
Abstract
The liver plays a crucial role in the control of glucose homeostasis and is therefore of great interest in the investigation of the development of type 2 diabetes. Hepatic glucose uptake (HGU) can be measured through positron emission tomography (PET) imaging with the tracer [18F]-2-fluoro-2-deoxy-d-glucose (FDG). HGU is dependent on many variables (e.g. plasma glucose, insulin and glucagon concentrations), and the metabolic state for HGU assessment should be chosen with care and coherence with the study question. In addition, as HGU is influenced by many factors, protocols and measurement conditions need to be standardised for reproducible results. This review provides insights into the protocols that are available for the measurement of HGU by FDG PET and discusses the current state of knowledge of HGU and its impairment in type 2 diabetes. Overall, a scanning modality that allows for the measurement of detailed kinetic information and influx rates (dynamic imaging) may be preferable to static imaging. The combination of FDG PET and insulin stimulation is crucial to measure tissue-specific insulin sensitivity. While the hyperinsulinaemic–euglycaemic clamp allows for standardised measurements under controlled blood glucose levels, some research questions might require a more physiological approach, such as oral glucose loading, with both advantages and complexities relating to fluctuations in blood glucose and insulin levels. The available approaches to address HGU hold great potential but await more systematic exploitation to improve our understanding of the mechanisms underlying metabolic diseases. Current findings from the investigation of HGU by FDG PET highlight the complex interplay between insulin resistance, hepatic glucose metabolism, NEFA levels and intrahepatic lipid accumulation in type 2 diabetes and obesity. Further research is needed to fully understand the underlying mechanisms and potential therapeutic targets for improving HGU in these conditions.
Graphical Abstract

Similar content being viewed by others







Introduction
The liver plays a crucial role in the control of glucose homeostasis and is therefore of great interest in the investigation of the development of diseases that involve dysregulation of blood glucose levels, such as type 2 diabetes and non-alcoholic fatty liver disease (NAFLD). While the investigation of hepatic endogenous glucose production (EGP) and of its regulation by insulin has been extensive, much less is known regarding the regulation of hepatic glucose uptake (HGU). This is mainly because of the complexity of its measurement and interpretation. Considering that HGU plays a key role in the regulation of blood glucose homeostasis, and is known to be dysregulated in insulin resistance, for example, [1] a better understanding of the mechanisms of HGU (dys)regulation could pave the way for targeted therapies based on specific pathways that are affected in type 2 diabetes, thus improving the management of type 2 diabetes and related metabolic disorders; and [2] the definition and harmonisation of HGU imaging protocols, differentiating requirements under diverse metabolic states (e.g. steady state in insulin clamp studies, unsteady state under meal or glucose loading), could expand its clinical usefulness. The measurement of HGU is mainly challenging because of the difficulty in accessing the hepatic blood supply for invasive measurement of glucose uptake. With advances in positron emission tomography (PET) technology, a growing number of studies have made use of non-invasive imaging with the radioactive tracer [18F]-2-fluoro-2-deoxy-d-glucose (FDG) to measure liver-specific glucose uptake. These studies have provided valuable insights into the mechanisms that lead to impairments in HGU. However, as HGU is dependent on many variables, such as plasma glucose and portal vein insulin and glucagon levels, additional efforts are required to standardise protocols and conditions for measuring HGU with FDG PET, and to achieve full understanding of its role in the pathophysiology of type 2 diabetes [1,2,3,4]. In this review, we aim to provide insights into the protocols that have been made available for the measurement of HGU by FDG PET. We will also discuss the current state of knowledge of HGU and its impairment in type 2 diabetes.
Fate of FDG in the liver
The fate of FDG in the liver is described in Fig. 1. Uptake of FDG into hepatocytes is mediated by the highly abundant GLUT2 glucose transporter, which is not saturated even when glucose levels are high [5]. Therefore, the rate and direction of movement of glucose (and by extension FDG) is strongly influenced by the relative concentrations of glucose inside and outside the cell. In the postprandial state, glucokinase (responsible for the phosphorylation of FDG into FDG-6-phosphate) is activated through its translocation from the nucleus to the cytoplasm. This translocation is mainly stimulated by increasing concentrations of glucose (and/or fructose) [6]. In the majority of tissues except for the liver, once FDG is taken up and phosphorylated into FDG-6-phosphate, it mostly becomes ‘trapped’. The liver expresses glucose-6-phosphatase (G6Pase), which is responsible for the dephosphorylation of glucose-6-phosphate (G6P) into glucose. Liver dephosphorylation of FDG within the typical time window of most imaging protocols may be limited but should be considered, especially in fasted or insulin-resistant states. This has consequences in terms of the mathematical model that should be employed to appropriately interpret FDG PET-derived HGU.

FDG kinetics in the liver. FDG enters tissues via glucose transporters, mainly GLUT2 in the case of the liver. Once inside cells, FDG is phosphorylated to FDG-6-phosphate (FDG-6-P). This reaction is catalysed by the enzyme glucokinase [50]. The liver expresses glucose-6-phosphatase (G6Pase), which is responsible for the dephosphorylation of glucose-6-phosphate (G6P) into glucose and by extension of FDG-6-P into FDG. G6Pase activity is to some extent suppressed by insulin. The model representing FDG kinetics in the liver most typically includes three compartments, namely FDG in inflowing vessels, FDG in hepatocytes and FDG-6-P in hepatocytes. The rate constants k1–k4 describe the exchange of FDG between blood and tissue (k1 for influx, k2 for efflux) and within tissue (k3 for phosphorylation, k4 for dephosphorylation). Additional compartments can be added to take into account FDG entering the pentose phosphate pathway (PPP), in which G6P is converted to ribulose-5-phosphate, generating NADPH, and the incorporation of FDG into glycogen. The amount of FDG that enters into the PPP pathway is considered to be quite small, with the amount incorporated into glycogen being even smaller. Compartmental modelling addresses the illustrated intracellular pathways, beyond the overall FDG influx (Ki), and is well suited to insulin-stimulated studies and fasting studies. The Patlak model allows for the estimation of Ki but not its (k) components and is better suited to insulin-stimulated states than fasting states. This figure is available as part of a downloadable slideset
Scanning modalities and analysis for studying HGU
Static and dynamic FDG PET
The advantages and disadvantages of static imaging and the standardised uptake value (SUV) (Text box 1) for HGU research have been extensively discussed by others [7]. Static scanning provides limited information on glucose fluxes, as FDG kinetics cannot be described, and uptake and phosphorylation cannot be dissected from dephosphorylation. The selection of a single appropriate time point to represent ‘net’ glucose uptake is challenging. If blood radioactivity over time is independently measured (by repeated blood sampling and gamma counting), a fractional uptake rate can also be calculated from a static scan, which is more quantitative than the SUV [8, 9]. Dynamic imaging (Text box 2 [2, 10]) provides a more informative and robust description of hepatic FDG kinetics [11]. After image reconstruction and data extraction, dynamic data (time–activity curves) allow the calculation of detailed kinetic information and fluxes using mathematical modelling. The most common models are discussed in the following sections. An overview of studies employing dynamic scanning to investigate HGU is presented in Table 1 and the pros and cons of each technique used are summarised in Table 2.
Compartmental models
To describe FDG kinetics in the liver, a three-compartment model was suggested, first described by Sokoloff et al [12]. The model and rate constants (k1–k4) are described in Fig. 1. While in most tissues the rate of dephosphorylation (k4) is null, the liver has high levels of G6Pase activity, especially in the fasted condition, as insulin levels are low and hepatic glucose output is high. Consequently, k4 cannot be assumed to be zero in the liver. From the single rate constants (Fig. 1), the tissue fractional extraction rate constant for FDG (Ki) can be calculated, that is, k1×k3/(k2+k3). FDG can also enter the pentose phosphate pathway (PPP) to some extent, in which G6P is converted to ribulose-5-phosphate, generating NADPH. This is mostly apparent 45–180 min following FDG injection; therefore, one could consider adding more compartments to the kinetic model in studies with an extended duration [13]. Furthermore, FDG can also be incorporated into glycogen, but to a much smaller extent, which is therefore mostly considered negligible, at least in pigs under fasting or hyperinsulinaemic–euglycaemic clamp (HEC) states [13, 14].
Patlak model
The Patlak model (or plot) is a sophisticated approach, leading to a simplified graphical analysis [15]. This approach assumes that the behaviour of the tracer can be approximated by two main compartments: a reversible compartment that is in rapid equilibrium with plasma (e.g. FDG in cytoplasm) and an irreversible compartment, where tracer enters and is trapped in tissue during the measurement time (FDG-6-phosphate). Using a graphical technique combining FDG activity curves in the tissue of interest (e.g. liver) and in plasma, a curve is generated, reaching linearity at time=t*. A regression line can be fitted to the data after t*. The slope of the line represents the net influx rate constant of the tracer in the irreversible compartment, that is, influx of FDG (Ki) from plasma to tissue.
The metabolic state studied (fasted, insulin stimulation) should be considered in the design and interpretation of Patlak analysis in the liver, and a corrective algorithm for the loss of tracer from irreversible compartments has been proposed to overcome the underestimation of Ki when some tracer is actually dephosphorylated and not trapped [16]. Under insulin stimulation, rates of dephosphorylation should be considerably suppressed and the Patlak model is commonly adopted without such correction; it is the most commonly used method for the investigation of HGU using FDG PET (Table 1). From the Ki value and blood glucose levels, the glucose metabolic rate (MRGlu) can be determined (Text box 3 [14, 17]).
Input function
To quantify FDG tissue uptake from dynamic imaging, knowledge of the input function is required (Text box 4 [18,19,20]). Under physiological circumstances, the liver receives 80% or more of its blood from the portal vein and 20% from the arterial circulation [21, 22]. Ideally, the dual input function supplying the liver should be known. This is complicated by the fact that the injected tracer is first distributed to the intestines, spleen and pancreas before reaching the portal vein entering the liver. The tracer concentration in the portal vein differs from that in the arteries, and the portal vein is very difficult to access for sampling. To date, measuring the image-derived input function (IDIF) from the portal vein has seldom been attempted and could result in high noise and partial volume effects [23]. Some authors have proposed methods to approximate the portal venous input [24, 25] mathematically or by adding the gut as a compartment in modelling [26, 27]. Studies of FDG PET in pigs in fasted and insulin-stimulated states [14, 28] have consistently shown that three-compartment analysis using a single arterial input does lead to an underestimation of rate constants and HGU [14, 28]. However, this underestimation seems to be systematic, as highlighted by a strong correlation between the HGU calculated from dual input functions and that calculated from single input functions, regardless of whether in a fasted or insulin simulated state [14]. Patlak analysis may be less dependent on the dual vs arterial input function to estimate the net influx rate (Ki) [28]. However, with advances in scanner technology, such as time-of-flight imaging, improvements in image quality and resolution have allowed an IDIF to be obtained from the portal vein [29], which is expected to increase precision in HGU quantification. Overall, the use of a single arterial input is informative for HGU, provided that results are viewed in light of the limitation regarding a potential underestimation of HGU [14, 28].
Measuring HGU under different conditions
When designing a study involving the measurement of HGU by FDG PET, one needs to decide under which metabolic state participants will be characterised (e.g. fasted, fed, insulin-stimulated). The choice will depend on the research question and population of interest. An overview of the advantages and disadvantages of each state is provided in Table 2.
Fasted state
Several metabolic studies have investigated HGU in the fasted state, during dynamic or static acquisition protocols [4, 30]. Because examinations in the fasted state are most common in routine clinical contexts, one advantage is that large numbers of participant data are available for retrospective or prospective analysis compared with more demanding studies, that is, studies carried out under stimulated conditions [1, 4]. In terms of pathophysiology, the fasted state is best suited to examine glucose output rather than uptake. In addition, detection of liver inflammation via FDG uptake may benefit from low hepatocyte FDG retention under fasting conditions. Recent studies have shown the feasibility of a dynamic protocol with kinetic modelling for the assessment of hepatic inflammation against histopathological inflammation grades from biopsies [31, 32]. Additionally, the k1 rate constant has been shown to be inversely correlated with hepatic inflammation scores in individuals with NAFLD or non-alcoholic steatohepatitis (NASH) [32]. In a preclinical study, HGU measured by FDG PET was shown to discriminate histology-proven NAFLD from initial NASH [33] in rats. Importantly, fasting measures are essential to demonstrate the magnitude of a stimulus, for example comparing fasted and insulin-stimulated states to quantify the relevance of insulin regulation. Some studies have shown that differences in HGU between participants with and without type 2 diabetes are detectable during insulin stimulation and not in the fasted state [30].
Hyperinsulinaemic–euglycaemic clamp
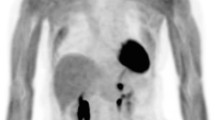
By combining the HEC (Text box 5 [34]) with FDG PET, one can quantify tissue-specific insulin-stimulated glucose uptake in any tissue that is in the tomographic field of view. Compared with the fasting state, a HEC inhibits dephosphorylation and stimulates phosphorylation, lipogenesis and glycogen synthesis, amplifying differences in HGU between individuals and providing relevant information on metabolic diseases such as type 2 diabetes, insulin resistance, obesity and NAFLD. Throughout the PET acquisition, insulin is continuously infused and euglycaemia is maintained. The FDG injection and PET acquisition are started at the time of steady state, which typically occurs at 60–90 min into the clamp, depending on the protocol (Table 1). Acquisition often consists of one or two dynamic bed positions, over the liver/thoracic area, to obtain the input function from the cardiac left ventricle. As mentioned, sometimes multiple bed positions over additional regions of interest (skeletal muscle, adipose tissue, brain) and/or whole-body dynamic protocols in combination with a HEC have also been performed [2, 10] (Table 1). The multi-organ approach allows estimation of the relative contribution of each organ to whole-body tissue-specific insulin resistance. HEC protocols in combination with FDG PET have a typical duration of 2–3 h in total, with around 40-60 min of PET acquisition (Table 1). An example of a whole-body PET map of glucose uptake and a Ki map (image generated by the calculation of Ki for every pixel) from a HEC FDG PET scan is provided in Fig. 2.

(a) Transaxial fused Ki map and CT image. Brighter areas (white-yellow) indicate a higher net influx rate constant (Ki). (b) Coronal view of a whole-body PET map of glucose uptake. Darker areas represent higher glucose uptake. This figure is available as part of a downloadable slideset
Peripheral infusion of insulin and glucose: advantages and disadvantages
During a HEC, glucose and insulin are infused through the systemic circulation, which is considerably different from what happens physiologically in response to consuming a meal. Under physiological conditions, glucose is absorbed by the gut and undergoes first pass extraction in the liver before reaching the systemic circulation. The same is true for insulin, which reaches the liver directly through the portal vein after secretion by the pancreas. During the first pass, 40–80% of this insulin is cleared by the liver and the rest is released into the systemic circulation [35, 36]. The liver is therefore always exposed to much higher insulin and glucose concentrations than other organs. Recent studies in unconscious dogs [37] clearly showed that peripheral insulin infusion during a clamp led to under-stimulation of HGU compared with portal insulin infusion, which can lead to misinterpretation of the role of the liver compared with other organs [37, 38]. This highlights that the route of insulin and glucose delivery is key to the assessment of HGU, and one should be aware of the potential underestimation of HGU in the absence of portal delivery. This concept is further supported by studies in rats [39] in which hepatic glycogen synthesis was lower under systemic than portal insulin infusion [39]. Importantly, this bias cannot be corrected by simply adjusting the dose of the peripheral infusion of insulin. Indeed, increasing the insulin levels would enhance the amount of insulin reaching the liver but would also stimulate skeletal muscle glucose uptake proportionally [37], perpetuating the imbalance.
Oral glucose load (and FDG)
Performing a scan after oral ingestion of glucose can be a more physiological alternative to a HEC or hyperglycaemic–hyperinsulinaemic clamp [40, 41]. In fact, an oral glucose load was shown to produce a threefold higher HGU than similar levels of blood glucose and insulinaemia achieved through peripheral glucose infusion [38, 42]. This may be a first pass effect, as glucose concentrations in the portal vein are a lot higher than in the peripheral circulation during a glucose bolus, enhancing HGU. However, an oral glucose load also presents its own challenges. In insulin-dependent organs such as the myocardium [43], image quality from FDG PET was superior during a HEC than 1 h after a glucose load, mainly because of the lower competition during a HEC between glucose and FDG levels for organ entry, together with protracted insulin stimulation in the HEC as insulin is continuously infused. In line with this, the Patlak slope (Ki) was clearly different after a glucose load compared with the clamp, but glucose uptake (accounting for blood glucose levels) was similar [43]. Notably, however, the image quality in the case of the heart and liver is not affected by the same factors, as insulin is a major driver of cardiac glucose uptake (less prominent for HGU), whereas glucose is the primary stimulus for HGU. Consequently, an oral glucose load should maximise HGU and improve the quality of liver imaging. An oral glucose load generates variable levels of glucose and insulin, either over time or between individuals, introducing complexity in modelling, but with the advantage that the test is personalised to the real-life situation of each individual. Measurement of the portal vein glucose concentration as an input function also remains challenging. In spite of these technical complexities, the combination of an oral glucose load and FDG injection has been performed for investigation of the effects of glucose-lowering medications on glucose uptake in the brain, liver and adipose tissue [40, 41]. In these studies, an oral glucose load of 75 g, which is the standard dose for OGTTs, was ingested at the same time as injection of FDG, following drug or placebo administration. The modelling procedure was refined to adjust for glucose variations. Overall, the combination of oral glucose ingestion and FDG PET seems to be a viable option to investigate the physiological response, reflecting real-life interpersonal differences.
Interestingly, a recent study also showed the feasibility of oral administration of FDG for the investigation of HGU in animals [44]. Oral administration of FDG may reflect the physiology of HGU more closely than peripheral administration. To our knowledge, this approach has rarely been used in humans, and outside of the current context, but may in fact be key to the understanding of normal physiology.
Current FDG PET data on insulin-stimulated HGU in type 2 diabetes and metabolic research
An overview of studies that have used FDG PET imaging during insulin stimulation to specifically investigate HGU is given in Table 3. Insulin has been shown to stimulate net glucose uptake in the liver compared with the fasted state [1] and this glucose uptake was impaired in individuals with type 2 diabetes and in those without diabetes with morbid obesity [3, 10, 30, 45] (Table 3). Commonly used medication for the treatment of type 2 diabetes, such as metformin and rosiglitazone, significantly improved insulin-stimulated HGU after 26 weeks of treatment [46]. Bariatric surgery increased insulin-stimulated HGU and suppressed EGP in morbidly obese individuals after 6 months [30] whereas a diet very low in energy (very low calorie diet) of shorter duration only reduced EGP [47]. While it is clear that insulin-stimulated HGU is impaired in type 2 diabetes and morbid obesity, it is not fully understood where the defects occur and whether they extend to less extreme insulin-resistant conditions [1, 10]. It has been suggested that these defects originate from changes in glucokinase activity and/or expression and an impaired ability of insulin to suppress G6Pase activity. Indirect factors such as high circulating NEFA levels as a result of adipose tissue insulin resistance seem to also play a major role in impaired suppression of HGU by insulin; high NEFA levels are also key in hepatic lipid accumulation, which is a hallmark of type 2 diabetes and has been shown to impair HGU. The cellular pathways involved in the (dys)regulation of hepatic glucose metabolism have been extensively described [38, 48, 49] and are beyond the scope of this review; some cellular pathways cannot be specifically measured using FDG PET and others require a range of kinetic parameters (e.g. see the following section), which have been addressed in a few studies and should be better exploited in the future. A summary of the findings from clinical trials investigating insulin-stimulated HGU with FDG PET is provided in Fig. 3

Summary of the findings from clinical trials investigating insulin-stimulated HGU with FDG PET. An increase in insulin-stimulated HGU was found for rosiglitazone [46, 58], metformin [46] and bariatric surgery [30]. A negative correlation with insulin-stimulated HGU was found for NEFA levels during a HEC [61] and type 2 diabetes [45]; however, the picture was less clear for insulin-resistant individuals who do not yet have type 2 diabetes [1]. Several studies have shown an inverse association between IHL levels and insulin-stimulated HGU [3, 58, 59]; however, some studies have found no change in insulin-stimulated HGU with changes in IHL levels or weight loss [47]. T2D, type 2 diabetes. This figure is available as part of a downloadable slideset
Glucokinase and G6Pase
The glucokinase gene (GCK) was one of the first genes identified in association with diabetes [50] and changes in glucokinase activity and expression are associated with type 2 diabetes and hepatic steatosis [50]. Some studies have shown a 50% decrease in glucokinase activity in individuals with type 2 diabetes and obesity compared with obese or lean individuals without type 2 diabetes [51]. In line with this, studies investigating the combination of hyperglycaemia and hyperinsulinaemia on splanchnic glucose uptake [52, 53] have provided further evidence that the defect in glucose uptake occurs at a proximal metabolic step. This evidence supports the involvement of glucokinase in the impairment of insulin-mediated HGU observed in type 2 diabetes by FDG PET.
A three-compartment model was used to determine FDG kinetics in the liver in insulin-resistant and insulin-sensitive individuals without diabetes [1], providing detailed information on each transfer rate constant (k1, k2, k3, k4). The authors found that, on insulin stimulation, k3 values were similar between insulin-sensitive and insulin-resistant participants, but the k3/k4 ratio (phosphorylation/dephosphorylation) was significantly lowered in the insulin-resistant group. This suggests an impaired ability of insulin to suppress dephosphorylation (thus G6Pase activity) in insulin-resistant individuals without diabetes, whereas phosphorylation remains normally stimulated. Further FDG PET studies, including participants with more advanced forms of insulin resistance, with or without fatty liver involvement, may allow the sequence of progressive impairments in hepatic metabolism contributing to type 2 diabetes or liver damage to be determined. The k3/k4 ratio could be a valuable measure of hepatic insulin resistance, which has been shown to correlate with whole-body insulin-stimulated glucose disposal [1]. In addition, the estimation of EGP by FDG plasma kinetics, as validated in pigs [54] and implemented in the human studies described above [30, 47], may corroborate k3/k4 findings and the resulting HGU vs EGP balance.
Plasma NEFA and NAFLD
The observed inability of insulin to suppress the dephosphorylation of FDG (k4) in insulin-resistant individuals could be an indirect effect of circulating levels of NEFAs. Plasma NEFA levels have been shown to be inversely related to net HGU under fasting conditions [55] and to directly impair insulin-stimulated net HGU in healthy men [55], highlighting that plasma NEFAs play a key role in the regulation of HGU. The relationship between high NEFA levels (from unsuppressed adipose tissue lipolysis) and impaired EGP suppression by insulin has been reviewed extensively [48, 49]. Higher NEFA levels in insulin-resistant individuals may stimulate G6Pase activity [56] and could explain the observed lack of suppression of k4 in these individuals [1]. Another effect of elevated plasma NEFA levels in insulin-resistant conditions is the accumulation of intrahepatic lipids (IHLs). Indeed, circulating NEFAs are the main source (60%) of fatty acids accumulating in the liver [57]. High liver fat and NAFLD are strongly related to hepatic insulin resistance [48]. Several studies have shown an inverse association between IHLs and insulin-stimulated HGU [3, 58, 59]. Additionally, interventions such as weight loss through a very low calorie diet [47] reduce IHLs and at the same time improve insulin-stimulated HGU (non-significantly) and EGP suppression by insulin (significantly) in individuals with obesity and without type 2 diabetes (Fig. 3). Likewise, after bariatric surgery the strong decrease in IHL was paralleled by significant improvements in insulin-stimulated HGU and EGP suppression [30]. This correlative evidence suggests a relationship between adipose tissue insulin resistance, IHL content and impaired insulin-stimulated HGU. It would therefore be of value to further investigate the mechanistic interplay between these factors in future studies.
Defect in the ability of glucose to promote its own uptake
HGU is stimulated by both hyperinsulinaemia and hyperglycaemia. Typically, in a clamp, euglycaemia is maintained whereas postprandial insulin levels are induced to specifically understand the role of insulin. As mentioned previously, this approach neglects the important action of hyperglycaemia in the regulation of HGU. A (non-imaging) study by Nielsen et al [60] investigated whether glucose-induced stimulation of HGU was abnormal in type 2 diabetes. To this end, they performed a clamp in which insulinaemia was kept at baseline levels and glucose levels were progressively increased. They found a defect in glucose-stimulated HGU in type 2 diabetes, which was also seen at high insulin levels [60]. Interestingly, the difference in glucose-stimulated HGU between participants with type 2 diabetes and healthy control participants was removed at glucose levels lower than 7.2 mmol/l. To date, studies of this kind using FDG PET have not been reported, but glucose sensitivity should be addressed to comprehensively understand HGU pathophysiology, as disregarding this factor may lead to pathophysiologically important abnormalities in HGU being underestimated or missed.
Conclusion
In conclusion, understanding the unique physiology and metabolic complexity of the liver is essential in designing protocols to measure HGU. In metabolic research, a scanning modality that allows for the provision of detailed kinetic information and influx rates (dynamic imaging) may be preferable to static imaging. The combination of FDG PET and the HEC provides a precise method to measure tissue-specific insulin sensitivity. However, the HEC technique can misinterpret the muscle/HGU balance because of the unnatural route and patterns of insulin and glucose delivery. While the HEC allows for standardised measurement under controlled blood glucose levels, many research questions (other than those focusing on insulin action per se) require a more ‘physiological’ approach, such as oral glucose loading, which has both advantages and disadvantages relating to fluctuations in blood glucose and insulin levels. For selected research questions, the use of a hyperglycaemic–hyperinsulinaemic clamp may be a practical compromise. When analysing the data, one should be aware of possible G6Pase and PPP activity and hence explore whether a simplified Patlak analysis is valid; otherwise, a three-compartment model may be used instead. In addition, choosing the right input function (arterial, portovenous or mixed) should be considered when analysing the data. Overall, after over 20 years of research in this field, the available approaches and technologies to address HGU using FDG PET hold great potential but require more intensive and systematic exploitation to improve our understanding of the mechanisms underlying metabolic diseases and to guide the development of new treatments.
Change history
23 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s00125-024-06112-9
Abbreviations
- EGP:
-
Endogenous glucose production
- FDG:
-
[18F]-2-fluoro-2-deoxy-d-glucose
- G6P:
-
Glucose-6-phosphate
- G6Pase:
-
Glucose-6-phosphatase
- HEC:
-
Hyperinsulinaemic–euglycaemic clamp
- HGU:
-
Hepatic glucose uptake
- IDIF:
-
Image-derived input function
- IHL:
-
Intrahepatic lipid
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- PET:
-
Positron emission tomography
- PPP:
-
Pentose phosphate pathway
- SUV:
-
Standardised uptake value
References
Iozzo P, Geisler F, Oikonen V et al (2003) Insulin stimulates liver glucose uptake in humans: an 18F-FDG PET study. J Nucl Med 44(5):682–689
Johansson E, Lubberink M, Heurling K et al (2018) Whole-body imaging of tissue-specific insulin sensitivity and body composition by using an integrated PET/MR system: a feasibility study. Radiology 286(1):271–278. https://doi.org/10.1148/radiol.2017162949
Rijzewijk LJ, van der Meer RW, Lubberink M et al (2010) Liver fat content in type 2 diabetes: relationship with hepatic perfusion and substrate metabolism. Diabetes 59(11):2747–2754. https://doi.org/10.2337/db09-1201
Keramida G, Peters AM (2017) Fasting hepatic glucose uptake is higher in men than women. Physiol Rep 5(11):e13174. https://doi.org/10.14814/phy2.13174
Thorens B (2015) GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58(2):221–32. https://doi.org/10.1007/s00125-014-3451-1
Agius L, Peak M (1993) Intracellular binding of glucokinase in hepatocytes and translocation by glucose, fructose and insulin. Biochem J 296(Pt 3):785–796. https://doi.org/10.1042/bj2960785
Keramida G, Peters AM (2020) FDG PET/CT of the non-malignant liver in an increasingly obese world population. Clin Physiol Funct Imaging 40(5):304–319. https://doi.org/10.1111/cpf.12651
Ishizu K, Nishizawa S, Yonekura Y et al (1994) Effects of hyperglycemia on FDG uptake in human brain and glioma. J Nucl Med 35(7):1104–1109
Thie JA (1995) Clarification of a fractional uptake concept. J Nucl Med 36(4):711–712
Boersma GJ, Johansson E, Pereira MJ et al (2018) Altered glucose uptake in muscle, visceral adipose tissue, and brain predict whole-body insulin resistance and may contribute to the development of type 2 diabetes: a combined PET/MR study. Horm Metab Res 50(8):627–639. https://doi.org/10.1055/a-0643-4739
Trägårdh M, Møller N, Sørensen M (2015) Methodologic considerations for quantitative 18F-FDG PET/CT studies of hepatic glucose metabolism in healthy subjects. J Nucl Med 56(9):1366–1371. https://doi.org/10.2967/jnumed.115.154211
Sokoloff L, Reivich M, Kennedy C et al (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28(5):897–916. https://doi.org/10.1111/j.1471-4159.1977.tb10649.x
Bender D, Munk OL, Feng H-Q, Keiding S (2001) Metabolites of 18F-FDG and 3-O-11C-methylglucose in pig liver. J Nucl Med 42(11):1673–1678
Iozzo P, Jarvisalo MJ, Kiss J et al (2007) Quantification of liver glucose metabolism by positron emission tomography: validation study in pigs. Gastroenterology 132(2):531–542. https://doi.org/10.1053/j.gastro.2006.12.040
Patlak CS, Blasberg RG, Fenstermacher JD (1983) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 3(1):1–7. https://doi.org/10.1038/jcbfm.1983.1
Patlak CS, Blasberg RG (1985) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab 5(4):584–590. https://doi.org/10.1038/jcbfm.1985.87
Graham MM, Muzi M, Spence AM et al (2002) The FDG lumped constant in normal human brain. J Nucl Med 43(9):1157–1166
Horsager J, Munk OL, Sorensen M (2015) Metabolic liver function measured in vivo by dynamic (18)F-FDGal PET/CT without arterial blood sampling. EJNMMI Res 5:32. https://doi.org/10.1186/s13550-015-0110-6
de Geus-Oei LF, Visser EP, Krabbe PF et al (2006) Comparison of image-derived and arterial input functions for estimating the rate of glucose metabolism in therapy-monitoring 18F-FDG PET studies. J Nucl Med 47(6):945–949
Brooks DC, Black PR, Arcangeli MA, Aoki TT, Wilmore DW (1989) The heated dorsal hand vein: an alternative arterial sampling site. J Parenter Enteral Nutr 13(1):102–105. https://doi.org/10.1177/0148607189013001102
Gaiani S, Bolondi L, Li Bassi S, Santi V, Zironi G, Barbara L (1989) Effect of meal on portal hemodynamics in healthy humans and in patients with chronic liver disease. Hepatology 9(6):815–819. https://doi.org/10.1002/hep.1840090604
Thomsen C, Ståhlberg F, Henriksen O (1993) Quantification of portal venous blood flow during fasting and after a standardized meal — a MRI phase-mapping study. Eur Radiol 3(3):242–247. https://doi.org/10.1007/BF00425902
Zuo Y, Sarkar S, Corwin MT, Olson K, Badawi RD, Wang G (2019) Structural and practical identifiability of dual-input kinetic modeling in dynamic PET of liver inflammation. Phys Med Biol 64(17):175023. https://doi.org/10.1088/1361-6560/ab1f29
Brix G, Ziegler SI, Bellemann ME et al (2001) Quantification of [18F] FDG uptake in the normal liver using dynamic PET: impact and modeling of the dual hepatic blood supply. J Nucl Med 42(8):1265–1273
Kudomi N, Järvisalo MJ, Kiss J et al (2009) Non-invasive estimation of hepatic glucose uptake from [18F]FDG PET images using tissue-derived input functions. Eur J Nucl Med Mol Imaging 36(12):2014–2026. https://doi.org/10.1007/s00259-009-1140-y
Garbarino S, Vivaldi V, Delbary F et al (2015) A new compartmental method for the analysis of liver FDG kinetics in small animal models. EJNMMI Res 5(1):1–9
Vivaldi V, Garbarino S, Caviglia G, Piana M, Sanbuceti G (2013) Compartmental analysis of nuclear imaging data for the quantification of FDG liver metabolism. arXiv:13057435 (Preprint). 31 May. Available from https://doi.org/10.48550/arXiv.1305.7435
Munk OL, Bass L, Roelsgaard K, Bender D, Hansen SB, Keiding S (2001) Liver kinetics of glucose analogs measured in pigs by PET: importance of dual-input blood sampling. J Nucl Med 42(5):795–801
Gu F, Wu Q (2023) Quantitation of dynamic total-body PET imaging: recent developments and future perspectives. Eur J Nucl Med Mol Imaging 50:3538–3557. https://doi.org/10.1007/s00259-023-06299-w
Immonen H, Hannukainen JC, Iozzo P et al (2014) Effect of bariatric surgery on liver glucose metabolism in morbidly obese diabetic and non-diabetic patients. J Hepatol 60(2):377–383. https://doi.org/10.1016/j.jhep.2013.09.012
Wang G, Corwin MT, Olson KA, Badawi RD, Sarkar S (2018) Dynamic PET of human liver inflammation: impact of kinetic modeling with optimization-derived dual-blood input function. Phys Med Biol 63(15):155004. https://doi.org/10.1088/1361-6560/aac8cb
Sarkar S, Corwin MT, Olson KA et al (2019) Pilot study to diagnose nonalcoholic steatohepatitis with dynamic (18)F-FDG PET. AJR Am J Roentgenol 212(3):529–537. https://doi.org/10.2214/AJR.18.20012
Guzzardi MA, Guiducci L, Campani D et al (2022) Leptin resistance before and after obesity: evidence that tissue glucose uptake underlies adipocyte enlargement and liver steatosis/steatohepatitis in Zucker rats from early-life stages. Int J Obes 46(1):50–58. https://doi.org/10.1038/s41366-021-00941-z
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237(3):E214–E223. https://doi.org/10.1152/ajpendo.1979.237.3.E214
Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19(5):608–624. https://doi.org/10.1210/edrv.19.5.0349
Eaton RP, Allen RC, Schade DS (1983) Hepatic removal of insulin in normal man: dose response to endogenous insulin secretion. J Clin Endocrinol Metab 56(6):1294–1300. https://doi.org/10.1210/jcem-56-6-1294
Edgerton DS, Scott M, Farmer B et al (2019) Targeting insulin to the liver corrects defects in glucose metabolism caused by peripheral insulin delivery. JCI Insight 4(7):e126974. https://doi.org/10.1172/jci.insight.126974
Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD (2012) Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr 3(3):286–294. https://doi.org/10.3945/an.112.002089
Farmer TD, Jenkins EC, O’Brien TP et al (2015) Comparison of the physiological relevance of systemic vs. portal insulin delivery to evaluate whole body glucose flux during an insulin clamp. Am J Physiol Endocrinol Metab 308(3):E206–E222. https://doi.org/10.1152/ajpendo.00406.2014
Gastaldelli A, Gaggini M, Daniele G et al (2016) Exenatide improves both hepatic and adipose tissue insulin resistance: a dynamic positron emission tomography study. Hepatology 64(6):2028–2037. https://doi.org/10.1002/hep.28827
Daniele G, Iozzo P, Molina-Carrion M et al (2015) Exenatide regulates cerebral glucose metabolism in brain areas associated with glucose homeostasis and reward system. Diabetes 64(10):3406–3412. https://doi.org/10.2337/db14-1718
Ishida T, Chap Z, Chou J et al (1983) Differential effects of oral, peripheral intravenous, and intraportal glucose on hepatic glucose uptake and insulin and glucagon extraction in conscious dogs. J Clin Investig 72(2):590–601. https://doi.org/10.1172/JCI111007
Knuuti MJ, Nuutila P, Ruotsalainen U et al (1992) Euglycemic hyperinsulinemic clamp and oral glucose load in stimulating myocardial glucose utilization during positron emission tomography. J Nucl Med 33(7):1255–1262
Guzzardi MA, La Rosa F, Campani D et al (2022) Evidence of a gastro-duodenal effect on adipose tissue and brain metabolism, potentially mediated by gut-liver inflammation: a study with positron emission tomography and oral (18)FDG in mice. Int J Mol Sci 23(5):2659. https://doi.org/10.3390/ijms23052659
Iozzo P, Hallsten K, Oikonen V et al (2003) Insulin-mediated hepatic glucose uptake is impaired in type 2 diabetes: evidence for a relationship with glycemic control. J Clin Endocrinol Metab 88(5):2055–2060. https://doi.org/10.1210/jc.2002-021446
Iozzo P, Hallsten K, Oikonen V et al (2003) Effects of metformin and rosiglitazone monotherapy on insulin-mediated hepatic glucose uptake and their relation to visceral fat in type 2 diabetes. Diabetes Care 26(7):2069–2074. https://doi.org/10.2337/diacare.26.7.2069
Viljanen AP, Iozzo P, Borra R et al (2009) Effect of weight loss on liver free fatty acid uptake and hepatic insulin resistance. J Clin Endocrinol Metab 94(1):50–55. https://doi.org/10.1210/jc.2008-1689
Petersen MC, Vatner DF, Shulman GI (2017) Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 13(10):572–87. https://doi.org/10.1038/nrendo.2017.80
Petersen MC, Shulman GI (2018) Mechanisms of insulin action and insulin resistance. Physiol Rev 98(4):2133–2223. https://doi.org/10.1152/physrev.00063.2017
Agius L (2016) Hormonal and metabolite regulation of hepatic glucokinase. Annu Rev Nutr 36:389–415. https://doi.org/10.1146/annurev-nutr-071715-051145
Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL (1995) Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res 27(1):19–22. https://doi.org/10.1055/s-2007-979899
Basu A, Basu R, Shah P et al (2001) Type 2 diabetes impairs splanchnic uptake of glucose but does not alter intestinal glucose absorption during enteral glucose feeding: additional evidence for a defect in hepatic glucokinase activity. Diabetes 50(6):1351–1362. https://doi.org/10.2337/diabetes.50.6.1351
Basu A, Basu R, Shah P et al (2000) Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes 49(2):272–283. https://doi.org/10.2337/diabetes.49.2.272
Iozzo P, Gastaldelli A, Järvisalo MJ et al (2006) 18F-FDG assessment of glucose disposal and production rates during fasting and insulin stimulation: a validation study. J Nucl Med 47(6):1016–1022
Iozzo P, Lautamaki R, Geisler F et al (2004) Non-esterified fatty acids impair insulin-mediated glucose uptake and disposition in the liver. Diabetologia 47(7):1149–1156. https://doi.org/10.1007/s00125-004-1443-2
Lam TK, van de Werve G, Giacca A (2003) Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 284(2):E281–E290. https://doi.org/10.1152/ajpendo.00332.2002
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Investig 115(5):1343–1351. https://doi.org/10.1172/JCI23621
Borra R, Lautamaki R, Parkkola R et al (2008) Inverse association between liver fat content and hepatic glucose uptake in patients with type 2 diabetes mellitus. Metabolism 57(10):1445–1451. https://doi.org/10.1016/j.metabol.2008.05.015
Eriksson JW, Visvanathar R, Kullberg J et al (2021) Tissue-specific glucose partitioning and fat content in prediabetes and type 2 diabetes: whole-body PET/MRI during hyperinsulinemia. Eur J Endocrinol 184(6):879–889. https://doi.org/10.1530/EJE-20-1359
Nielsen MF, Basu R, Wise S, Caumo A, Cobelli C, Rizza RA (1998) Normal glucose-induced suppression of glucose production but impaired stimulation of glucose disposal in type 2 diabetes: evidence for a concentration-dependent defect in uptake. Diabetes 47(11):1735–1747. https://doi.org/10.2337/diabetes.47.11.1735
Honka MJ, Latva-Rasku A, Bucci M et al (2018) Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: a positron emission tomography study. Eur J Endocrinol 178(5):523–531. https://doi.org/10.1530/EJE-17-0882
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
VS-H and JB were supported by a European Research Council Starting Grant (grant no. 759161 ‘MRS in diabetes’).
Authors’ relationships and activities
VS-H is a member of the editorial board of Diabetologia. The authors declare that there are no other relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
All authors were responsible for drafting the article and reviewing it critically for content. All authors approved the version to be published.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Basset-Sagarminaga, J., van de Weijer, T., Iozzo, P. et al. Advances and challenges in measuring hepatic glucose uptake with FDG PET: implications for diabetes research. Diabetologia 67, 407–419 (2024). https://doi.org/10.1007/s00125-023-06055-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-06055-7