
Abstract
Aims/hypothesis
Glucose-dependent insulinotropic polypeptide (GIP) and xenin, regulatory gut hormones secreted from enteroendocrine K cells, exert important effects on metabolism. In addition, xenin potentiates the biological actions of GIP. The present study assessed the actions and therapeutic utility of a (DAla2)GIP/xenin-8-Gln hybrid peptide, in comparison with the parent peptides (DAla2)GIP and xenin-8-Gln.
Methods
Following confirmation of enzymatic stability, insulin secretory activity of (DAla2)GIP/xenin-8-Gln was assessed in BRIN-BD11 beta cells. Acute and persistent glucose-lowering and insulin-releasing effects were then examined in vivo. Finally, the metabolic benefits of twice daily injection of (DAla2)GIP/xenin-8-Gln was determined in high-fat-fed mice.
Results
All peptides significantly (p < 0.05 to p < 0.001) enhanced in vitro insulin secretion from pancreatic clonal BRIN-BD11 cells, with xenin (and particularly GIP)-related signalling pathways, being important for this action. Administration of (DAla2)GIP or (DAla2)GIP/xenin-8-Gln in combination with glucose significantly (p < 0.05) lowered blood glucose and increased plasma insulin in mice, with a protracted response of up to 4 h. All treatments elicited appetite-suppressive effects (p < 0.05), particularly (DAla2)GIP/xenin-8-Gln and xenin-8-Gln at elevated doses of 250 nmol/kg. Twice-daily administration of (DAla2)GIP/xenin-8-Gln or (DAla2)GIP for 21 days to high-fat-fed mice returned circulating blood glucose to lean control levels. In addition, (DAla2)GIP/xenin-8-Gln treatment significantly (p < 0.05) reduced glycaemic levels during a 24 h glucose profile assessment. Neither of the treatment regimens had an effect on body weight, energy intake or circulating insulin concentrations. However, insulin sensitivity was significantly (p < 0.001) improved by both treatments. Interestingly, GIP-mediated glucose-lowering (p < 0.05) and insulin-releasing (p < 0.05 to p < 0.01) effects were substantially improved by (DAla2)GIP and (DAla2)GIP/xenin-8-Gln treatment. Pancreatic islet and beta cell area (p < 0.001), as well as pancreatic insulin content (p < 0.05), were augmented in (DAla2)GIP/xenin-8-Gln-treated mice, related to enhanced proliferation and decreased apoptosis of beta cells, whereas (DAla2)GIP evoked increases (p < 0.05 to p < 0.01) in islet number.
Conclusions/interpretation
These studies highlight the clear potential of GIP/xenin hybrids for the treatment of type 2 diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A defect in the postprandial insulin-secretory incretin response, mediated by the gut hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), is a specific pathophysiological characteristic of type 2 diabetes [1]. The main impairments are recognised as reduced postprandial GLP-1 secretion and defective GIP receptor signalling [1]. The inadequacy in the GLP-1 arm of the incretin effect can be easily overcome through administration of exogenous GLP-1, which significantly amplifies circulating concentrations [2, 3]. In contrast, pharmacological augmentation of circulating GIP levels fails to evoke an effective increase in insulin secretion in patients with type 2 diabetes [4]. As such, it seems unlikely that stand-alone GIP-based drugs would have therapeutic value for type 2 diabetes. Notwithstanding this, strategies to overcome defective GIP action in type 2 diabetes would be of considerable interest.
Near normalisation of blood glucose levels has been shown to restore the insulin-secretory effect of GIP in both animal models of type 2 diabetes [5] and in humans [6] with this condition, providing evidence that defective GIP receptor signalling is reversible. In addition, co-administration of GIP with a sulfonylurea restores pancreatic beta cell sensitivity to GIP [7], although this could be linked to uncoupling of incretin glucose dependency by sulfonylureas [8]. More encouraging, recent studies have highlighted the possibility that xenin, a hormone co-secreted with GIP from a subset of enteroendocrine K cells, could amplify the insulin-secretory response of GIP [9]. In agreement, observations from our laboratory and others confirm the GIP-potentiating effects of xenin under normal and type 2 diabetes conditions [10–13]. Furthermore, there is also evidence to suggest that xenin acts as a satiety hormone in animals [10, 14–17] and humans [18]. As such, therapeutic interventions that combine the biological actions of xenin and GIP, and potentially restore GIP action in type 2 diabetes, would have particularly exciting potential. There has been a recent upsurge in interest focused on generating designer hybrid peptides that can modulate multiple regulatory peptide hormone receptor pathways [19–22].
Successful generation of hybrid peptides has been achieved through fusion of the key bioactive amino acid sequences of the parent peptides [19–22]. This increases the therapeutic applicability of gut-hormone-based drugs by facilitating formulation and dosing with a single molecule, rather than co-injection of separate parent peptide forms. For xenin, the naturally occurring C-terminal fragment, known as xenin-8, retains biological activity at the level of the endocrine pancreas [13, 23]. Moreover, we have also shown that a stable analogue of xenin-8, namely xenin-8-Gln, is biologically active and has a spectrum of beneficial metabolic effects in vitro and in vivo [24]. For GIP, the first 14 N-terminal amino acid residues contain the bioactive domain important for insulin-secretory function [25, 26]. Based on this knowledge, we constructed a novel GIP/xenin hybrid peptide, (DAla2)GIP/xenin-8-Gln, by linking GIP(1-14) to xenin-8-Gln, retaining the regions of each peptide known to be important for biological activity (see electronic supplementary material [ESM] Table 1). Importantly, since GIP is a substrate for dipeptidyl peptidase-4 (DPP-4) [27], the hybrid peptide includes substitution of the naturally occurring alanine l isomer residue by a d isomer at position 2 [28, 29]. The results reveal that GIP/xenin hybrid molecules require further consideration as a treatment option for type 2 diabetes.
Methods
Peptide synthesis
All peptides (ESM Table 1) were purchased from GL Biochem (Shanghai, China; greater than 95% purity). Peptides were characterised in-house using HPLC and matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry (ESM Table 2), as described previously [29].
Assessment of DPP-4 degradation
Peptide (20 μg) DPP-4 degradation profiles (5 μl purified DPP-4, 5 mU; Sigma-Aldrich, UK) were assessed as described previously [29]. Xenin-based peptides have been shown to be resistant to DPP-4 [10, 24].
In vitro insulin secretion
BRIN-BD11 cells were used to assess the insulin-releasing activity of test peptides, as described previously [30]. This hybrid cell line (ECACC 10033003) was originally generated in-house by electrofusion of a primary culture of New England Deaconess Hospital (NEDH) rat pancreatic islets with RINm5F (a cell line derived from an NEDH rat insulinoma), and has been mycoplasma eradicated. For details of experimental conditions please see ESM Methods. Insulin was measured by radioimmunoassay [31]. We assessed the impact of GIP and neurotensin receptors on (DAla2)GIP/xenin-8-Gln-induced insulin secretion in BRIN-BD11 cells. Cells were incubated in 5.6 mmol/l glucose with (DAla2)GIP/xenin-8-Gln (10−6 mol/l) alone or in combination with the GIP receptor antagonist GIP(6-30)Cex-K40[Pal] [32] (where Pal is palmitate) or the neurotensin receptor antagonist SR142948A (Sigma, Poole, UK), and insulin secretion determined as described above. In addition, to determine mechanisms, intracellular Ca2+ was also measured in BRIN-BD11 cells following treatment with test peptides (10−6 mol/l), using a Flexstation scanning fluorometer (FLIPR Calcium 5 assay kit; Molecular Devices, Sunnyvale, CA, USA), as described previously [10].
Animals
Acute and persistent dose-dependent animal studies were conducted in male albino NIH outbred Swiss mice (Envigo, Huntingdon, UK), that were derived from a nucleus colony obtained from the National Institutes of Health, Bethesda, Maryland. Longer-term experiments were conducted with male high-fat-fed NIH Swiss mice. For further details of mice age and maintenance please see ESM.
Acute in vivo effects in lean mice
Specific information on acute feeding, glucose homeostasis and insulin-secretory studies is given in ESM Methods. To assess the impact of GIP and neurotensin receptor activation on (DAla2)GIP/xenin-8-Gln-induced glucose-lowering and insulin-secretory actions in mice, (DAla2)GIP/xenin-8-Gln was administered by i.p. injection (25 nmol/kg body weight) in combination with glucose (18 mmol/kg) in the absence and presence of GIP(6-30)Cex-K40[Pal] and SR142948A (both at 25 nmol/kg), and glucose and insulin were measured as described above. In a final series of experiments, test peptides (25 nmol/kg) or saline vehicle (0.9% wt/vol. NaCl) were injected 4 h before a glucose load (18 mmol/kg) in non-fasted mice and blood glucose and plasma insulin measured as above.
Subchronic in vivo studies in high-fat-fed mice
Twice-daily (09:30 and 17:30 hours) i.p. injections of saline vehicle, (DAla2)GIP or (DAla2)GIP/xenin-8-Gln hybrid (both at 25 nmol/kg) were administered for 21 days in high-fat-fed mice. Energy intake and body weight were monitored daily and non-fasting blood glucose and plasma insulin concentrations were assessed at 3 day intervals. On day 21, a non-fasted 24 h glucose profile was conducted. At the end of the treatment period, i.p. glucose tolerance (18 mmol/kg), biological response to GIP (18 mmol/kg glucose in combination with native GIP [25 nmol/kg], i.p.) and insulin sensitivity (15 U/kg; i.p.) tests were performed. Terminal analysis included measurement of total body fat and lean mass by dual-energy X-ray absorptiometry (DEXA) scanning (Piximus Densitometer; Inside Outside Sales, Fitchburg, WI, USA) and extraction of pancreatic tissue for analysis as detailed below.
Immunohistochemistry
Pancreas tissue was excised, divided longitudinally and either snap frozen for extraction of insulin using acid ethanol (5 ml/g), as described previously [22], or processed for immunohistochemical examination. Immunohistochemistry was performed as described previously [33]. For specific details, please see ESM Methods.
Biochemical analysis
Blood samples were collected from the cut tip on the tail vein of conscious mice into chilled fluoride/heparin glucose micro-centrifuge tubes (Sarstedt, Numbrecht, Germany). Blood glucose was measured directly using a hand-held Ascencia Contour blood glucose meter (Bayer Healthcare, Newbury, UK). Plasma and pancreatic insulin was assayed by a modified dextran-coated charcoal radioimmunoassay [31].
Statistical analysis
Statistical analysis was performed using GraphPad PRISM (La Jolla, CA, USA; version 5). For in vitro, islet histology and pancreatic insulin content studies, all samples were numbered and blinded. For acute and subchronic metabolic tests, there was no blinding. There were no inclusion and exclusion criteria applied. Results are expressed as means ± SEM and data were compared using repeated measures ANOVA followed by the Student–Newman–Keuls post hoc test. Groups of data were considered to be significantly different if p < 0.05.
Results
In vitro studies
In contrast to native GIP, (DAla2)GIP/xenin-8-Gln and (DAla2)GIP remained fully intact when incubated in the presence of DPP-4 for up to 12 h (ESM Table 2). All peptides significantly (p < 0.05 to p < 0.001) stimulated insulin secretion from BRIN-BD11 cells at a concentration of 10−6 mol/l (Fig. 1a, b). Only (DAla2)GIP evoked a significant (p < 0.01 to p < 0.001) increase in insulin release at 10−8 mol/l when compared with respective glucose controls (Fig. 1a, b). As expected, the GIP receptor inhibitor GIP(6-30)Cex-K40[Pal] completely annulled (DAla2)GIP-mediated elevation of insulin secretion, while the neurotensin receptor inhibitor SR142948A had a similar, but less prominent, effect on xenin-8-Gln (Fig. 1c). As such, insulin concentrations were still significantly elevated (p < 0.05) following co-culture of xenin-8-Gln and SR142948A when compared with 5.6 mmol/l glucose control (Fig. 1c). The insulinotropic effect of (DAla2)GIP/xenin-8-Gln was completely inhibited (p < 0.001) by GIP(6-30)Cex-K40[Pal] and partially reversed (p < 0.05) by SR142948A (Fig. 1c). Neither, (DAla2)GIP, xenin-8-Gln nor (DAla2)GIP/xenin-8-Gln had any effect on intracellular Ca2+ concentrations (Fig. 1d).

Effects of peptides on insulin release and intracellular Ca2+ concentrations in BRIN-BD11 cells and on cumulative food intake in lean control mice. (a, b) BRIN-BD11 cells were incubated (20 min) with test peptides in the presence of 5.6 mmol/l glucose (a) or 16.7 mmol/l glucose (b). Light-grey bars, glucose controls; white bars, xenin-8-Gln; dark-grey bars, (DAla2)GIP; black bars, (DAla2)GIP/xenin-8-Gln. (c) Effects of the GIP and neurotensin receptor antagonists, GIP(6-30)Cex-K40[Pal] and SR142948A, respectively, on (DAla2)GIP-, xenin-8-Gln- and (DAla2)GIP/xenin-8-Gln-mediated (20 min) insulin release in BRIN-BD11 cells. White bars, incubation in 5.6 mmol/l glucose alone; black bars, glucose plus GIP(6-30)Cex-K40[Pal] (10−6 mol/l); grey bars, glucose plus SR142948A (10−6 mol/l). (d) BRIN-BD11 cells were incubated with 5.6 mmol/l glucose in the presence of test peptides (10−6 mol/l) and intracellular Ca2+ concentrations were assessed over a 5 min period, with alanine (10 mmol/l) as positive control. AUC (0–300 s) data is shown in the inset. Black circles and light-grey bars, 5.6 mmol/l glucose control; black squares and striped bars, 10 mmol/l alanine; white circles and white bars, xenin-8-Gln; white squares and dark-grey bars, (DAla2)GIP; black triangles and black bars, (DAla2)GIP/xenin-8-Gln. (e–g) Cumulative food intake was measured after i.p. injection of peptides at 25 (e), 100 (f) and 250 nmol/kg (g) in overnight-fasted (18 h) lean control mice. Black bars, saline control; diagonally striped bars, xenin-8; dark-grey bars, xenin-25; horizontally striped bars, xenin-8-Gln; light-grey bars, (DAla2)GIP; white bars, (DAla2)GIP/xenin-8-Gln. Values represent means ± SEM (n = 8). *p < 0.05, **p < 0.01 and ***p < 0.001 compared with respective glucose (a–d) or saline controls (e–g); † p < 0.05 and ††† p < 0.001 compared with incubations in the absence of GIP or neurotensin receptor antagonists, as appropriate
Acute in vivo food-intake studies
At 25 nmol/kg, (DAla2)GIP/xenin-8-Gln induced a significant (p < 0.05) reduction in food intake at 30 min post injection when compared with saline controls (Fig. 1e). None of the peptides had significant appetite-suppressive effects when administered at 100 nmol/kg (Fig. 1f). At a supraphysiological dose of 250 nmol/kg, all peptides except (DAla2)GIP induced significant (p < 0.05 to p < 0.01) reductions in food intake at 180 min post injection (Fig. 1g). In addition, at 250 nmol/kg, the appetite-suppressive effect of xenin was significantly (p < 0.05) superior to control mice at 90 min post injection, while xenin-8-Gln and (DAla2)GIP/xenin-8-Gln also evoked significant (p < 0.05) reductions in food intake at 120 min (Fig. 1g).
Acute and persistent glucose-lowering and insulin-releasing effects in lean mice
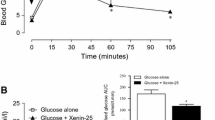
Administration of xenin-8-Gln, (DAla2)GIP or (DAla2)GIP/xenin-8-Gln concomitantly with glucose resulted in significantly (p < 0.05) lowered blood glucose values at 30 min post injection, culminating in significantly (p < 0.05) decreased overall AUC blood glucose values when compared with controls (Fig. 2a). (DAla2)GIP also induced a significant (p < 0.05) reduction in blood glucose levels 15 min post injection (Fig. 2a). Corresponding glucose-induced plasma insulin concentrations were not altered between groups in terms of individual values, although overall glucose-stimulated plasma insulin levels were significantly (p < 0.05) increased in all treatment groups compared with controls (Fig. 2b). The beneficial (p < 0.01) acute glucose-lowering effect of (DAla2)GIP/xenin-8-Gln was blocked by both GIP(6-30)Cex-K40[Pal] and SR142948A (Fig. 2c), whereas only GIP(6-30)Cex-K40[Pal] impaired (DAla2)GIP/xenin-8-Gln-induced insulinotropic actions (Fig. 2d). When administered 4 h prior to a glucose load, xenin-8-Gln was devoid of glucose-lowering or insulin-releasing effects (Fig. 2e, f). However, when administered 4 h previously, both (DAla2)GIP and (DAla2)GIP/xenin-8-Gln significantly reduced the overall AUC for blood glucose (p < 0.05) and increased plasma insulin AUC (p < 0.05) following a glucose challenge (Fig. 2e, f).

Acute and persistent glucose-lowering and insulin-releasing effects of (DAla2)GIP, xenin-8-Gln and (DAla2)GIP/xenin-8-Gln in lean control mice. (a, b) Blood glucose (a) and plasma insulin (b) concentrations were measured before and after i.p. injection of glucose alone (18 mmol/kg) or in combination with peptides (each at 25 nmol/kg) in non-fasted mice. AUCs are shown in insets. (c, d) Overall AUCs for blood glucose (c) and plasma insulin (d) were measured before and after i.p. injection of (DAla2)GIP/xenin-8-Gln. (25 nmol/kg) in combination with glucose (18 mmol/kg) in the absence or presence of GIP(6-30)Cex-K40[Pal] and SR142948A (both at 25 nmol/kg) in non-fasted mice. (e, f) Blood glucose (e) and plasma insulin (f) concentrations were measured following an i.p. glucose load (18 mmol/kg) in non-fasted mice injected with saline vehicle or peptides (each at 25 nmol/kg) 4 h previously. AUCs are shown in insets. Black circles and white bars, glucose alone (a–d) or saline control (e, f); white triangles and dark-grey bars, glucose (a, b) or saline (e, f) in combination with (DAla2)GIP; white squares and light-grey bars, glucose (a, b) or saline (e, f) in combination with xenin-8-Gln; black squares and black bars, glucose (a–d) or saline (c, d) in combination with (DAla2)GIP/xenin-8-Gln; horizontal striped bars, (DAla2)GIP/xenin-8-Gln in combination with GIP(6-30)Cex-K40[Pal]; diagonally striped bars, (DAla2)GIP/xenin-8-Gln in combination with SR142948A. Values represent means ± SEM for 7 or 8 mice. *p < 0.05 and **p < 0.01 compared with glucose alone (a, b) or saline control (c, d); † p < 0.05 and †† p < 0.01 compared with (DAla2)GIP/xenin-8-Gln group
Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on energy intake, body weight, non-fasted blood glucose and plasma insulin in high-fat-fed mice
Twice-daily administration of (DAla2)GIP or (DAla2)GIP/xenin-8-Gln for 21 days to high-fat-fed mice had no significant effect on cumulative energy intake or body weight compared with high-fat-fed controls (Fig. 3a, c). In addition, total body fat and lean mass were also unaltered after the 21 day regimen (Fig. 3b). However, all high-fat fed mice exhibited significantly increased (p < 0.05 to p < 0.001) body weight and cumulative energy intake compared with lean controls (Fig. 3a, c). Non-fasting blood glucose levels progressively declined in (DAla2)GIP/xenin-8-Gln-treated high-fat fed mice over the 21 days, being significantly (p < 0.05 to p < 0.01) reduced on observation days 3, 6 and 15 compared with levels in control high-fat mice (Fig. 3d). Both (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated mice had glucose levels that were not significantly different from lean controls from day 12 onwards (Fig. 3d). Analysis of non-fasting 24 h glucose profile on day 21 revealed that (DAla2)GIP/xenin-8-Gln treated mice had similar blood glucose levels to lean controls at each observation point and significantly (p < 0.05) reduced levels compared with high-fat-fed controls at 17:00 hours (Fig. 3e). Blood glucose levels in (DAla2)GIP-treated mice were also reduced, but not to the same extent as in (DAla2)GIP/xenin-8-Gln mice (Fig. 3e). Circulating plasma insulin levels were generally elevated in all high-fat-fed mice compared with lean controls (Fig. 3f). Mice treated twice daily with either (DAla2)GIP or (DAla2)GIP/xenin-8-Gln did have reduced plasma insulin levels compared with high-fat-fed controls on day 21 but the difference did not reach statistical significance (Fig. 3f).

Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on body weight, body composition, cumulative energy intake, non-fasted blood glucose, 24 h blood glucose profile and non-fasted plasma insulin in high-fat-fed mice. (a, c, d, f) Variables were measured for 3 days before and 21 days during (indicated by black horizontal line) twice-daily treatment with saline vehicle, (DAla2)GIP or (DAla2)GIP/xenin-8-Gln hybrid (each at 25 nmol/kg). (b, e) Total fat and lean mass (b) and 24 h glucose profile (e) were assessed on day 21. Arrows indicate timing of normal twice-daily injections. White squares and light-grey bars, saline control; white circles and dark-grey bars, (DAla2)GIP; black circles and black bars, (DAla2)GIP/xenin-8-Gln; black triangles and white bars, lean control. Values represent means ± SEM for 6–8 mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat-fed controls. † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with lean controls
Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on glucose tolerance and metabolic response to GIP in high-fat-fed mice
Treatment with (DAla2)GIP or (DAla2)GIP/xenin-8-Gln for 21 days reduced overall AUC for blood glucose levels of high-fat-fed mice following a glucose load but the difference failed to reach statistical significance (Table 1). Similarly, glucose-stimulated plasma insulin AUCs were reduced non-significantly by (DAla2)GIP or (DAla2)GIP/xenin-8-Gln treatment compared with high-fat controls (Table 1). All high-fat-fed mice had a significantly (p < 0.05 to p < 0.001) increased glycaemic excursion and overall insulin-secretory response compared with lean controls (Table 1). As illustrated in Table 1, (DAla2)GIP and (DAla2)GIP/xenin-8-Gln treatment significantly (p < 0.05) improved the glucose-lowering and insulin-releasing actions of native GIP; overall blood glucose AUC values were significantly (p < 0.05) decreased by 36% and 30%, respectively, compared with saline-treated control. Moreover, AUCs for glycaemic values were not different when compared with lean controls in response to administration of GIP in combination with glucose (Table 1). Corresponding GIP-induced elevations of plasma insulin concentrations were also significantly (p < 0.05 to p < 0.01) augmented in (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice compared with high-fat-fed controls (Table 1).
Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on insulin sensitivity and pancreatic insulin content in high-fat fed mice
Individual blood glucose levels were reduced, albeit non-significantly, in (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice following administration of exogenous insulin (Fig. 4a). However, the overall glucose-lowering effect of insulin was significantly (p < 0.001) improved in (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated mice compared with control high-fat mice (Fig. 4b). The pancreatic insulin content of (DAla2)GIP/xenin-8-Gln-treated mice was augmented (p < 0.05) compared with that of lean controls but was not significantly different from that of high-fat controls (Fig. 4c). In contrast, the pancreatic insulin content of (DAla2)GIP-treated mice was similar to that of lean control mice (Fig. 4c).

Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on insulin sensitivity and pancreatic insulin content in high-fat-fed mice. Insulin (15 U/kg) was given by i.p. injection (t = 0) in non-fasted mice following 21 days treatment with saline vehicle, (DAla2)GIP or (DAla2)GIP/xenin-8-Gln hybrid (each at 25 nmol/kg). Blood glucose was measured (a), area above the curve (AAC) was calculated using identical baseline subtraction for each group (b) and pancreatic insulin content was measured by RIA (c). White squares and light-grey bars, saline control; white circles and dark-grey bars, (DAla2)GIP; black circles and black bars, (DAla2)GIP/xenin-8-Gln; black triangle and white bars, lean control. Values represent means ± SEM for 6–8 mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat controls; † p < 0.05 compared with lean controls
Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on pancreatic islet histology in high-fat-fed mice
Representative images of pancreatic islets from each mouse group are shown in Fig. 5a–d. High-fat-fed mice had significantly (p < 0.01) increased islet area compared with lean controls (Fig. 5e), giving rise to significant (p < 0.01) increases in both beta and alpha cell areas (Fig. 5f, g). Treatment with (DAla2)GIP had no significant effect on overall pancreatic islet area or beta cell area but resulted in decreased (p < 0.001) alpha cell area compared with high-fat-fed controls (Fig. 5e–g). Similarly, 21 days of twice-daily treatment with (DAla2)GIP/xenin-8-Gln significantly (p < 0.001) reduced pancreatic alpha cell area compared with high-fat-fed controls (Fig. 5g). It also increased (p < 0.001) pancreatic islet and beta cell area compared with lean controls (Fig. 5e, f). The number of islets per mm2 was significantly (p < 0.05 to p < 0.01) augmented in (DAla2)GIP-treated mice compared with all other groups (Fig. 5h). This effect appeared to be related to an increase in number of small-sized islets (Fig. 5i). Thus, high-fat feeding significantly (p < 0.001) decreased the number of small islets and increased the number of large islets, compared with lean controls (Fig. 5i). (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice also had a decreased (p < 0.001) number of small- and medium-sized islets, and fewer (p < 0.001) large islets, when compared with lean controls (Fig. 5i). Representative images of Ki67 and TUNEL immunostained pancreatic islets from each group of mice are shown in Fig. 6a–h. Proliferation and apoptosis rates of pancreatic islet cells appeared to be relatively high in this strain of mouse. The rate of beta cell proliferation in (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice was significantly (p < 0.05 to p < 0.001) increased when compared with the rate in all other groups (Fig. 6i). In addition, the frequency of beta cell apoptosis was significantly (p < 0.01) decreased in (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice compared with high-fat-fed controls, and was similar to levels in lean control mice (Fig. 6j).

Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on pancreatic histology. (a–d) Representative images of islets showing insulin (red) and glucagon (green) immunoreactivity from pancreatic tissues extracted from high-fat-fed saline control mice (a), (DAla2)GIP- (b) and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice (c) and lean control mice (d). (e–i) Variables were assessed using CellF image analysis software after 21 days of twice-daily i.p injections of saline vehicle or peptides (each at 25 nmol/kg) in high-fat-fed mice. Light-grey bars, saline control; dark-grey bars, (DAla2)GIP; black bars, (DAla2)GIP/xenin-8-Gln; white bars, lean control (e–h). White bars, small islets (<10,000 μm2); grey bars, medium islets (10,000–25,000 μm2); black bars, large islets (>25,000 μm2) (i). Values are means ± SEM of 6–8 mice. Scale bar, 50 μm. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat-fed controls. † p < 0.05 and ††† p < 0.001 compared with lean controls; ‡‡ p < 0.01 and ‡‡‡ p < 0.001 compared with the (DAla2)GIP group

Effects of twice-daily administration of (DAla2)GIP and (DAla2)GIP/xenin-8-Gln on beta cell proliferation and apoptosis. (a–d) Representative images showing insulin (green) and Ki67 (red, indicated by arrows) in islets from high-fat-fed saline control mice (a), (DAla2)GIP- (b) and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice (c) and lean control mice (d). (e–h) Representative images showing insulin (red) and TUNEL (green, indicated by arrows) immunoreactivity in islets from high-fat-fed saline control mice (e), (DAla2)GIP- (f) and (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice (g) and lean control mice (h). (i, j) Quantification of beta cell proliferation (i) and apoptosis frequency (j) expressed as a percentage of beta cells analysed. Light-grey bars, saline control; dark-grey bars, (DAla2)GIP; black bars, (DAla2)GIP/xenin-8-Gln; white bars, lean control. Approximately 5000 beta cells per experimental group were counted. Values are means ± SEM of 6–8 mice. Scale bar, 50 μm. **p < 0.01 compared with high-fat-fed controls; ††† p < 0.001 compared with lean controls; ‡ p < 0.05 compared with (DAla2)GIP group
Discussion
Despite encouraging preclinical data and the success of DPP-4 inhibitors [34, 35], progression of enzyme-resistant GIP-based drugs to the type 2 diabetes clinic is lacking. Therefore, in the present study we have evaluated the biological actions and therapeutic applicability of a novel GIP/xenin hybrid peptide, namely (DAla2)GIP/xenin-8-Gln.
In harmony with previous studies [28, 29], (DAla2)GIP and the hybrid peptide (DAla2)GIP/xenin-8-Gln were completely resistant to degradation by DPP-4, indicating that the N-terminal d-Ala2 modification employed effectively masked the DPP-4 binding site. As expected, the parent peptides, (DAla2)GIP and xenin-8-Gln, stimulated insulin release from clonal pancreatic BRIN-BD11 beta cells at physiological and elevated glucose concentrations [24, 29]. To determine the influence of both GIP and xenin pathways on induction of bioactivity by (DAla2)GIP/xenin-8-Gln, we utilised the specific GIP receptor antagonist GIP(6-30)Cex-K40[Pal] [32] and the commercially available neurotensin receptor antagonist, SR142948A. Reassuringly, (DAla2)GIP/xenin-8-Gln was equally effective in vitro as an insulin secretagogue as either of the parent peptides. Its insulinotropic effects appeared to be more dependent on activation of GIP receptor-related cell signalling pathways, when compared with xenin. In full agreement, the acute in vivo insulinotropic actions of (DAla2)GIP/xenin-8-Gln were also chiefly dependent upon activation of GIP receptor pathways. Interestingly, the acute glucose-lowering actions of (DAla2)GIP/xenin-8-Gln appeared to be associated with activation of both GIP and xenin pathways, suggesting possible insulin-independent glucose-lowering effects of xenin [12]. However, the main effect of the xenin component of (DAla2)GIP/xenin-8-Gln likely relates to enhancement of GIP-mediated biological actions, as demonstrated here and previously [10, 11]. Full clarification of the mechanism of (DAla2)GIP/xenin-8-Gln-induced insulin release is necessary, but similar (DAla2)GIP/xenin-8-Gln-induced acute insulin-secretory effects were also noted in lean control mice, confirming biological efficacy. Our previous studies using clonal beta cells have shown that (DAla2)GIP activates adenylate cyclase pathways, whereas xenin-based peptides likely operate through the action of phospholipase C [10]. Further to this, we have shown that neither (DAla2)GIP/xenin-8-Gln nor its parent peptides significantly affected intracellular Ca2+ levels in BRIN-BD11 beta cells.
In accordance with preserved bioactivity of the hybrid peptide, we observed satiety effects of (DAla2)GIP/xenin-8-Gln even at the lowest dose examined and especially at elevated doses. This is in agreement with previous findings suggesting that elevated doses of xenin are required to impart significant appetite-suppressive activity, whereas lower doses have beneficial metabolic effects [10, 13]. The persistence of the metabolic actions of (DAla2)GIP/xenin-8-Gln was then assessed. Even 4 h after a single injection of (DAla2)GIP/xenin-8-Gln or (DAla2)GIP, glucose-lowering and insulin-releasing actions were clearly evident. This presumably relates to enhanced enzymatic stability of (DAla2) modified GIP-forms and corresponds well with the findings of other studies [28, 29]. Development of a specific assay to directly measure (DAla2)GIP/xenin-8-Gln in plasma would be useful to determine the pharmacokinetic profile of the hybrid peptide. Moreover, methodology such as peptide acylation or PEGylation may help to extend the biological half-life of (DAla2)GIP/xenin-8-Gln [29, 36]. Notably, the biological effects of (DAla2)GIP/xenin-8-Gln were equal, or indeed enhanced, when compared with either parent peptide administered at the same dose. This indicates that the hybrid peptide still fully retained the ability to activate GIP- and xenin-related cellular signalling pathways involved in glucose homeostasis, insulin secretion and appetite suppression [13, 29]. Taken together, these data provided a strong basis for the subsequent 21 day twice-daily injection regimen conducted in high-fat-fed mice.
Chronic treatment of high-fat-fed mice with (DAla2)GIP resulted in sustained and significant reductions of circulating blood glucose levels, both in terms of individual observation points and during assessment of a 24 h blood glucose profile [28, 29]. Crucially, (DAla2)GIP/xenin-8-Gln had equal or improved beneficial effects on glycaemic status in high-fat mice. Indeed, reductions in glucose levels were much more apparent in mice treated with (DAla2)GIP/xenin-8-Gln during assessment of the more-in-depth 24 h blood glucose profile. Interestingly, significantly greater reductions in insulin-induced blood glucose levels were observed in (DAla2)GIP/xenin-8-Gln- and (DAla2)GIP-treated mice. This clearly points towards beneficial effects of both treatments that are additional to effects on pancreatic beta cell function. This improvement of insulin action was not a consequence of reduced adipose tissue mass. Thus, it presumably reflects alleviation of glucotoxicity, or the actions of GIP to improve insulin resistance [29, 37]. However, further investigations, including euglycaemic–hyperinsulinaemic clamps, glucose uptake and gene and protein expression studies, are required to fully address the molecular mechanisms underpinning this benefit. Somewhat surprisingly, at the dose tested, (DAla2)GIP/xenin-8-Gln had no effect on energy intake, although similar observations have been reported previously with xenin-based drugs [38]. This limited satiety effect presumably reflects the requirement of supraphysiological doses of xenin to impart appetite-suppressive effects [10]. However, it could also be related to factors such as the palatability of the high-fat diet, the age and strain of mice employed or the duration of the study.
As expected, a key component of the beneficial action of (DAla2)GIP/xenin-8-Gln concerned the stimulation of beta cell function [13, 29]. Circulating and glucose-induced plasma insulin concentrations were significantly elevated compared with those in lean control mice, despite non-fasting glucose levels being similar. Moreover, the pancreatic insulin content was persistently higher in (DAla2)GIP/xenin-8-Gln-treated high-fat-fed mice. In agreement, pancreatic islet and beta cell areas were increased in (DAla2)GIP/xenin-8-Gln-treated mice, in line with the proliferative and anti-apoptotic beta cell actions of (DAla2)GIP/xenin-8-Gln, as has previously been reported for GIP [39, 40]. Consistent with our observations, the positive actions of GIP on islet and beta cell architecture have previously been shown to improve beta cell function and glycaemic control in animal models of type 2 diabetes [41]. Interestingly, there is a recent suggestion that xenin may inhibit GLP-1 secretion under certain circumstances [42]. However, such observations still need to be fully confirmed and the current findings would not indicate any obvious detrimental effects linked to reduced GLP-1 secretion or action by (DAla2)GIP/xenin-8-Gln. Moreover, GIP is known to stimulate GLP-1 secretion from the gut [43] as well as pancreatic alpha cells [44], affording GIP/xenin hybrids a clear advantage over xenin monotherapies. In keeping with improved beta cell dynamics in (DAla2)GIP/xenin-8-Gln-treated mice, the insulin-secretory and subsequent glucose-lowering effects of native GIP were substantially enhanced in these mice. As expected, the biological action of GIP was severely perturbed in high-fat-fed mice, akin to the situation in patients with type 2 diabetes [4]. It would be more than reasonable to link a major part of the positive therapeutic effects of (DAla2)GIP/xenin-8-Gln directly to augmentation of GIP action, as previously observed with other xenin-related compounds [10, 38]. Thus, the main rationale for inclusion of the xenin component of our novel hybrid peptide was essentially to enhance GIP-mediated actions [10, 38]. However, normalisation of blood glucose levels, as clearly demonstrated in both (DAla2)GIP- and (DAla2)GIP/xenin-8-Gln-treated mice, has also been shown to independently restore GIP insulin-secretory function in type 2 diabetes [5, 6]. In addition, we have already confirmed that xenin has significant glucose-lowering activity independent of the GIP receptor [38]. Therefore, further studies are required to systematically assess the direct and indirect beneficial effects of (DAla2)GIP/xenin-8-Gln in restoring GIP action in type 2 diabetes.
Interestingly, both activation and inhibition of GIP receptors has been postulated as a possible treatment option for type 2 diabetes and obesity [45]. Thus, GIP receptor agonism augments beta cell-induced glucose-dependent insulin secretion, akin to the actions of clinically approved GLP-1 mimetics [3]. This approach is very encouraging given that the issue of GIP sensitivity in type 2 diabetes is now being addressed. However, GIP is also believed to play a role in lipid metabolism and fat deposition [45]. Accordingly, GIP receptor antagonism can potentially protect against or even reverse many of the obesity-associated abnormalities of type 2 diabetes [46–48]. Similar to the conundrum regarding the clinical use of GIP receptor agonists or antagonists, both genetic knockout [49] and overexpression [50] of GIP has been shown to protect against obesity and development of diabetes. Taken together, this likely reflects the plasticity of cellular pathways linked to the regulation of energy balance. Nonetheless, it is clear from the current study that novel treatment options aimed at overcoming GIP resistance in type 2 diabetes have therapeutic potential.
In conclusion, the present study has demonstrated that the novel hybrid peptide analogue (DAla2)GIP/xenin-8-Gln has an impressive profile of beneficial metabolic effects in high-fat-fed mice, including improvement of glucose tolerance, insulin resistance and pancreatic islet morphology. Importantly, there was a clear augmentation of the biological action of native GIP in high-fat-fed mice, suggestive of restored GIP effectiveness by (DAla2)GIP/xenin-8-Gln. Further studies are required to fully evaluate the molecular mechanisms and full range of actions of (DAla2)GIP/xenin-8-Gln. However, it is evident that this stable GIP/xenin hybrid may represent an attractive potential new therapeutic for type 2 diabetes.
Abbreviations
- DPP-4:
-
Dipeptidyl peptidase 4
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide 1
- Pal:
-
Palmitate
References
Holst JJ, Knop FK, Vilsbøll T, Krarup T, Madsbad S (2011) Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 34:S251–S257
Zander M, Madsbad S, Madsen JL, Holst JJ (2002) Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 359:824–8230
Drucker DJ (2015) Deciphering metabolic messages from the gut drives therapeutic innovation: the 2014 Banting lecture. Diabetes 64:317–326
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307
Piteau S, Olver A, Kim SJ et al (2007) Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun 362:1007–1012
Højberg PV, Vilsbøll T, Rabøl R et al (2009) Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52:199–207
Meneilly GS, Bryer-Ash M, Elahi D (1993) The effect of glyburide on beta-cell sensitivity to glucose-dependent insulinotropic polypeptide. Diabetes Care 16:110–114
de Heer J, Holst JJ (2007) Sulfonylurea compounds uncouple the glucose dependence of the insulinotropic effect of glucagon-like peptide 1. Diabetes 56:438–443
Althage MC, Ford EL, Wang S, Tso P, Polonsky KS, Wice BM (2008) Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high-fat diet. J Biol Chem 283:18365–18376
Taylor AI, Irwin N, McKillop AM, Patterson S, Flatt PR, Gault VA (2010) Evaluation of the degradation and metabolic effects of the gut peptide xenin on insulin secretion, glycaemic control and satiety. J Endocrinol 207:87–93
Wice BM, Wang S, Crimmins DL et al (2010) Xenin-25 potentiates glucose-dependent insulinotropic polypeptide action via a novel cholinergic relay mechanism. J Biol Chem 285:19842–19853
Chowdhury S, Reeds DN, Crimmins DL et al (2014) Xenin-25 delays gastric emptying and reduces postprandial glucose levels in humans with and without type 2 diabetes. Am J Physiol Gastrointest Liver Physiol 306:G301–G309
Martin CM, Parthsarathy V, Pathak V, Gault VA, Flatt PR, Irwin N (2014) Characterisation of the biological activity of xenin-25 degradation fragment peptides. J Endocrinol 221:193–200
Cline MA, Nandar W, Rogers JO (2007) Xenin reduces feed intake by activating the ventromedial hypothalamus and influences gastrointestinal transit rate in chicks. Behav Brain Res 179:28–32
Cooke JH, Patterson M, Patel SR et al (2009) Peripheral and central administration of xenin and neurotensin suppress food intake in rodents. Obesity (Silver Spring) 17:1135–1143
Kim ER, Lew PS, Spirkina A, Mizuno TM (2016) Xenin-induced feeding suppression is not mediated through the activation of central extracellular signal-regulated kinase signaling in mice. Behav Brain Res 312:118–126
Leckstrom A, Kim ER, Wong D, Mizuno TM (2009) Xenin, a gastrointestinal peptide, regulates feeding independent of the melanocortin signaling pathway. Diabetes 58:87–94
Schiavo-Cardozo D, Lima MM, Pareja JC, Geloneze B (2013) Appetite-regulating hormones from the upper gut: disrupted control of xenin and ghrelin in night workers. Clin Endocrinol (Oxf) 79:807–811
Bhat VK, Kerr BD, Vasu S, Flatt PR, Gault VA (2013) A DPP-IV-resistant triple-acting agonist of GIP, GLP-1 and glucagon receptors with potent glucose-lowering and insulinotropic actions in high-fat-fed mice. Diabetologia 56:1417–1424
Gault VA, Bhat VK, Irwin N, Flatt PR (2013) A novel glucagon-like peptide-1 (GLP-1)/glucagon hybrid peptide with triple-acting agonist activity at glucose-dependent insulinotropic polypeptide, GLP-1, and glucagon receptors and therapeutic potential in high-fat-fed mice. J Biol Chem 288:35581–35591
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Irwin N, Pathak V, Flatt PR (2015) A novel CCK-8/GLP-1 hybrid peptide exhibiting prominent insulinotropic, glucose-lowering and satiety actions with significant therapeutic potential in high-fat fed mice. Diabetes 64:2996–3009
Silvestre RA, Rodríguez-Gallardo J, Egido EM, Hernández R, Marco J (2003) Stimulatory effect of xenin-8 on insulin and glucagon secretion in the perfused rat pancreas. Regul Pept 115:25–29
Martin CM, Parthsarathy V, Hasib A et al (2016) Biological activity and antidiabetic potential of C-terminal octapeptide fragments of the gut-derived hormone xenin. PLoS One 11:e0152818
Hinke SA, Manhart S, Pamir N et al (2001) Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta 1547:143–155
Hinke SA, Manhart S, Speck M, Pederson RA, Demuth HU, McIntosh CH (2004) In depth analysis of the N-terminal bioactive domain of gastric inhibitory polypeptide. Life Sci 75:1857–1870
Deacon CF (2005) What do we know about the secretion and degradation of incretin hormones? Regul Pept 128:117–124
Hinke SA, Gelling RW, Pederson RA et al (2002) Dipeptidyl peptidase IV-resistant [D-Ala(2)]glucose-dependent insulinotropic polypeptide (GIP) improves glucose tolerance in normal and obese diabetic rats. Diabetes 51:652–661
Martin CM, Irwin N, Flatt PR, Gault VA (2013) A novel acylated form of (d-Ala(2))GIP with improved antidiabetic potential, lacking effect on body fat stores. Biochim Biophys Acta 1830:3407–3413
McClenaghan NH, Barnett CR, Ah-Sing E et al (1996) Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electrofusion. Diabetes 45:1132–1140
Flatt PR, Bailey CJ (1981) Abnormal plasma-glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Pathak V, Vasu S, Gault VA, Flatt PR, Irwin N (2015) Sequential induction of beta cell rest and stimulation using stable GIP inhibitor and GLP-1 mimetic peptides improves metabolic control in C57BL/KsJ db/db mice. Diabetologia 58:2144–2153
Vasu S, Moffett RC, Thorens B, Flatt PR (2014) Role of endogenous GLP-1 and GIP in beta cell compensatory responses to insulin resistance and cellular stress. PLoS One 9:e101005
Irwin N, Gault V, Flatt PR (2010) Therapeutic potential of the original incretin hormone glucose-dependent insulinotropic polypeptide: diabetes, obesity, osteoporosis and Alzheimer’s disease? Expert Opin Investig Drugs 19:1039–1048
Irwin N, Flatt PR (2013) Enteroendocrine hormone mimetics for the treatment of obesity and diabetes. Curr Opin Pharmacol 13:989–995
Martin CM, Gault VA, McClean S, Flatt PR, Irwin N (2012) Degradation, insulin secretion, glucose-lowering and GIP additive actions of a palmitate-derivatised analogue of xenin-25. Biochem Pharmacol 84:312–319
Irwin N, McClean PL, Flatt PR (2007) Comparison of the subchronic antidiabetic effects of DPP IV-resistant GIP and GLP-1 analogues in obese diabetic (ob/ob) mice. J Pept Sci 13:400–405
Gault VA, Martin CM, Flatt PR, Parthsarathy V, Irwin N (2015) Xenin-25[Lys(13)PAL]: a novel long-acting acylated analogue of xenin-25 with promising antidiabetic potential. Acta Diabetol 52:461–471
Trümper A, Trümper K, Hörsch D (2002) Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol 174:233–246
Kim SJ, Nian C, Widenmaier S, McIntosh CH (2008) Glucose-dependent insulinotropic polypeptide-mediated up-regulation of beta-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol Cell Biol 28:1644–1656
Widenmaier SB, Kim SJ, Yang GK et al (2010) A GIP receptor agonist exhibits beta-cell anti-apoptotic actions in rat models of diabetes resulting in improved beta-cell function and glycemic control. PLoS One 5:e9590
Sterl K, Wang S, Oestricker L et al (2016) Metabolic responses to xenin-25 are altered in humans with Roux-en-Y gastric bypass surgery. Peptides 82:76–84
Hansen L, Holst JJ (2002) The effects of duodenal peptides on glucagon-like peptide-1 secretion from the ileum. A duodeno–ileal loop? Regul Pept 110:39–45
Timper K, Dalmas E, Dror E et al (2016) Glucose-dependent insulinotropic peptide stimulates glucagon-like peptide 1 production by pancreatic islets via interleukin 6, produced by α cells. Gastroenterology 151:165–179
Irwin N, Flatt PR (2009) Therapeutic potential for GIP receptor agonists and antagonists. Best Pract Res Clin Endocrinol Metab 23:499–512
Irwin N, McClean PL, OʼHarte FP, Gault VA, Harriott P, Flatt PR (2007) Early administration of the glucose-dependent insulinotropic polypeptide receptor antagonist (Pro3)GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia 50:1532–1540
Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR (2007) Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diets. Diabetologia 50:1752–1762
McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR (2007) GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am J Physiol Endocrinol Metab 293:E1746–E1755
Miyawaki K, Yamada Y, Ban N et al (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8:738–742
Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH (2012) GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS One 7:e40156
Acknowledgements
Some of the data were presented as an abstract at the 52nd EASD Annual Meeting in Munich in September 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
All data are available on request from the authors.
Funding
These studies were supported by a University of Ulster Vice-Chancellor Research Scholarship (to AH), University of Ulster Proof of Principle Funding Programme, the European Foundation for the Study of Diabetes and a research grant from Invest Northern Ireland Proof of Concept funding.
Duality of interest
NI, VAG and PRF are named on patents filed by the University of Ulster for exploitation of incretin-based drugs and other peptide therapeutics. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
NI, VAG and PRF conceived the study, participated in the analysis and interpretation of data, drafted the manuscript and revised it critically for intellectual content. AH, MTN, DK and VP participated in the analysis and interpretation of data and drafted the manuscript and revised it critically for intellectual content. All authors approved the final version of the manuscript. AH is the guarantor of this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 95 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hasib, A., Ng, M.T., Gault, V.A. et al. An enzymatically stable GIP/xenin hybrid peptide restores GIP sensitivity, enhances beta cell function and improves glucose homeostasis in high-fat-fed mice. Diabetologia 60, 541–552 (2017). https://doi.org/10.1007/s00125-016-4186-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4186-y