately 15 km2 in size, ellipsoidal and principally composed of pyroxenite, with variable amounts of phlogopite, magnetite and apatite (Verwoerd 1967). Dolomite carbonatite is the predominant carbonatite type and occurs as dykes, irregular veins and sills which dip towards the centre of the complex. Calcite carbonatite is subordinate and occurs towards the edge of the complex. Minor, late-stage, REE-bearing carbonatite stringer veins occur throughout the complex. Exposure is poor, and weathering is extensive, with karst-like dissolution cavities reported down to depths of approximately 30 m (Verwoerd 1967) and oxidation of pyrite in carbonatite occurring to depths of 150 m (van der Walt et al. 2012).
The Glenover phosphate deposit is located near the centre of the complex (Fig. 1), in an area of elevated radioactivity, and is composed of a ferruginous apatite-rich breccia body, approximately 200 by 250 m in size. The breccia body is likely to be bowl-shaped, and shallow in depth, with a sharply defined contact with the carbonatite (Fig. 2e) occurring at a maximum depth of approximately 150–170 m (Verwoerd 1967, 1986; van der Walt et al. 2012). The apatite-rich breccia is a dark red heterogeneous rock consisting of iron oxides, haematite after magnetite and small apatite grains, termed ‘consolidated ferruginous earth’ by Verwoerd (1967; Fig. 2f). Clasts in the breccia include fine-grained pale-red/purple fragments, predominantly composed of apatite mixed with Fe oxides, cemented by very fine-grained colloform white apatite (Verwoerd 1967). The apatite-rich material is now mined out, but samples matching the above description were acquired from adjacent dumps. For comparison, a sample of weathered carbonatite was also collected from a drill core (HTAC_008).
Catalão I, Brazil
Catalão I is located about 10 km NE of the town of Catalão in Goiás state and is part of the Late Cretaceous Alto Paranaíba Alkaline Province (Gomes and Comin-Chiaramonti 2005). The intrusion is approximately 6 km in diameter and is composed of a core of dolomite carbonatite, nelsonite and phoscorite dykes, surrounded by pyroxenite, glimmerite and serpentinised peridotite (Cordeiro et al. 2010, 2011; Fig. 1).
Phosphate mineralisation is hosted in both laterite and saprolite, concentrated above the carbonatite, and may range between 15 and 250 m in thickness (Morteani and Preinfalk 1996; Fig. 2g). Economic mineralisation is located in the north and central parts of the complex, with grades reaching 30% P2O5 (Carvalho and Bressan 1989). The laterite is divided into different zones by different authors (cf. Morteani and Preinfalk 1996; Oliveira and Imbernon 1998; Toledo et al. 2004), but a simplified sequence from the surface down can be summarised as follows: (a) barren material / laterite, (b) saprolite and (c) fresh rock (Fig. 2g). The thickness and extent of these zones is very irregular. The top-most zone is considered overburden, which includes alluvium and soil, and apatite grades are low. The saprolite is somewhat heterogeneous, with lenses of mica- or haematite-rich material, ‘silicicrete’ and fragments of unweathered rock. The sample analysed in this study is from the saprolite (Fig. 2g) and is brilliant white in colour, being predominantly composed of massive apatite, with minor residual grains of mica (Fig. 2h). For comparison, a sample of weathered carbonatite was also collected from a drill core (BRR3).
Analytical techniques
Textural characterisation of the samples was undertaken using a standard petrographic microscope, CITL Mk3 and Mk5 cold-cathodoluminescence (CL) equipment, and an FEI Quanta 600 scanning electron microscope (SEM). The CL was typically operated with a beam current of ~300–500 μA and a corresponding voltage of 8–12 kV.
Electron probe micro analyses (EPMA) were undertaken at Camborne School of Mines using a JEOL JXA 8200 instrument, operated with a 10 nA, 15 kV beam defocussed to a 5-μm spot. Peak counting times were 20 s for CaO, Cl, SO3, La2O3, Ce2O3, MnO and FeO; 30 s for P2O5, F and SrO; 40 s for SiO2 and Y2O3; and 60 s for Na2O, with background times half those of the peaks. The primary standards used for La, Ce, Nd and Y were synthetic REE glasses from the University of Edinburgh; Ca, P and F were standardised against MKII Durango fluorapatite, while all other elements were standardised using Astimex mineral standards. X-ray counts were converted to oxide concentrations using the inbuilt Jeol φρz correction programme. Astimex fluorapatite and in-house Durango and Odegaarden apatite grains were used as secondary standards, with results within 2% of published data from these localities.
Trace elements were analysed, using LA-ICP-MS, at the University of Leeds and Camborne School of Mines. Ablation at Leeds was carried out using a Lambda Physik 193 nm ArF excimer laser with a 100 μm spot and a repetition rate of 5 Hz, maintaining a fluence of approximately 4 J cm−2. This was coupled to an Agilent 7500c ICP-MS operating in reaction mode, with a flow rate of 2.5 ml min−1 H2, to suppress 40Ar+ interference on 40Ca. Ablation at Camborne School of Mines utilised a New Wave Research 213 nm Nd-YAG laser coupled to an Agilent 7700 ICP-MS. The laser was operated with a 100 μm spot and a repetition rate of 10 Hz, resulting in a fluence of 3–4 J cm−2. In both cases, median Ca concentrations for the apatite generation analysed, obtained by EPMA, were used as the internal standard composition and NIST SRM 610 was used for calibration. SRM 612, and at Camborne School of Mines, in-house Durango and Odegaarden apatites were used as a secondary standards with the concentrations of all analysed elements falling within 10% of published values.
Infrared spectra were collected for polished surfaces using a ThermoNicolet i10 FTIR spectrometer fitted with a Ge-tipped attenuated total reflectance head, housed at the University of Bristol. In total, 128 scans were collected from 650 to 4000 cm−1 at a resolution of 4 cm−1 using a mercury cadmium telluride detector and a KBr beam-splitter in absorbance mode. The effective collection area ranged in size from approximately 30 to 10 μm squares.
Results
Apatite textures
Textural details for apatite from each locality are presented in Figs. 3, 4, 5, 6 and 7, grouped by locality. In total, five different apatite habits are recognised. These habits can be summarised as follows: (1) ovoid grains, (2) epitactic overgrowths, (3) massive, (4) colloform and (5) late euhedral. Not all habits occur in apatite from each example, with apatite from Bukusu and Glenover notably not exhibiting epitactic overgrowths or colloform types. The different habits are interpreted as ‘primary’ (ovoid grains residually derived from a cognate carbonatite melt) and ‘secondary’ (overgrowths, massive, colloform and late euhedral), which could be derived from either hydrothermal or weathering processes. The term ‘secondary’ is not intended to convey information about the apatite genesis, but is used here to describe apatite forming after ovoid grains.

CL (a, b) and BSE images (c–f) of different primary and secondary apatite generations from Sokli. a, b Primary apatite grains hosted in a massive apatite groundmass, cut by fractures cemented with colloform apatite and haematite. Note the muted blue luminescence in the massive apatite, and the lack of luminescence in the colloform apatite. c A primary apatite grain overgrown by euhedral zoned secondary apatite, hosted in a massive apatite groundmass. d, e Primary apatite grains with spongey secondary apatite overgrowths, possibly after euhedral zoned secondary apatite (d) is fractured and infilled with a darker generation of spongey secondary apatite. f Primary apatite with a small epitactic secondary apatite rim, overgrown by colloform apatite formed of 20–30 μm wide parallel apatite needles

Primary and secondary apatite from Kovdor. a CL image of primary apatite grains hosted in massive apatite, locally preserving remnant cleavage after carbonate, and overgrown by colloform apatite. Note the dark blue luminescence of the massive apatite, and the sky blue luminescence in colloform apatite. b Fractured primary apatite grains overgrown by epitactic carbonate-bearing fluorapatite, hosted in a massive apatite groundmass. c Massive carbonate-bearing fluorapatite, with remnant cleavage planes after carbonate minerals. d Colloform carbonate-bearing fluorapatite overgrown by blister-like apatite and, subsequently, crandallite. Holes and lines are ablation pits

Apatite grains from Catalão I. a CL image of primary apatite grains hosted in a cement of secondary carbonate. b Euhedral skeletal secondary apatite, BSE image

Apatite grains from Bukusu. a CL image of apatite hosted in massive carbonate-bearing fluorapatite, overgrown by colloform carbonate-bearing fluorapatite. Note the non-luminescence of the carbonate-bearing fluorapatite. b, c Close-up BSE images of grains in a showing epitactic zoning in the carbonate-bearing fluorapatite and a fracture of carbonate-bearing fluorapatite crossing a primary grain

CL (a, d, e) and BSE (b, c, e) image of primary and secondary apatite from Glenover. a–c Primary rounded grains, hosted in secondary apatite (a, c) and in a rock composed of secondary Fe oxides and calcite (b). Note the breakdown and replacement of primary apatite grains in a, c to secondary, yellow-/non-luminescent apatite and the presence of bright grains of Ca-rich monazite associated with the massive apatite. d Colloform apatite, transitioning into late euhedral apatite. e, f Late euhedral apatite, associated with small euhedral grains of Ca-rich monazite. Note the yellow luminescence corresponds with spongey apatite while non-spongey areas do not luminesce
Ovoid grains are typically within the centre of ‘clasts’ and are locally associated with magnetite and biotite. Oscillatory zoning within the ovoid grains is evident in CL images, ranging in colour from yellowish to purple. Zoned, ovoid grains are ubiquitous in plutonic carbonatites and are considered to crystallise early in a carbonatite melt with rounding likely forming due to subsequent mechanical or chemical abrasion during emplacement (Chakhmouradian et al. 2017). Based on this common association, ovoid apatite is almost-certainly derived from a carbonatite melt. Zoning within the grains is not used here to differentiate the ovoid apatite into further sub-types. Grains from Glenover and Catalão I are partially fragmented and cemented by calcite and Fe oxide minerals, un-associated with carbonate-bearing apatite (Figs. 5a and 7b).
Epitactic overgrowths are distinct from zoning within the ovoid grains as indicated by a clear change from an ovoid habit to euhedral hexagonal overgrowths. This change is coupled with a decrease in BSE intensity, substantially reduced CL intensity, and a drop in RI compared with the original ovoid grain (Figs. 3c, 4b and 6b). Epitactic overgrowths typically exhibit fine euhedral oscillatory zoning in BSE images, with no corresponding CL variation apparent. Epitactic overgrowths are commonly overprinted by massive apatite, with the original overgrowth only partly preserved (Figs. 3c, f and 4b), or with just the crystal outline retained (Fig. 3d, e).
Massive apatite forms the groundmass of all the carbonate-bearing fluorapatite rocks studied. It grows around and cross-cuts earlier ovoid and euhedral grains (Figs. 3d and 6c). Locally, the massive apatite retains linear features intersecting at ~60° and ~ 120°, suggesting replacement of carbonate minerals (Fig. 4a, c). The massive structure is turbid in thin sections and composed of many fine-grained interlocking needles between 1 and 10 μm wide, but up to several hundred micrometres in length (Fig. 3f). Locally, the massive apatite is spongey, with spongey areas commonly corresponding to lower BSE intensity (Figs. 3c and 7c). The spongey texture is likely to be indicative of partial apatite dissolution (Putnis 2002, 2009). In most samples, a locally precipitated reaction product is not evident but massive apatite from Glenover is also associated with Ca-rich monazite-(Ce) and cerianite-(Ce) (Fig. 7c). Samples from Bukusu and Sokli do not exhibit any luminescence in CL images, but massive apatite from Kovdor luminesces a muted blue colour while massive apatite from Glenover luminesces yellow.
Colloform apatite is the most distinctive megascopic apatite habit (Fig. 2). The samples in this study are cream-white in colour, but turquoise coloured samples have been reported (Chakhmouradian et al. 2017). In thin section, colloform apatite is first formed of late euhedral apatite needles radiating perpendicular to a growth surface (Fig. 3a, b). The needles vary in thickness, between 1 and 50 μm, although they most commonly are towards the lower end of this range. Euhedral apatite from Catalão I is considered here as part of the late euhedral stage, although it is subtly different from the other examples (Fig. 5b). For instance, original ovoid grains are not commonly found in the cores of the ‘overgrowths’ although their presence was demonstrated by Toledo et al. (2004) in their type 5 apatite. Additionally, while fine oscillatory zoning is not present, similar changes in the crystallisation environment are manifested by lamellar, or skeletal, growth. Euhedral apatite overgrowths at Catalão I also exhibit subtle colour variation in CL images.
Up to three generations of colloform apatite occur at the different sites and these vary in turbidity, needle thickness and colour. Early generations at Sokli are composed of thinner needles than later stages and are commonly darker in colour (Fig. 3f). In the final growth stages at Glenover, the needle-like form is lost, and the apatite is more equant, forming late euhedral apatite (Fig. 7d–f). The needle-like structure is also absent in later colloform generations at Kovdor, and apatite forms a blister-like texture (Fig. 4d).
Colloform apatite is commonly followed by the growth of REE-bearing crandallite group minerals as well as iron oxides (Figs. 3b and 4d). Additionally, at Glenover, cerianite and Ca-rich monazite also occur in pore spaces, locally associated with quartz, after late euhedral apatite. Luminescence is absent in colloform apatite with the exceptions of Kovdor, where it is muted blue to green in colour (Fig. 4a), and Glenover where the yellow luminescence present in the massive apatite is retained. However, yellow luminescence at Glenover is only found in spongey, altered apatite, and where unaltered secondary apatite is preserved, it is non-luminescent (Fig. 7e).
Compositional variation
FTIR spectra indicate similarities between carbonate-bearing fluorapatite types from the studied localities. Spectra from all samples indicate the presence, to varying extents, of CO32− in the apatite structure, as indicated by V3 CO3 peaks between 1400 and 1550 cm−1 (Fig. 8). Nonetheless, there is considerable variety between the magnitude and shape of the CO3 peaks. Ovoid apatite grains exhibit the lowest magnitude V3 CO3 peaks, compared with other apatite types analysed. They are additionally characterised by a distinctive peak at approximately 720–730 cm−1, and a small, but consistent, OH peak at ~3530 cm−1 (Fig. 8). The 720–730 cm−1 peak is probably related to vibrational OH bands that are weakly hydrogen bonded to F ions (Fowler 1973). In contrast to the grains, colloform apatite does not exhibit peaks at 720–730 and ~ 3530 cm−1, but does show a broad stretch between 2800 and 3600 cm−1, related to molecular (i.e. not structurally bound) H2O, as well as a peak at around 2900 cm−1 (Fig. 8). The V3 CO3 peaks are consistently higher than those in the ovoid grains. Euhedral overgrowths from Bukusu and massive apatite from Kovdor exhibit the same spectral features as colloform apatite (Fig. 8). In grains from Catalão I and Kovdor, as well as in massive apatite at Kovdor, a small peak is apparent at 2350 cm−1, corresponding to molecular CO2, which also correlates with a broad H2O peak (2800–3600 cm−1). These peaks are probably due to fluid inclusions in the area analysed.

IR spectra of apatite from the study sites. Note the presence of V3 CO3 peaks in all samples, but the elevated size of those from secondary apatite. Primary apatite has a distinct OH peak at 3530 cm−1 and an OH-F peak around 720 cm−1. Secondary apatite has a broad stretch between 2800 and 3600 cm−1. Spectra are normalised to the maximum PO4 intensity, and intensity units are arbitrary. PO4 peaks are trimmed in order to emphasise detail in smaller peaks. Dashed spectra indicate possible mixed signal with fluid inclusion(s). Data in ESM Table S5
Apatite major and trace element compositions are presented in ESM Tables S1–4. The composition of the ovoid grains is typical of apatite derived from a carbonatite, with relatively high F, Sr, Na and REE contents, and low Cl and Mn, compared to apatite from other rock types (e.g. granitoids; Hogarth 1989; Belousova et al. 2002; Broom-Fendley et al. 2017). All grains exhibit almost-straight REE patterns, with a negative Y anomaly, although grains from Glenover exhibit a small hump around Ce–Pr (Fig. 9). Thorium and uranium form a positive correlation, with grains from Glenover reaching the highest Th contents, up to ~300 μg/g (Fig. 10a). The concentration of U in some Sokli grains is decoupled from the Th contents, possibly as a result of the ablation of inclusions or contamination from later surrounding apatite generations.

Chondrite-normalised REE distribution of primary apatite grains from the study sites. Note: Tm and Lu are not analysed in some samples. Chondrite values from McDonough and Sun (1995)

Composition of ovoid grains and secondary apatite from the study sites. Coloured arrows indicate the compositional trends for apatite from individual complexes. Error bars are derived from 2 se of the mean counts. Grey dotted bars indicate the whole-rock composition of Moroccan Phosphate Rock (MPR) standard BCR-032 (Serrini and Haemers 1980), except for that of La, which represents the median composition of 765 globally distributed phosphorite samples (Emsbo et al. 2015). Grey dashed bar illustrates the maximum Cd content for fertiliser sold in the EU from 2032 (Ulrich 2019)
Secondary apatite has notably lower EPMA totals than the primary grains (ESM Table S2), with the lowest from epitactic overgrowths from Kovdor and Sokli, reaching ~90%. The decreasing total corresponds to an under-full PO4 site (Fig. 10b), and almost-certainly reflects the substitution of CO3 into the structure, as supported by the presence of CO3 peaks from the FTIR data (Fig. 8).
Minor and trace element concentrations are notably lower in secondary apatite than primary apatite grains, typically below the EPMA detection limit and, in some cases, the LA-ICP-MS detection limit too. Although only a limited number of analyses are available, epitactic overgrowths are the closest in trace element contents to primary grains, with similar REE, Na and Sr concentrations (Fig. 10c, ESM Table S4). REE, Na and Sr contents in massive and colloform apatite drop steadily towards concentrations below the LA-ICP-MS detection limit (Fig. 10c, d). However, some trace element contents are elevated relative to earlier ovoid apatite. A key example is U, which can reach contents of over 1000 μg/g and, in samples from Kovdor, exhibits a weak negative correlation with REE contents (R2 = 0.7; Fig. 10e). Conversely, U and REE positively correlate in the euhedral overgrowths from Catalão I (R2 = 0.75; Fig. 10e). Transition metals, such as V, Cu, Zn and Cd, also exhibit elevated contents in the secondary apatite generations, relative to ovoid apatite. Moreover, with the exception of Cu, these elements also exhibit a weak positive correlation with U (Figs. 10f–h), although data for Zn are limited to only a few analyses. Similarly, data for Mo and Cr are restricted to a small number of data points and are close to the detection limit, but also exhibit a tentative positive correlation with U.
The REE distribution of non-magmatic apatite from Sokli, Kovdor and Glenover emulates that of the magmatic apatite grains in each example, but with lower overall REE contents (Fig. 11). The REE patterns for Catalão I and Bukusu, however, are notably flat, contrasting with the LREE-enriched pattern of the primary apatite grains from these localities. Secondary apatite from Catalão I and Bukusu both exhibit a negative Ce anomaly, and a positive Y anomaly, which is most pronounced in samples from Catalão I. Massive and colloform apatite from Sokli and Kovdor generally also have a strong negative Ce anomaly, but some analyses exhibit a small positive anomaly, or none at all. In addition, at Sokli and Kovdor a Y anomaly is typically absent, or slightly negative (Fig. 11).

Chondrite-normalised REE distributions of secondary apatite from the study sites. The primary apatite distribution is also shown (block colour) for reference. Dashed lines highlight different secondary apatite textures from the same locality. Massive and colloform apatite distributions from Sokli and Kovdor are merged, as there is no clear difference between the two. Chondrite values from McDonough and Sun (1995)
Compositional differences between different secondary apatite generations are limited to analyses from Kovdor, Sokli and Glenover. Massive and colloform secondary apatite from Sokli is compositionally identical, within the range of data for each apatite type (ESM Table S4). While there is also substantial overlap between different generations at Kovdor, some differences between the different apatite generations are apparent (Fig. 10). Massive apatite from Kovdor has the highest REE, Na and Sr contents, most reminiscent of ovoid apatite, compared to colloform apatite. Conversely, a later generation of colloform apatite has noticeably the lowest REE, Na and Sr contents, and the highest U and Cd concentration (Fig. 10g, h). There is no noticeable compositional change from the core to rim of colloform apatite, nor any substantial variation in composition with luminescence colour. At Glenover, massive apatite has a marked negative Ce anomaly which is absent in the late euhedral type (Fig. 11e).
Interpretation and discussion
Generalised paragenetic evolution
Figure 12 is a generalised paragenetic sequence of the different apatite generations. Ovoid magmatic fluorapatite grains (i) are fragmented and overgrown by epitactic carbonate-bearing fluorapatite (ii) which is enveloped in massive, fine-grained carbonate-bearing fluorapatite (iii). These rocks, when subsequently fragmented, show additional late euhedral (iv) and colloform (v) generations of apatite occurring along fractures and in vugs. Multiple colloform generations may also occur. REE minerals (vi) typically crystallise last. Locally, magmatic grains exhibit resorption / dissolution (i-a), relating to changing magmatic-hydrothermal conditions during carbonatite emplacement. Dissolution and resorption of epitactic carbonate-bearing fluorapatite (ii-a) and massive carbonate-bearing fluorapatite (iii-a) is common in most samples.

A generalised paragenetic sequence for carbonate-bearing fluorapatite deposits associated with carbonatites. Roman numerals denote the successive stages of apatite growth. The columns to the right of the paragenetic stages indicate deposits where the different stages have been identified. Black lines denote evidence from this study, while grey lines are inferred from other studies (Oliveira and Imbernon 1998; Tazaki et al. 1986, 1987; Dahanayake and Subasinghe 1988, 1989a, b; Potapoff 1989; Sage 1991; Araújo 2015). Numbers in square boxes refer to figures which show evidence for the interpreted stage. Numbers in circles indicate the equivalent stage as interpreted by previous researchers (Sokli: Vartiainen 1989; Kovdor: Krasnova 2003; Catalão I: Toledo et al. 2004; Mabounié: Boulingui 1997; Matongo: Decrée et al. 2016; Juquiá: Walter et al. 1995a)
Previous workers (Vartiainen 1989; Krasnova 2003; Toledo et al. 2004) have recognised the different successive generations of secondary apatite at the Sokli, Kovdor and Catalão I deposits, and these are included in Fig. 12 for comparison. At Sokli, Vartiainen (1989) recognised three generations of carbonate-bearing fluorapatite. Stage 1 and stage 2 carbonate-bearing fluorapatites form a cement, with stage 1 comprising a microcrystalline aggregate with goethite and crandallite and stage 2 forming small (<0.02 mm) individual fibrous crystals of carbonate-bearing fluorapatite, which typically occur as radial growths. Stage 3 is milky-white and occurs in fissure cracks and cavities with abundant dust-like inclusions. At Kovdor, Krasnova (2003) divides carbonate-bearing fluorapatite into four sub-types. These consist of (1) overgrowths on relict residual magmatic apatite grains, (2) fine-grained aggregates intermixed with Fe and Mn oxides, (3) crusts/kidney-like aggregates and (4) porcelaneous aggregates. At Catalão I, apatite in the laterite is present both as residual magmatic grains and as neo-formed apatite and is subdivided into 7 different sub-types by Toledo et al. (2004). Residual grains (1) are commonly strongly corroded and fractured, with corrosion typically located along fracture planes. Locally, the residual grains are rimmed (2), replaced or cross-cut (3) by secondary apatite. In places, overgrowths form small euhedral tabular prismatic crystals (4) or larger, lamellar grains (5). Sub-types 6 and 7 grow in fissures or aggregates in weathered material. They consist of fibrous crystals in aggregates growing perpendicular to the growth surface (6) or globules, locally linked together in botryoidal growth (7). Other phosphate minerals in the laterite include gorceixite, crandallite, florencite and, in ‘silicicrete’, monazite (Oliveira and Imbernon 1998).
In general, the paragenetic sequence presented in Fig. 12 is coincident with that of Toledo et al. (2004). Nonetheless, some subtle differences include the exclusion of their stage 3 (intracrystalline bands of acicular secondary apatite across grains of primary apatite), which we consider to be coincident with the formation of massive apatite as part of our stage iii (Figs. 3d and 6c). We also only recognise their stage 5 (lamellar secondary apatite surrounding primary apatite) in our sample from Catalão I and tentatively associate this with our late euhedral stage iv.
Localised dissolution of carbonate-bearing fluorapatite is prominent in some samples, especially from Sokli. This has the effect of masking some important paragenetic stages, such as the formation of epitactic overgrowths (stage ii; Fig. 3c–e), which may explain the absence of this stage in previous work (Vartiainen 1989). With the recognition of the presence of epitactic overgrowths at Sokli, it is clear that the different apatite stages from Kovdor, Sokli and Catalão I are texturally near-identical. While epitactic overgrowths at Bukusu and Glenover are not readily apparent, it is quite possible that these are either obscured by subsequent alteration or are missing as a consequence of the limited availability of sample material.
Some of the aspects of the paragenetic sequence presented in Fig. 12 have been previously noted by workers at several different deposits. In cases where work has focussed on different aspects of a deposit, information on the order of crystallisation is limited to noting the presence of residual apatite hosted in a secondary apatite groundmass (Cargill, Canada, [Sage 1991]; Ngualla, Tanzania, [Witt et al. 2019]; Araxá, Brazil [Mariano 1989]), the formation of secondary apatite on the rim of a primary apatite grain (Dorowa, Zimbabwe [Fernandes 1978]; Mt Weld, Australia [Lottermoser 1990]), or the formation of fibrous apatite in fractures (Anitapolis, Brazil, [Girard et al. 1993]). However, paragenetic descriptions of secondary apatite are available for the Mabounié, Matongo and Juquiá complexes (Walter et al. 1995a, b; Boulingui 1997; Decrée et al. 2016), and sufficiently detailed descriptions of apatite textures are reported for the Eppawala, Salitre I and Martison deposits such that a crystallisation sequence can be inferred (Tazaki et al. 1986, 1987; Dahanayake and Subasinghe 1988, 1989a, b; Potapoff 1989; Sage 1991; Araújo 2015). These localities have also been summarised in Fig. 12, with reference to ascribed paragenetic stages.
Notably, the textural evolution of other carbonate-bearing fluorapatite deposits bears a strong similarity to those studied in this contribution. For instance, epitactic apatite, equivalent to our stage ii (Fig. 12), occurs at Mabounié (described by Boulingui 1997 as type 2 ‘overgrowing apatite’), Matongo (as ‘prismatic apatite’; Decrée et al. 2016) and Juquiá (as ‘type 2 apatite’; Walter et al. 1995a, b). ‘Massive apatite’, equivalent to stage iii, occurs in most comparison examples with the possible exception of Juquiá where it is not explicitly described. At Mabounié, massive apatite exhibits similar remnant cleavage after carbonate, as is present at Kovdor (Boulingui 1997). Commonly, this stage of apatite growth is also associated with the formation of iron oxyhydroxide minerals (Salitre I, Mabounié, Martison), or siderite (Matongo, Tomtor; Kravchenko and Pokrovsky 1995). The development of late euhedral apatite (type iv) is ubiquitous, although at some localities it is unclear if this stage post-dates massive mineralisation, or if massive mineralisation is a further manifestation of this mineralisation type, with no pore space remaining. The development of subsequent colloform apatite generations (type v) is quite variable. At Juquiá, for instance, Walter et al. (1995b) report several generations of colloform and late euhedral apatite (their apatite types 3–6), with later bands cross-cutting earlier colloform generations. Similarly to the studied localities, the occurrence of different REE minerals late in the paragenesis, where reported, is quite variable. Neo-formed monazite (or rhabdophane) is commonly associated with carbonate-bearing fluorapatite (Matongo, Salitre I), but does not always occur at the end of the paragenetic sequence (Decrée et al. 2016). The presence of crandallite group minerals, such as florencite, notably varies depending on weathering depth (Kravchenko and Pokrovsky 1995; Oliveira and Imbernon 1998; Araújo 2015).
While, as highlighted above, there are subtle local textural differences between the carbonate-bearing fluorapatite deposits in this study and those previously investigated, we consider the paragenetic sequence in Fig. 12 to likely be applicable to carbonatite-hosted apatite deposits in general. Considering the textural similarities alone, a common process for the formation of these rocks, as suggested by other authors (Lapin and Lyagushkin 2014), seems highly probable. As several of the examples listed here (Catalão I, Mabounié, Salitre I) unequivocally formed by weathering, by extension, we propose that such a mechanism can account for carbonate-bearing fluorapatite deposits related to most carbonatites, even where modern weathering is not active (e.g. Glenover, Kovdor).
Compositional similarities and evolution
Compositionally, primary grains from each locality are identical to apatite in unaltered carbonatites (e.g. Kovdor, Zaitsev et al. 2015; Sokli, Hornig-Kjarsgaard 1998). While some grains texturally appear partially altered, their composition does not diverge from that of the primary grains (Fig. 10), suggesting that this small-scale alteration is within the compositional bounds of magmatic carbonatite-apatite.
Textural similarities between carbonate-bearing fluorapatite from the studied deposits are strongly borne out in the compositional data, especially those where samples are available for the full paragenetic sequence. The FTIR spectra and major element compositions for non-magmatic apatite from each locality are near-identical, with the exception of epitactic overgrowths (Fig. 8; ESM Table S2), demonstrating the presence of CO3 in the apatite structure, as inferred from low EPMA totals and noted in previous studies (Davies 1947). While the trace element contents vary somewhat between different deposits, many of the compositional trends are the same. For example, all secondary apatite varieties trend towards higher U, and lower REE, Na and Sr contents, than primary apatite from the same deposit (Fig. 10c–e). Importantly, this trend is broadly in harmony with the textural evolution of the apatite, with later paragenetic stages containing the highest U, and lowest REE, Na and Sr contents (Fig. 10c–e).
REE distribution and Ce anomalies
The shape of the REE distribution for colloform and massive carbonate-bearing fluorapatite from Sokli, Kovdor and Glenover broadly matches that of primary ovoid grains from these localities (Fig. 11a–c). In early stages from Kovdor, the distribution matches that of primary apatite from the same deposit, but with a negative Ce anomaly (Fig. 11a). In later generations, the REE concentration decreases linearly for each REE dropping off to below the LA-ICP-MS detection limit.
The lack of REE fractionation in later generations of carbonate-bearing fluorapatite at Sokli, Kovdor and Glenover is intriguing. For instance, it contrasts with the extreme REE fractionation observed in hydrothermal apatite from Tundulu, Kangankunde and Songwe (Malawi), which are enriched in the HREE relative to earlier ovoid igneous apatite from the same complexes (Broom-Fendley et al. 2016, 2017). Such a contrast circumstantially supports the textural evidence that the carbonate-bearing fluorapatite from the studied localities is not derived from a hydrothermal environment. However, in the Malawian examples, the change in REE distribution is considered to be caused by the differing stability of some REE anion complexes between the LREE and HREE (Migdisov and Williams-Jones 2014). Such a process is not unique to the hydrothermal environment and, depending on the REE complexes formed, can be absent in hydrothermally formed minerals. While it is beyond the scope of this study to comment on the composition of the REE-transporting agent in the studied examples, the lack of fractionation rules out a ligand which fractionates the REE, such as Cl (at elevated temperature; Migdisov et al. 2009), F, and, at ambient temperature, CO3 (Luo and Byrne 2000, 2004).
With few exceptions, all secondary apatite exhibits a negative Ce anomaly (Fig. 11), a feature also noted at the Eppawala, Juquiá and Kovdor complexes (Tazaki et al. 1987; Walter et al. 1995a, b; Chakhmouradian et al. 2017). The presence and magnitude of the Ce anomaly does not change with different paragenetic stages.
Anomalous Ce contents, relative to the other REE, are caused by the transition of Ce3+ to Ce4+ in highly oxidizing and alkaline pH conditions and, consequently, the different geochemical behaviour of Ce4+ compared with the other REE (Henderson 1984; Akagi and Masuda 1998). Such conditions are typically met during rock weathering and low-temperature surface processes (Braun et al. 1990, 1993; Leybourne and Johannesson 2008; Janots et al. 2015; Sanematsu and Watanabe 2016). For example, during the weathering of granitic rocks, acidic fluids, sourced from the breakdown of organic matter in the soil horizon, transport the REE as aqueous complexes down the weathering horizon. This process splits the weathering profile of a granite into an upper zone of REE depletion and a deeper zone of REE accumulation, where the REE precipitate due to an increase in pH caused by a change in mineralogy or the presence of groundwater (Sanematsu and Watanabe 2016). Under most ambient conditions, however, Ce4+ is less soluble than other REE3+ ions, resulting in Ce4+ preferentially accumulating in the upper REE depletion zone as a solid phase, such as cerianite-(Ce), or adsorbing onto Mn oxides, Fe oxides or clay minerals (Bau 1999; Takahashi et al. 2000; Bau and Koschinsky 2009). These solid phases, therefore, exhibit a positive Ce anomaly, while redeposited minerals in the REE accumulation zone, lower in the profile, exhibit a negative Ce anomaly. The negative Ce anomaly in the secondary carbonate-bearing fluorapatite is, therefore, consistent with formation from a fluid where Ce4+ has already partitioned into Ce-rich minerals higher in the weathering horizon. Elevated Ce4+ contents in the upper part of the weathering horizon have been indicated by the presence of CeO2-rich pyrochlore from the upper weathering levels at Mt Weld (Lottermoser and England 1988) and Lueshe (Wall et al. 1996). However, the processes of weathering and REE redistribution above a relatively P2O5-poor granite are most likely not representative of those above a P2O5-rich carbonatite. For instance, previous studies of the REE profiles of weathered carbonatites do not show Ce anomalies in the REE minerals or bulk rock from the upper levels of a carbonatite weathering zone (e.g. Lottermoser 1990; Walter et al. 1995b; Morteani and Preinfalk 1996; Oliveira and Imbernon 1998; Broom-Fendley et al. 2020). Furthermore, the remarkably low REE concentration of carbonate-bearing fluorapatite (ESM Table S4) contradicts the notion that it is forming in a REE accumulation zone, as apatite typically readily incorporates the REE. Moreover, at Kovdor, Bukusu, Glenover and Catalão I, Fe oxide and Ce4+-bearing minerals commonly occur after the formation of the final carbonate-bearing fluorapatite stage. There is, thus, no immediately obvious phase which is likely to retain and fractionate Ce4+ early in the genesis of these rocks, and, therefore, another mechanism to remove Ce4+ may be in operation.
While uncommon, negative Ce anomalies can occur in residual solid phases, and preferential transport of Ce4+ over the trivalent REEs has been described in certain conditions. A key example of a rock with a negative Ce anomaly is a marine phosphorite (McArthur and Walsh 1985). However, this is inherited from the REE pattern of seawater which, in turn, is derived from the retention of Ce4+ in weathered solids, akin to the mechanism discounted above. As an alternative example, organic ligands, especially siderophores, have been shown experimentally and in natural studies to preferentially complex with Ce4+ over the REE3+, and retain Ce4+ in solution (Pourret et al. 2007; Tanaka et al. 2010; Loges et al. 2012; Bau et al. 2013; Kraemer et al. 2015, 2017). Thus, preferential transport of Ce4+ could result in a negative Ce anomaly in the residual solid phase and may explain the negative anomaly in the carbonate-bearing fluorapatite deposits. Alternatively, anomalously positive Ce anomalies have also been documented in highly alkaline waters where Ce4+ forms stable complexes with carbonate (Möller and Bau 1993; Johannesson et al. 1994). Given the extreme dissolution and replacement of carbonate in the secondary apatite-rich rocks, high carbonate concentrations in the REE-transporting fluids are feasible and thus carbonate complexation may also explain the negative Ce anomalies in the residual secondary apatite. The presence of a Ce anomaly in these rocks is, therefore, not unequivocal evidence for formation in a weathering environment, as other processes can lead to Ce fractionation. However, the ubiquity of the Ce anomaly in the secondary carbonate-bearing fluorapatite does point towards formation in an oxic environment, and in most circumstances, this requires the presence of organic ligands which can only occur in the surface environment.
Implications of elevated U contents in secondary carbonate-bearing fluorapatite
In addition to the presence of a Ce anomaly, the increasing concentration of U in the secondary carbonate-bearing fluorapatite also points towards a strong redox control on the formation of this mineral. Oxidised U6+ is relatively soluble, compared to U4+, and can be transported as phosphate or carbonate complexes in neutral and alkaline solutions, respectively (Langmuir 1978). The transport and reprecipitation of uranium in weathering profiles is well documented, with uranium commonly adsorbing onto Fe oxide phases or reprecipitating as uranium-bearing phosphate minerals in the weathering horizon (Murakami et al. 1997; Jerden and Sinha 2006; Göb et al. 2013; Boekhout et al. 2015). Extensive experimental and natural studies have shown that hydroxylapatite and fluorapatite can be highly effective at immobilising aqueous U from groundwater (Fuller et al. 2002; Jerden and Sinha 2003; Ohnuki et al. 2004; Simon et al. 2008; Lammers et al. 2017). In these cases with high U contents in the groundwater, a dissolution reprecipitation process leads to the precipitation of secondary uranium phosphate minerals. However, for solutions with lower U (< 7000 ppm) and elevated Ca contents, which are more appropriate for a weathering carbonatite, the precipitation of uranium phosphates is suppressed, and instead U6+ adsorbs onto the surface of apatite (Fuller et al. 2002; Simon et al. 2008). Importantly, adsorption is partly reversible, such that successive batches of weathering fluids would be capable of removing U6+ from earlier apatite and re-fixing it onto later generations of apatite. With the continued input of newly weathered U from the breakdown of primary minerals from the host carbonatite, this process would progressively increase the U content of later apatite generations.
Deleterious and beneficial element contents
The content of U, Th and heavy metals, such as Cd, is an important criterion in the economic viability of potential phosphate sources for fertiliser as these elements are typically passed into the soil and, consequently, into the food chain (Roberts 2014). Regulation of the deleterious element content of fertiliser is becoming increasingly restrictive with the EU, for example, progressively limiting Cd contents from <60 to <20 mg/kg P2O5 by 2032 (Ulrich 2019). Phosphate from igneous sources, such as carbonatites, is low in U and Cd, but relatively high in Th (van Kauwenbergh 1997), and the composition of the primary grains analysed in this study matches this finding (Fig. 10).
As a reference point, the concentration of Moroccan Phosphate Rock (MPR) standard BCR-032 (Serrini and Haemers 1980) is included in Fig. 10. Since this is a bulk-rock standard, it is not a perfect comparison with apatite trace element contents but it provides an indication of the differing trace element content of phosphorites compared with igneous apatite sources. Notably, the Th concentration in secondary apatite from carbonatite is several orders of magnitude lower than that of the primary igneous grains from the same samples and is lower than the MPR phosphorite example (Fig. 10a). While this drop in Th content corresponds to an increase in U, the increase in U concentration is only approximately 1 order of magnitude, and it is only in the latest and most volumetrically minor stage that U contents exceed that of MPR (Fig. 10e). Increasing U contents in the carbonate-bearing fluorapatite also correspond to an increase in the concentration of other potentially deleterious elements such as Zn and Cd (Fig. 10). In particular, Cd in secondary apatite approaches and exceeds the mean MPR concentration although, as for U, this is only in the latest and most volumetrically minor stage (Fig. 10h).
Apatite is commonly viewed as a potential economic source of the REE, typically as a by-product or co-product of other commodities (Emsbo et al. 2015). In particular, hydrothermally formed carbonate-bearing fluorapatite can contain elevated contents of the HREE (Broom-Fendley et al. 2016). Morteani and Preinfalk (1996) suggested secondary carbonate-bearing apatite could be a major REE carrier in carbonatites. However, the samples analysed in this study indicate that such apatite has REE concentrations several orders of magnitude below the median content of phosphorites (Fig. 10c) and therefore appears not to represent an attractive source of the REE at this time.
Conclusions and implications
-
1
The textural, paragenetic and compositional similarity of carbonate-bearing fluorapatite from carbonatites indicates a common origin for the five, globally distributed examples investigated in this study. By textural comparison, an additional six carbonate-bearing fluorapatite deposits are also likely to have formed via the same mechanism.
-
2
The presence of a negative Ce anomaly and elevated contents of U in carbonate-bearing fluorapatite indicates formation in an oxidised environment which, based on comparison with other weathered rocks, is highly likely to indicate formation within the surface environment.
-
3
Considering the above two points, we infer that carbonate-bearing fluorapatite deposits which are not evidently caused by modern weathering (such as Glenover and Kovdor) formed instead during palaeo-weathering.
-
4
Progressive formation of later generations of carbonate-bearing fluorapatite increases the concentration of potentially deleterious elements for fertiliser production, such as Cd and U. However, only in the latest generation, comprising the smallest mass fraction of apatite, do these approach and exceed the corresponding mean contents observed in Moroccan phosphorites. Thus, carbonate-bearing fluorapatite could be an attractive feedstock for fertiliser production, especially in light of increasing regulatory limits on the deleterious element content of phosphate rock.
-
5
The concentration of potentially economic REE is low overall and decreases in successive generations of carbonate-bearing fluorapatite, suggesting such rocks have limited potential as a source of REE.
References
Akagi T, Masuda A (1998) A simple thermodynamic interpretation of Ce anomaly. Geochem J 32:301–314
Alloway BJ (2008) Micronutrients and crop production: an introduction. In: Alloway BJ (ed) Micronutrient deficiencies in global crop production. Springer, Berlin
Appleton JD (1994) Direct-application fertilizers and soil amendments—appropriate technology for developing countries? In: Mathers SJ, Northolt AJG (eds) Industrial minerals in developing countries, 18. AGID Report Series Geosciences in International Development, p 223–256
Appleton JD (2002) Local phosphate resources for sustainable development in sub-Saharan Africa. British Geological Survey Report, CR/02/121/N
Araújo IMC (2015) The weathering profile and the P, Ti and REE mineralization of the Salitre alkaline-carbonatite complex, Central Brazil. Thesis, University of Brasilia
Baldock JW (1971) Feldspathic vent agglomerates at Bukusu, Uganda—a reinterpretation. Geol Mag 108:407–412
Bau M (1999) Scavenging of dissolved yttrium and rare earths by precipitating iron oxyhydroxide: experimental evidence for Ce oxidation, Y-Ho fractionation, and lanthanide tetrad effect. Geochim Cosmochim Acta 63:67–77
Bau M, Koschinsky A (2009) Oxidative scavenging of cerium on hydrous Fe oxide: evidence from the distribution of rare earth elements and yttrium between Fe oxides and Mn oxides in hydrogenetic ferromanganese crusts. Geochem J 43:37–47
Bau M, Tepe N, Mohwinkel D (2013) Siderophore-promoted transfer of rare earth elements and iron from volcanic ash into glacial meltwater, river and ocean water. Earth Planet Sci Lett 364:30–36
Belousova E, Griffin W, O’Reilly SY, Fisher N (2002) Apatite as an indicator mineral for mineral exploration: trace-element compositions and their relationship to host rock type. J Geochem Explor 76:45–69
Boekhout F, Gérard M, Kanzari A, Michel A, Déjeant A, Galoisy A, Calas G, Descostes M (2015) Uranium migration and retention during weathering of a granitic waste rock pile. Appl Geochem 58:123–135
Boulingui, B (1997) Mineralogie et Geochime du Gisement Residuel de Phosphore et Niobium de Mabounié (Moyen-Ogooue, Gabon). Thesis, National Polytechnic Institute of Lorraine
Brasseur H, Herman P, Hubaux A (1961) Apatites de l’est du Congo et du Ruanda. Mémoires de l’Institut Géologique de Belgique, Liége 85:61–85
Braun J-J, Pagel M, Muller J-P, Bilong P, Michard A, Guillet B (1990) Cerium anomalies in weathering profiles. Geochim Cosmochim Acta 54:781–795
Braun J-J, Pagel M, Herbilln A, Rosin C (1993) Mobilization and redistribution of REEs and thorium in a syenitic lateritic profile: a mass balance study. Geochim Cosmochim Acta 57:4419–4434
Broom-Fendley S, Styles MT, Appleton JD, Gunn AG, Wall F (2016) Evidence for dissolution-reprecipitation of apatite and preferential LREE mobility in carbonatite-derived late-stage hydrothermal processes. Am Mineral 101:596–611
Broom-Fendley S, Brady AE, Wall F, Gunn AG, Dawes W (2017) REE minerals at the Songwe Hill carbonatite, Malawi: HREE-enrichment in late-stage apatite. Ore Geol Rev 81:23–41
Broom-Fendley S, Smith MP, Andrade MB, Ray S, Banks DA, Loye E, Atencio D, Pickles JR, Wall F (2020) Sulfur-bearing monazite-(Ce) from the Eureka carbonatite, Namibia: oxidation state, substitution mechanism, and formation conditions. Mineral Mag 84:35–48
Burton CCJ (1986) Investigation of the mineral resources of Cerro Manomó. Eastern Bolivia mineral exploration project ‘Proyecto Precambrico’ report number 19
Carvalho WT, Bressan SR (1989) The phosphate deposit of the ultramafic alkaline complex of Catalão I, Goias, Brazil. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Chakhmouradian AR, Reguir EP, Zaitsev AN, Coüeslan C, Xu C, Kynický J, Mumin AH, Yang P (2017) Apatite in carbonatitic rocks: compositional variation, zoning, element partitioning and petrogenetic significance. Lithos 274:188–213
Chebotarev DA, Doroshkevich AG, Klemd R, Karmanov NS (2017) Evolution of Nb-mineralization in the Chuktukon carbonatite massif, Chadobets upland (Krasnoyarsk territory, Russia). Period Mineral 86:99–118
Chen M, Graedel TE (2015) The potential for mining trace elements from phosphate rock. J Clean Prod 91:337–346
Cordeiro PFO, Brod JA, Dantas EL, Barbosa ESR (2010) Mineral chemistry, isotope geochemistry and petrogenesis of niobium-rich rocks from the Catalão I carbonatite–phoscorite complex, Central Brazil. Lithos 118:223–237
Cordeiro PFO, Brod JA, Palmieri M, Oliveira CG, Barbosa ESR, Santos RV, Gaspar JC, Assis LC (2011) The Catalão I niobium deposit, Central Brazil: resources, geology and pyrochlore chemistry. Ore Geol Rev 41:112–121
Cordell D, White S (2014) Life's bottleneck: sustaining the world's phosphorus for a food secure future. Annu Rev Environ Resour 39:161–188
Dahanayake K, Subasinghe SMND (1988) Development of recent stromatolitic strutures and phosphatic enrichment in Precambrian marble of Sri Lanka. Econ Geol 83:1468–1474
Dahanayake K, Subasinghe SMND (1989a) A modern terrestrial phosphorite— an example from Sri Lanka. Sediment Geol 61:311–316
Dahanayake K, Subasinghe SMND (1989b) Secondary phosphate mineralization in a karstic environment in Central Sri Lanka. Mineral Deposita 24:169–175
Davies KA (1947) The phosphate deposits of the Eastern Province, Uganda. Econ Geol 42:137–146
Davies KA (1956) The geology of part of South-East Uganda with special reference to the alkaline complexes. Geological Survey of Uganda Memoir number 8
de Jayawardena DE S (1989) The phosphate resource of the Eppawala carbonatite complex, northern Sri Lanka. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Decrée S, Boulvais P, Cobert C, Baele J-M, Midende G, Gardien V, Tack L, Nimpagaritse G, Demaiffe D (2015) Structurally-controlled hydrothermal alteration in the Upper Ruvubu Alkaline Plutonic Complex (Burundi): implications for REE and HFSE mobilities. Precambrian Res 269:281–295
Decrée S, Boulvais P, Tack L, André L, Baele J-M (2016) Fluorapatite in carbonatite-related phosphate deposits: the case of the Matongo carbonatite (Burundi). Mineral Deposita 51:453–466
Egerov LS (1991) Ijolite–Carbonatite Plutonism. Nedra Press, Lenigrad, USSR (In Russian)
Emsbo P, McLaughlin PI, Breit GN, du Bray EA, Koenig AE (2015) Rare earth elements in sedimentary phosphate deposits: solution to the global REE crisis? Gondwana Res 27:776–785
Erdosh G (1979) The Ontario Carbonatite Province and its phosphate potential. Econ Geol 74:331–338
Fernandes TRC (1978) Electron microscopy applied to the beneficiation of apatite ores of igneous origin. Trans Geol Soc S Afr 81:249–253
Fernandes TRC (1989) Dorowa and Shawa: late Palaeozoic to Mesozoic carbonatite complexes in Zimbabwe. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Ferrari VC, de Toledo MCM, Santos CN, Kahn H (2001) Aspectos cristaloquímicos, mineralógicos e tecnológicos da apatita de Tapira (MG). Geochim Bras 15:93–112
Fleet ME (2015) Carbonated hydroxyapatite: materials, synthesis, and application. CRC Press, Florida (Taylor and Francis Group)
Fletcher CJN, Litherland M (1981) The geology and tectonic setting of the Velasco alkaline province, eastern Bolivia. J Geol Soc Lond 138:541–548
Fowler BO (1973) Infrared studies of Apatites. I. Vibrational assignments for calcium, strontium, and barium hydroxyapatites utilizing isotopic substitution. Inorg Chem 13:194–207
Fuller CC, Bargar JR, Davis JA, Piana MJ (2002) Mechanisms of uranium interactions with hydroxyapatite: implications for groundwater remediation. Environ Sci Technol 36:158–165
Girard J-P, Flicoteaux R, Walter A-V, Savin SM, Nahon D (1993) Oxygen and carbon isotope composition of structural carbonate in weathering apatites from laterites, southern Brazil and western Senegal. Appl Geochem 8:617–632
Göb S, Gühring J-E, Bau M, Markl G (2013) Remobilization of U and REE and the formation of secondary minerals in oxidized U deposits. Am Mineral 98:530–548
Gomes CB, Comin-Chiaramonti P (2005) Some notes on the Alto Paranaíba igneous province. In: Comin-Chiaramonti P, Gomes CB (eds) Mezozoic to Cenozoic alkaline magmatism in the Brazilian platform. Edusp, São Paulo
Graupner T, Klemd R, Henjes-Kunst F, Goldmann S, Behnsen H, Gerdes A, Dohrmann R, Barton JM, Opperman R (2018) Formation conditions and REY enrichment of the 2060 Ma phosphorus mineralization at Schiel (South Africa): geochemical and geochronological constraints. Mineral Deposita 53:1117–1142
Henderson P (1984) Rare earth element geochemistry. Elsevier Science, Amsterdam
Hogarth D (1989) Pyrochlore, apatite and amphibole: distinctive minerals in carbonatite. In: Bell K (ed) Carbonatites: genesis and evolution. Unwin Hyman, London
Horner T, Finch E, Wyslouzil H (2016) Technical report on the Martison phosphate project, Ontario, Canada. Technical report, D57–R–178, for Fox River Resources Corp
Hornig-Kjarsgaard I (1998) Rare earth elements in sövitic carbonatites and their mineral phases. J Petrol 39:2105–2121
Ihlen PM, Schiellerup H, Gautneb H, Skår Ø (2014) Characterization of apatite resources in Norway and their REE potential, a review. Ore Geol Rev 58:126–147
Ivanyuk GY, Yakovenchuk VN, Pakhomovsky YA (2002) Kovdor. Laplandia Minerals, Apatity
Janots E, Bernier F, Brunet F, Muñoz M, Trcera N, Berger A, Lanson M (2015) Ce(III) and Ce(IV) (re)distribution and fractionation in a laterite profile from Madagascar: insights from in situ XANES spectroscopy at the Ce LIII-edge. Geochim Cosmochim Acta 153:134–148
Jerden JL, Sinha AK (2003) Phosphate based immobilization of uranium in an oxidizing bedrock aquifer. Appl Geochem 18:823–843
Jerden JL, Sinha AK (2006) Geochemical coupling of uranium and phosphorous in soils overlying an unmined uranium deposit: Coles Hill, Virginia. J Geochem Explor 91:56–70
Johannesson KH, Lyons WB, Bird DA (1994) Rare earth element concentrations and speciation in alkaline lakes from the western U.S.A. Geophys Res Lett 21:773–776
Kapustin YL (1983) Explosion pipes in carbonatite complexes. Int Geol Rev 25:1187–1198
Kraemer D, Kopf S, Bau M (2015) Oxidative mobilization of cerium and uranium and enhanced release of “immobile” high field strength elements from igneous rocks in the presence of the biogenic siderophore desferrioxamine B. Geochim Cosmochim Acta 165:263–279
Kraemer D, Tepe N, Pourret O, Bau M (2017) Negative cerium anomalies in manganese (hydr)oxide precipitates due to cerium oxidation in the presence of dissolved siderophores. Geochim Cosmochim Acta 196:197–208
Krasnova NI (2003) Kovdor apatite-francolite deposit as an example of explosive and phreatomagmatic endogenous activity in the ultramafic-alkaline and carbonatite complex (Kola Peninsula, Russia). In: Vladykin NV (ed) Plumes and problems of deep sources of alkaline magmatism. Russian Academy of Sciences, Moscow
Krasnova NI, Balaganskaya EG, Garcia D (2004) Kovdor— classic phoscorites and carbonatites. In: Wall F, Zaitsev AN (eds) Phoscorites and carbonatites from mantle to mine: the key example of the Kola Alkaline Province. Mineralogical Society, London
Kravchenko SM, Pokrovsky BG (1995) The Tomtor alkaline ultrabasic massif and related REE-Nb deposits, northern Siberia. Econ Geol 90:676–689
Lammers LN, Rasmussen H, Adilman D, deLemos JL, Zeeb P, Larson DG, Quicksall AN (2017) Groundwater uranium stabilization by a metastable hydroxyapatite. Appl Geochem 84:105–113
Langmuir D (1978) Uranium solution-mineral equilibria at low temperatures with applications to sedimentary ore deposits. Geochim Cosmochim Acta 42:547–569
Lapin AV, Lyagushkin AP (2014) The Kovdor apatite–francolite deposit as a prospective source of phosphate ore. Geol Ore Deposits 56:61–80
Lapin AV, Tolstov AV, Kulikova IM (2016) Distribution of REE, Y, Sc, and Th in the unique complex rare-metal ores of the Tomtor deposit. Geochim Int 54:1061–1078
Laval M, Johan V, Tourlière B (1988) La carbonatite de Mabounié: exemple de formation d’un gîte résiduel à pyrochlore. Chronique de la Recherche Minière 491:125–113
Lazareva EV, Zhmodik SM, Dobretsov NL, Tolstov AV, Shcherbov BL, Karmanov NS, Gerasimov EY, Bryanskaya AV (2015) Main minerals of abnormally high-grade ores of the Tomtor deposit (Arctic Siberia). Russ Geol Geophys 56:844–873
Lehr JR, McClellan GH (1972) A revised laboratory reactivity scale for evaluating phosphate rocks for direct application. Bulletin Y-43. Tennessee Valley Authority, National Fertilizer Development Center, Muscle Shoals, Alabama
Leybourne MI, Johannesson KH (2008) Rare earth elements (REE) and yttrium in stream waters, stream sediments, and Fe–Mn oxyhydroxides: fractionation, speciation, and controls over REE + Y patterns in the surface environment. Geochim Cosmochim Acta 72:5962–5983
Litherland M, Annells RN, Appleton JD, Berrangé JP, Bloomfield K, Burton CCJ, Darbyshire DPF, Fletcher DJN, Hawkins MP, Klinck BA, Llanos A, Mitchell WI, O’Connor EA, Pitfield PEJ, Power G, Webb BC (1986) The geology and mineral resources of the Bolivian Precambrian shield. Overseas Memoir of the British Geological Survey, No. 9
Loges A, Wagner T, Barth M, Bau M, Göb S, Markl G (2012) Negative Ce anomalies in Mn oxides: the role of Ce4+ mobility during water–mineral interaction. Geochim Cosmochim Acta 86:296–317
Lottermoser BG (1990) Rare-earth element mineralisation within the Mt. Weld carbonatite laterite, Western Australia. Lithos 24:151–167
Lottermoser BG, England BM (1988) Compositional variation in pyrochlores from the Mt Weld carbonatite laterite, Western Australia. Mineral Petrol 38:37–51
Luo Y-R, Byrne RH (2000) The ionic strength dependence of rare earth and yttrium chloride complexation at 25°C. J Solut Chem 29:1089–1099
Luo Y-R, Byrne RH (2004) Carbonate complexation of yttrium and the rare earth elements in natural waters. Geochim Cosmochim Acta 68:691–699
Mariano AN (1989) Nature and economic mineralisation in carbonatites and related rocks. In: Bell K (ed) Carbonatites: genesis and evolution. Unwin Hyman, London
McArthur JM, Walsh JN (1985) Rare-earth geochemistry of phosphorites. Chem Geol 47:191–220
McClellan GH, Van Kauwenbergh SJ (1990) Mineralogy of sedimentary apatites. Geol Soc Lond, Spec Publ 52:23–31
McConnell D, Gruner JW (1940) The problem of the carbonate-apatites. III. Carbonate-apatite from Magnet Cove, Arkansas. Am Mineral 25:157–167
McDonough WF, Sun SS (1995) The composition of the earth. Chem Geol 120:223–253
Mgonde F (1994) Mobilization and redistribution of phosphate and rare earth elements in the weathering zone above Panda Hill carbonatite, SW Tanzania. Thesis, Carleton University
Migdisov AA, Williams-Jones A (2014) Hydrothermal transport and deposition of the rare earth elements by chlorine-bearing aqueous liquids. Mineral Deposita 49:987–997
Migdisov AA, Williams-Jones A, Wagner T (2009) An experimental study of the solubility and speciation of the rare earth elements (III) in chloride-and chloride-bearing aqueous solutions at temperatures up to 300°C. Geochim Cosmochim Acta 73:7087–7109
Mikhailova JA, Kalashnikov AO, Sokharev VA, Pakhomovsky YA, Konopleva NG, Yakovenchuk VN, Bazai AV, Goryainov PM, Ivanyuk GY (2016) 3D mineralogical mapping of the Kovdor phoscorite–carbonatite complex (Russia). Mineral Deposita 51:131–149
Möller P, Bau M (1993) Rare-earth patterns with positive cerium anomaly in alkaline waters from Lake Van, Turkey. Earth Planet Sci Lett 117:671–676
Morteani G, Preinfalk C (1996) REE distribution and REE carriers in laterites formed on the alkaline complexes of Araxá and Catalão (Brazil). In: Jones AP, Wall F, Williams CT (eds) Rare earth minerals. Mineralogical Society, London
Murakami T, Ohnuki T, Isobe H, Sato T (1997) Mobility of uranium during weathering. Am Mineral 82:888–899
Northolt AJG, Sheldon RP, Davidson DF (1989) Phosphate deposits of the world, volume 2, phosphate rock resources. Cambridge University Press, Cambridge
O’Brien H, Hyvönen E (2015) The Sokli Carbonatite complex. In: Maier WD, Lahtinen R, O’Brien H (eds) Mineral deposits of Finland. Elsevier, Amsterdam
Ohnuki T, Kozai N, Samafam M, Yasuda R, Yamamoto S, Narumi K, Naramoto H, Murakami T (2004) The formation of autunite (Ca(UO2)2(PO4)2nH2O) within the leached layer of dissolving apatite: incorporation mechanism of uranium by apatite. Chem Geol 211:1–14
Oliveira SMB, Imbernon RAL (1998) Weathering alteration and related REE concentration in the Catalão I carbonatite complex, Central Brazil. J S Am Earth Sci 11:379–388
Oliveira ÍL, Brod JA, Cordeiro PFO, Dantas EL, Mancini LH (2017) Insights into the late-stage differentiation processes of the Catalão I carbonatite complex in Brazil: New Sr–Nd and C-O isotopic data in minerals from niobium ores. Lithos 274–275:214–224
Pasero M, Kampf AR, Ferraris C, Pekov IV, Rakovan J, White TJ (2010) Nomenclature of the apatite supergroup minerals. Eur J Mineral 22:163–179
Ponomarchuk VA, Dobretsov NL, Lazareva EV, Zhmodik SM, Karmanov NS, Tolstov AV, Pyryaev AN (2020) Evidence of microbial-induced mineralization in rocks of the Tomtor Carbonatite Complex (Arctic Siberia). Dokl Earth Sci 490:33–38
Potapoff P (1989) The Martison carbonatite deposit, Ontario. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Pourret O, Davranche M, Gruau G, Dia A (2007) Rare earth elements complexation with humic acid. Chem Geol 243:128–141
Pressacco R (2001) Geology of the Cargill township residual carbonatite-associated phosphate deposit, Kapuskasing, Ontario. Explor Min Geol 10:77–84
Prins P (1973) Apatite from African carbonatites. Lithos 6:133–143
Pufahl PK, Groat LA (2017) Sedimentary and igneous phosphate deposits: formation and exploration. Econ Geol 112:483–516
Putnis A (2002) Mineral replacement reactions: from macroscopic observations to microscopic mechanisms. Mineral Mag 66:689–708
Putnis A (2009) Mineral replacement reactions. Rev Mineral Geochem 70:87–124
Puustinen K, Kauppinen H (1989) The Siilinjärvi carbonatite complex, eastern Finland. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Richardson DG, Birkett TC (1996) Peralkaline rock-associated rare metals. In: Eckstrand OR, Sinclair WD, Thorpe RI (eds) Geology of Canadian mineral deposit types. Geology of Canada, no. 8. Geological Survey of Canada, Ottawa, pp 523–540
Roberts TL (2014) Cadmium and phosphorous fertilizers: the issues and the science. Procedia Eng 83:52–59
Sage RP (1988a) Geology of carbonatite-alkali rock complexes in Ontario: Cargill Township Carbonatite Complex, District of Cochrane. Ontario Geological Survey, Study 36, p 92
Sage RP (1988b) Geology of carbonatite-alkali rock complexes in Ontario: Schryburt Lake Carbonatite Complex, District of Kenora. Ontario Geological Survey, Study 50:43
Sage RP (1991) Geology of the Martison Carbonatite Complex; Ontario Geological Survey, Open File Report, 5420, p 74
Sanchez PA (2002) Soil fertility and hunger in Africa. Science 295:2019–2020
Sandell EB, Hey MH, McConnell D (1939) The composition of francolite. Mineral Mag 25:395–401
Sandvik PO, Erdosh G (1977) The geology of the Cargill phosphate deposit, Northern Ontario; Canadian Institute of Mining and Metallurgy Bulletin 69:90–96
Sanematsu K, Watanabe Y (2016) Characteristics and genesis of ion adsorption-type rare earth element deposits. Rev Econ Geol 18:55–79
Serrini G, Haemers L (1980) The certification of calcium, phosphorus, carbonates, fluorine, silicon, total sulphur, aluminium, magnesium and iron contents in a natural Moroccan phosphate rock (phosphorite). Commission of the European Communities, Community Bureau of Reference report EUR 6807EN
Simon FG, Biermann V, Peplinski B (2008) Uranium removal from groundwater using hydroxyapatite. Appl Geochem 23:2137–2145
Slukin AD (1994) Bauxite deposits with unusually high concentrations of REE, Nb, Ti, and Th, Chadobets uplift, Siberian platform. Int Geol Rev 36:179–193
Soubiès F, Melfi AJ, Autefage EF (1991) Comportamento geoquímico dos elementos terras raras nos alteritos da jazida de fosfato e titânio de Tapira (Minas Gerais, Brasil): a importância dos fosfatos. Rev Bras Geosci 21:3–16
Subasinghe ND (2013) Observations and interpretations on past microbial activities at Eppawala phosphate deposit. Journal of the Geological Society of Sri Lanka 15:99–110
Takahashi Y, Shimizu H, Usui A, Kagi H, Nomura M (2000) Direct observation of tetravalent cerium in ferromanganese nodules and crusts by X-ray-absorption near-edge structure (XANES). Geochim Cosmochim Acta 64:2929–2935
Tanaka K, Tani Y, Takahashi Y, Tanimizu M, Suzuki Y, Kozai N, Ohnuki T (2010) A specific Ce oxidation process during sorption of rare earth elements on biogenic Mn oxide produced by Acremonium sp. strain KR21-2. Geochim Cosmochim Acta 74:5463–5477
Tazaki K, Fyfe WS, Dissanayake CB (1986) Weathering of phosphatic marble to exploitable apatite deposit, Sri Lanka. Appl Geochem 1:287–300
Tazaki K, Fyfe WS, Dissanayake CB (1987) Weathering of apatite under extreme conditions of leaching. Chem Geol 60:151–162
Toledo MCM, Lenharo SLR, Ferrari VC, Fontan F, Parseval P, Leroy G (2004) The compositional evolution of apatite in the weathering profile of the Catalão I alkaline-carbonatitic complex, Goias, Brazil. Can Mineral 42:1139–1158
Tulsidas H, Gabriel S, Kiegiel K, Haneklaus N (2019) Uranium resources in EU phosphate rock imports. Res Policy 61:151–156
Ulrich AE (2019) Cadmium governance in Europe's phosphate fertilizers: not so fast? Sci Total Environ 650:541–545
Ulrich AE, Schnug E, Prasser H-M, Frossard E (2014) Uranium endowments in phosphate rock. Sci Total Environ 478:226–234
van der Walt GN, Dabrowski FA, Richards D, Siegfried PR (2012) Geological report and resource estimate for the Glenover Carbonatite project. Geo-Consult International (Pty) Ltd
Van Kauwenbergh SJ (1997) Cadmium and other minor elements in world resources of phosphate rock. Proceedings of the Fertiliser Society, 400, p 1–41
Van Straaten P (2002) Rocks for crops: agrominerals of sub-Saharan Africa. ICRAF, Nairobi
Vartiainen H (1980) The petrography, mineralogy and petrochemistry of the Sokli carbonatite massif, northern Finland. Geological Survey of Finland Bulletin 313:1–126
Vartiainen H (1989) The phosphate deposits of the Sokli carbonatite complex, Finland. In: Northolt AJG, Sheldon RP, Davidson DF (eds) Phosphate deposits of the world. Cambridge University Press, Cambridge
Vartiainen H, Paarma H (1979) Geological characteristics of the Sokli Carbonatite complex, Finland. Econ Geol 74:1296–1306
Vartiainen H, Melnikov I, Sulimov B (1990) The francolite ore deposits of Kovdor and Sokli. Proceedings of the Finnish-Soviet Symposium held in Helsinki. November 14–15. Research Report TKK-IGE A13, 7–14
Verwoerd WJ (1966) South African carbonatites and their probable mode of origin. Annale Universiteit van Stellenbosch 41:1–233
Verwoerd WJ (1967) The carbonatites of South Africa and South West Africa. Handbook 6. Department of Mines and Geological Survey of South Africa, p 452
Verwoerd WJ (1986) Mineral deposits associated with carbonatites and alkaline rocks. In: Anhaeusser CR, Maske S (eds) Mineral deposits of Southern Africa 1–2. Geological Society of South Africa, Johannesburg, pp 2173–2191
Wall F, Williams CT, Woolley AR, Nasraoui M (1996) Pyrochlore from weathered carbonatite at Lueshe, Zaire. Mineral Mag 60:731–750
Walter A-V, Flicoteaux R, Parron C, Loubet M, Nahon D (1995a) Rare-earth elements and isotopes (Sr, Nd, O, C) in minerals from the Juquiá carbonatite (Brazil): tracers of a multistage evolution. Chem Geol 120:27–44
Walter A-V, Nahon D, Flicoteaux R, Girard JP, Melfi A (1995b) Behaviour of major and trace elements and fractionation of REE under tropical weathering of a typical apatite-rich carbonatite from Brazil. Earth Planet Sci Lett 136:591–602
Witt WK, Hammond DP, Hughes M (2019) Geology of the Ngualla carbonatite complex, Tanzania, and origin of the weathered bastnaesite zone REE ore. Ore Geol Rev 105:28–54
Zaitsev AN, Williams CT, Jeffries TE, Strekopytov S, Moutte J, Ivashchenkova OV, Spratt J, Petrov SV, Wall F, Seltmann R, Borozdin AP (2015) Rare earth elements in phoscorites and carbonatites of the Devonian Kola Alkaline Province, Russia: examples from Kovdor, Khibina, Vuoriyarvi and Turiy Mys complexes. Ore Geol Rev 64:477–498
Zanin YN, Zamirailova AG (2009) Rare earth elements in supergene phosphorites. Geochim Int 47:282–296
Acknowledgements
The organisers of the 2015 ‘Alkaline Magmatism of the Earth and Related Critical Metal Deposits’ workshop arranged a visit to Kovdor and A. Zaitsev helped export samples. S. Gehör and L. Anttonen arranged access to visit Sokli. M. Howe and E. Vaccaro organised sample loans. B. Boulingui and S. Blancher provided unpublished papers on the Mabounié complex. M. Spence, S. MacGowan, H. McAllister, D. Morris, R. Parr, J. Pearn and W. Pulido Rodriguez helped acquire some of the data. P. Cordeiro and an anonymous reviewer, as well as editorial comments from R. Linnen and G. Beaudoin, helped improve this manuscript.
Funding
This study was funded by a Natural Environment Research Council (NERC) Industrial Innovation Fellowship NE/R013403/1 (SBF), the NERC SoS RARE consortium NE/M011429/1 (SBF, FW, DB); the EU H2020 HiTechAlkCarb programme grant agreement no. 689909 (PS, FW), and NERC grant NE/R011389/1 (RB and EF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: R. Linnen
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 1854 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Broom-Fendley, S., Siegfried, P.R., Wall, F. et al. The origin and composition of carbonatite-derived carbonate-bearing fluorapatite deposits. Miner Deposita 56, 863–884 (2021). https://doi.org/10.1007/s00126-020-01010-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-020-01010-7